

Abstract
The structure of l-amino acid oxidase (LAAO) from Calloselasma rhodostoma has been determined to 2.0 Å resolution in the presence of two ligands: citrate and o-aminobenzoate (AB). The protomer consists of three domains: an FAD-binding domain, a substrate-binding domain and a helical domain. The interface between the substrate-binding and helical domains forms a 25 Å long funnel, which provides access to the active site. Three AB molecules are visible within the funnel of the LAAO–AB complex; their orientations suggest the trajectory of the substrate to the active site. The innermost AB molecule makes hydrogen bond contacts with the active site residues, Arg90 and Gly464, and the aromatic portion of the ligand is situated in a hydrophobic pocket. These contacts are proposed to mimic those of the natural substrate. Comparison of LAAO with the structure of mammalian d-amino acid oxidase reveals significant differences in their modes of substrate entry. Furthermore, a mirror-symmetrical relationship between the two substrate-binding sites is observed which facilitates enantiomeric selectivity while preserving a common arrangement of the atoms involved in catalysis.
Keywords: l-amino acid oxidase/crystal structure/flavoenzyme/glycosylation/inhibitor complex
Introduction
l-amino acid oxidase (LAAO) is a dimeric flavoprotein first described by Zeller in 1944 (Zeller and Maritz, 1944). It contains non-covalently bound FAD as cofactor, and is present in various species (Curti et al., 1992). LAAO catalyzes the stereospecific oxidative deamination of an l-amino acid substrate to an α-keto acid along with the production of ammonia and hydrogen peroxide via an imino acid intermediate (Scheme 1). LAAO enzymes purified from the venoms of a variety of snake species are the best studied members of this family of enzyme. The enzyme is present at significantly high concentrations in venom and is postulated to be a toxin (Li et al., 1994; Torii et al., 1997). These enzymes exhibit a marked preference for hydrophobic amino acids including phenylalanine, tryptophan, tyrosine and leucine. Although the mode of toxicity of snake venom LAAO is not known, it has been shown that the enzymes from Crotalus adamanteus and Crotalus atrox can associate specifically with mammalian endothelial cells (Suhr and Kim, 1996). Furthermore, snake venom LAAO can induce apoptosis in mammalian endothelial cells, possibly through the production of highly localized concentrations of hydrogen peroxide (Suhr and Kim, 1996, 1999). We recently have determined the amino acid sequence of LAAO from Calloselasma rhodostoma (P.Macheroux, personal communication); this protein shares a high degree of sequence identity with the two other known ophidian LAAOs from C.adamanteus (83%) and C.atrox (83%). Also, these proteins share significant similarity (>30% identity) with the murine interleukin 4-inducible Fig1 protein (Raibekas and Massey, 1998). This protein is predicted to be a flavoprotein homologous to monoamine oxidase (MAO) and may be involved in the allergic inflammatory response (Chu and Paul, 1997).




Scheme 1..
A peculiarity of LAAO from C.adamanteus is the reversible inactivation observed upon freezing (–20°C) or raising the pH to above neutrality (Curti et al., 1968; Coles et al., 1977). Reactivation of freeze- or pH-inactivated enzyme is achieved by heat treatment (37°C) at pH 5. This reversible process is associated with small changes in the absorbance spectrum of the FAD cofactor with no change in the CD spectrum of the protein (Coles et al., 1977). The structural basis for the inactivation/reactivation of C.adamanteus LAAO remains elusive.
The structures of three enzymes functionally similar to LAAO have been determined recently: d-amino acid oxidase (DAAO) from pig kidney (Mattevi et al., 1996; Mizutani et al., 1996; Miura et al., 1997), DAAO from the yeast Rhodotorula gracilis (Umhau et al., 1999) and polyamine oxidase (PAO) from Zea mays (Binda et al., 1999). These structural studies reveal that the overall folds of DAAOs and PAO are similar; however, the mode of substrate entry differs between them.
Here we present the first high-resolution structure of an LAAO. The enzyme was purified from C.rhodostoma (Malayan pit viper) and solved to 2.0 Å resolution. The enzyme has been crystallized in the presence of citrate and o-aminobenzoate (AB), two ligands that are inhibitory to LAAO activity. Analysis of the LAAO structure in the presence of these ligands, and in conjunction with the structures of DAAO and PAO, provides us with valuable insights into: (i) how these enzymes are related in terms of modes of substrate entry; (ii) how the structural factors dictate enantiomeric specificity; and (iii) the relative arrangement of atoms involved in the chemical catalysis within their respective active sites.
Results and discussion
Overall structure
The structure of the LAAO–citrate (CIT) complex from Malayan pit viper has been determined at 2.0 Å resolution by the method of multiple isomorphous replacement (MIR) combined with solvent flipping and 4-fold averaging. This model was then used to determine the structure of the LAAO–AB complex to 2.0 Å resolution by the method of molecular replacement. Table I gives the final refinement statistics for each of the structures. The Ramachandran plot for both complexes shows that 99.3% of the residues fall within favorable regions.
Table I. Refinement statistics.
| Model refinement statistics | LAAO–citrate complex | LAAO–AB complex |
|---|---|---|
| Resolution range (Å) | 50.0–2.0 | 50.0–2.0 |
| Total reflections used in refinement | 139 206 | 283 071 |
| R-factora | 0.185 | 0.205 |
| Rfreeb | 0.210 | 0.225 |
| R.m.s.d. bond lengths (Å) | 0.007 | 0.007 |
| R.m.s.d. bond angles (°) | 1.26 | 1.25 |
| No. of non-hydrogen atoms | ||
| protein | 15 376 | 30 792 |
| FAD | 212 | 424 |
| citrate | 52 | – |
| carbohydrate | 208 | 112 |
| o-aminobenzoate | – | 240 |
| water | 1791 | 2378 |
| Average B-factors (Å2) | ||
| overall | 24.2 | 19.0 |
| protein atoms | 23.1 | 18.9 |
| FAD atoms | 14.0 | 10.4 |
| citrate atoms | 66.3 | – |
| carbohydrate atoms | 53.6 | 37.7 |
| o-aminobenzoate atoms | – | 43.5 |
| water molecules | 32.2 | 21.9 |
aR-factor = ΣΣh||Fobs (h)| – k|Fc(h)||/Σh|Fobs(h)|
bRfree was calculated using a10% randomly selected subset of the total number of reflections.
Each protomer includes the FAD cofactor, the N-linked oligosachharide chains at Asn172 and Asn361, and the ligand (CIT or AB). The composition of the sugars making up the oligosaccharide has been determined and the sequence of glycosylation is being confirmed by NMR analysis (O.Geyer, P.Pawelek, P.Fitzpatrick, K.Kitzing, A.Vrielink, S.Ghisla and P.Macheroux, in preparation). Only the proximal sugar residues within the N-linked oligosaccharides are clearly evident in the electron density and have been included in the model.
LAAO from Malayan pit viper is a dimeric enzyme of 55 kDa per protomer. Each protomer consists of 15 α-helices and 22 β-strands that fold into three well-defined domains (Figure 1). The FAD-binding domain consists of three discontinuous regions of the structure: residues 35–64, 242–318 and 446–486. The main structural feature of this domain is a six-stranded β-pleated sheet sandwiched between three α-helices and a four-stranded β-pleated sheet. The motif makes up the classical nucleotide-binding fold seen in many FAD- and NAD(P)-binding enzymes. The substrate-binding domain is made up of residues 5–25, 73–129, 233–236 and 323–420. Finally, an entirely helical domain is made up of residues 130–230. This domain comprises one side of a funnel-shaped entrance to the active site of the enzyme.
Fig. 1. Stereo ribbon representation of the secondary structure elements for l-amino acid oxidase (produced with the program Molscript; Kraulis, 1991) (A) The functional dimer. The individual protomers are colored red and green. (B) A single protomer of l-amino acid oxidase. (C) Topology diagram. The β-strands are depicted as arrows and the α-helices as cylinders. The strands and helices are numbered in the order in which they appear in the primary sequence. The FAD-binding domain is shown in red, the substrate-binding domain in green and the helical domain in blue. The FAD and citrate molecules as well as the glycosylation sites are shown as a ball-and-stick representation.
The topology of LAAO most closely resembles that of PAO (Binda et al., 1999) with further similarities to p-hydroxybenzoate hydroxylase (Wierenga et al., 1979), cholesterol oxidase (Vrielink et al., 1991), glucose oxidase (Hecht et al., 1993) and DAAO (Mattevi et al., 1996; Mizutani et al., 1996; Umhau et al., 1999). A structure superposition with PAO reveals an r.m.s. deviation of 2.9 Å with 414 Cα atoms included in the alignment. Given the high degree of topological similarity, analysis of the sequence identity between LAAO and PAO is rather low at only 26% (19% for structurally equivalent residues), with the major structural difference localized in the helical domain. In LAAO, this domain comprises six α-helices (α4–α9). Four of these helices correspond to ones found in the PAO structure (Sα1, Sα2, Sα3 and Sα4); however, in PAO, these helices have been identified as part of the substrate-binding domain and do not comprise a separate domain. Furthermore, the relative orientations of these helices are different between the two enzymes. The average temperature factors for the helical domain in both complexes of LAAO are higher than for other domains in the structure, suggesting that the helical domain may be more flexible than other regions of the protein.
Biochemical studies have shown that LAAO is active as a dimer. In the crystal structure of LAAO–CIT we observe four molecules in the asymmetric unit arranged as a dimer of dimers, and in the structure of LAAO–AB the asymmetric unit contains two dimers of dimers. Figure 1A shows the secondary structure representation for a single closely associated dimer in the asymmetric unit. The 2-fold non-crystallographic rotation axis relating the two protomers with a single dimer results in positioning of the isoalloxazine rings of the two FAD molecules along a single plane, with the remaining portions of the prosthetic group extending in opposite directions. Furthermore, the entrance to the active site for each protomer is located on opposite surfaces of the dimer.
Studies of LAAO have shown that the enzyme is inactivated reversibly upon freezing or increasing the pH to above neutrality, and reactivation can be achieved by heat treatment at pH 5.0 (Wellner, 1966). The LAAO enzyme that was crystallized had been frozen and thawed prior to crystallization and thus was in the inactive form. However, crystals of the enzyme were obtained at pH 4.5, suggesting that the protein may have been reactivated during the course of the crystallization experiment. In order to verify whether the crystallized enzyme was active, crystals dissolved in 50 mM Tris buffer (pH 7.4) were assayed and the solubilized enzyme was shown to be active (data not shown). In addition, crystals were transferred to a mother liquor solution (at pH 7.4) containing 10 mM phenylalanine. Visual observation of these crystals revealed cracking and a color change from yellow to colorless and back to yellow again within 40 min, indicating a change in the redox state of LAAO-bound FAD within the crystal. A similar experiment, conducted in acetate-buffered mother liquor (pH 4.5), yielded similar observations, however at a substantially slower rate. The enzyme crystallized at pH 4.5 is therefore in the active form. Furthermore, the bleaching experiments support our data suggesting that the catalytic efficiency of the enzyme is significantly lower at pH 4.5 and that a conformational change upon substrate binding may occur, resulting in crystal cracking.
Glycosylation sites
The protein used for crystallization and subsequent structure solution was that isolated from the native source (snake venom) and is glycosylated. Indeed, studies by Macheroux and co-workers have shown that the enzyme contains up to 3.7 kDa of glycosylation per protomer (Macheroux et al., 1999). LAAO from C.adamanteus has also been shown to be highly glycosylated (de Kok and Rawitch, 1969). From the electron density map of the LAAO–CIT complex, two N-glycosylation sites have been identified in agreement with biochemical analysis: Asn172, located in the loop region between helices α5 and α6 of the helical domain, and Asn361, located in the loop region between β18 and β19 of the substrate-binding domain (Figure 1B).
Inspection of the glycosylation sites within the dimer reveals that the carbohydrates connected to Asn172 lie along a single molecular surface (the distance between the sugar moieties is ∼35 Å) whereas those connected to Asn361 lie on opposite surfaces of the dimer (separated by ∼75 Å). In the case of Asn172, three carbohydrate residues are visible in the electron density and have been included in the model: C1 of N-acetylglucosamine (Nag523) is linked to the amide nitrogen of Asn172. This Nag has a further two sugar molecules connected to it: a second Nag (Nag524) in a βO4–C1 linkage and a fucose molecule (Fuc525) in an αO6–C1 linkage.
Chemical studies suggest that the composition and branching of the sugar chains at Asn172 and Asn361 are similar (O.Geyer, P.Pawelek, P.Fitzpatrick, K.Kitzing, A.Vrielink, S.Ghisla and P.Macheroux, in preparation). The electron density is of sufficient definition at Asn361 to reveal only a single Nag molecule (Nag522), but small amounts of positive difference electron density are seen near to O4 and O6 of Nag522, indicating branching similar to that observed at Asn172. The structure of PAO also shows glycosylation; however, in contrast to LAAO, only a single glycosylation site (Asn77) is present. Superposition of the structures indicates that the glycosylation sites are located at different positions in the overall structures of LAAO and PAO. However, the sequence of the first three carbohydrates at Asn172 in LAAO and Asn77 in PAO are identical, but with differing linkages.
FAD interactions
The FAD prosthetic group is buried deeply within the enzyme and makes extensive interactions with protein atoms and with conserved water molecules. The FAD adopts a conformation similar to that seen in a number of other FAD enzymes. The interactions made by the FAD are shown for a single protomer in Figure 2A. All protomers for both complexes show identical interactions between the protein and the prosthetic group. In addition, the water molecules involved in hydrogen bond interactions are structurally conserved for the protomers in the asymmetric unit. Of the residues involved in side chain interactions with FAD, three are identical in PAO (these residues are Glu457–Glu430, Arg71–Arg43 and Glu63–Glu35 coinciding to LAAO–PAO).


Fig. 2. Stereoview of the LAAO–citrate complex in the region of (A) the FAD prosthetic group and (B) the citrate ligand. The protein main chain is shown as a green coil, and specific amino acid residues and the FAD molecule are depicted as ball-and-stick models with yellow and gray bonds, respectively. Water molecules are represented as red spheres. The hydrogen bond contacts are shown as black dashed lines.
The isoalloxazine ring is positioned at the interface between the FAD-binding domain and the substrate-binding domain in a similar fashion to that observed for a number of FAD-binding enzymes. The dimethylbenzene ring is surrounded by a number of hydrophobic residues (Ile374, Trp420 and Ile430) (Figure 2A). An identical environment is observed in the structure of PAO with amino acid conservation seen for Ile374 (Thr350 in PAO), Trp420 (Trp393 in PAO) and Ile430 (Phe403 in PAO).
In the structure of PAO, Mattevi and co-workers have identified a water molecule interacting with N5 of the isoalloxazine ring and with the side chain amino group of a lysine residue (Lys300). They have suggested that this water molecule may be important in the hydrolytic attack on the imino intermediate. In the structure of LAAO, we also see a water molecule (Wat605) hydrogen bonded to N5 as well as to the side chain amino group of Lys326 in the β16 strand of the substrate-binding domain. A comparison of the sequences of LAAO and PAO around this conserved lysine residue reveals significant identity (TKIFL) along the β16-strand. Furthermore, the environment surrounding the side chain of Lys326 reveals a large number of hydrophobic residues (see Figure 2A) (Phe328, Ile370, Tyr372, Tyr356, Met89 and Leu86) similar to that observed around Lys300 in PAO. This suggests that the Lys326 may have a decreased pKa and may act as a base to increase the nucleophilicty of Wat605 for attack on the imino intermediate. Neither a water molecule nor a lysine residue is observed at the identical positions in the structures of DAAO. Thus, if hydrolysis of the imino acid was to be mediated by the protein, this might suggest that in LAAO and DAAO it occurs through different mechanisms. Indeed, with DAAO, hydrolysis is thought to occur non-enzymatically.
LAAO–CIT complex
The active site of the enzyme is located at the base of a long funnel extending 25 Å from the surface into the interior of the protein. The entrance to the funnel is bounded by the side chains of residues within four α-helices in the helical domain: α4, α5, α8 and α9, as well as residues in the β6 strand. The wide entrance to the funnel is filled with water molecules and gradually narrows to a constricted region located ∼15 Å into the interior of the molecule and formed by the side chains of His223 and Glu209. This constriction is not complete since a continuous solvent-accessible channel is observed from the surface to the active site. However, the opening is not large enough to allow the passage of either substrate or α-keto acid product. Interior to the constricted region of the funnel, near to the N5 of the isoalloxazine ring of FAD, a widened internal cavity constitutes the substrate-binding site.
Inspection of the residual electron density in the active site region as well as the crystallization conditions for the original LAAO structure indicate that a citrate molecule is positioned ∼17 Å from the external surface of the protein, blocking the entrance to the active site. Extensive hydrogen bond interactions are made between the citrate and protein side chains and with water molecules located in the active site cleft as shown in Figure 2B; however, there are no interactions between citrate and the FAD cofactor. In all four protomers, the temperature factors for the citrate molecules are high (average 70 Å2) and extensive residual difference electron density indicates that the citrate molecules are not tightly bound to the enzyme and are highly mobile. Steady-state kinetic studies performed on LAAO indicate that citrate is an inhibitor at pH 4.6 but does not inhibit the enzyme at pH 7.4 (data not shown). At pH 4.5, its balance of charge is less negative than at physiological pH which may facilitate more favorable contacts with LAAO. Increasing the pH will result in deprotonation of the ligand and will adversely affect its binding in the active site of the enzyme.
Extending from the main cavity where the citrate molecule is bound is a pocket filled with water molecules and bounded by helix α9 and the loop between β22 and α15. An alternative entrance to this pocket, from the external surface of the molecule, is restricted by the N-terminal region of the structure, specifically the main chain of residues 10–14. In PAO, this region of the structure forms one of the entrances to the U-shaped tunnel. The N-terminal region of LAAO is not present in the structure of PAO and thus the tunnel is only open at one end. In LAAO, this N-terminal region is held to the helical domain by hydrogen bond interactions between the side chain of Asn5 and Asp225 as well as van der Waals interactions between the side chain of Phe11 and those of Leu221, and the alkyl portion of Lys222 along the α9 helix.
LAAO–AB complex
AB is an inhibitor of C.adamanteus LAAO (de Kok and Veeger, 1968). Through preliminary steady-state kinetic assays, we have confirmed that AB is also a mixed inhibitor of C.rhodostoma LAAO at pH 4.6 (data not shown). We were able to obtain crystals of the complex of LAAO with AB, which grew in a crystal form different from those of the original LAAO–CIT crystals. The change in cell dimensions results in a doubling of the asymmetric unit such that four dimers are present instead of two as in the case of the LAAO–CIT crystals. The structure of the LAAO–AB complex was determined using molecular replacement techniques and refined to 2.0 Å resolution (Table I). The LAAO–AB structure shows the ligand bound in three discrete positions within each of the eight protomers in the asymmetric unit (Figure 3A) (AB1, AB2 and AB3). A comparison of the temperature factors of AB1, AB2 and AB3 averaged across the eight protomers in the asymmetric unit indicates that AB1 has the most well ordered position since its Bavg is 35.7 Å2 compared with 43.3 Å2 for AB2 and 41.1 Å2 for AB3. This is consistent with both the well-defined electron density and the increased number of contacts observed for AB1. Although the density is weaker for AB2 and AB3, one can still clearly model the ligand into the density and orient both the carboxylate moiety and the amino group with confidence in all eight protomers within the asymmetric unit. Interestingly, the recently solved structure of yeast DAAO (yDAAO) indicates the presence of two AB molecules bound within its active site funnel (Umhau et al., 1999). The yDAAO funnel is substantially shallower than that seen for LAAO, and could not accommodate a third AB molecule.
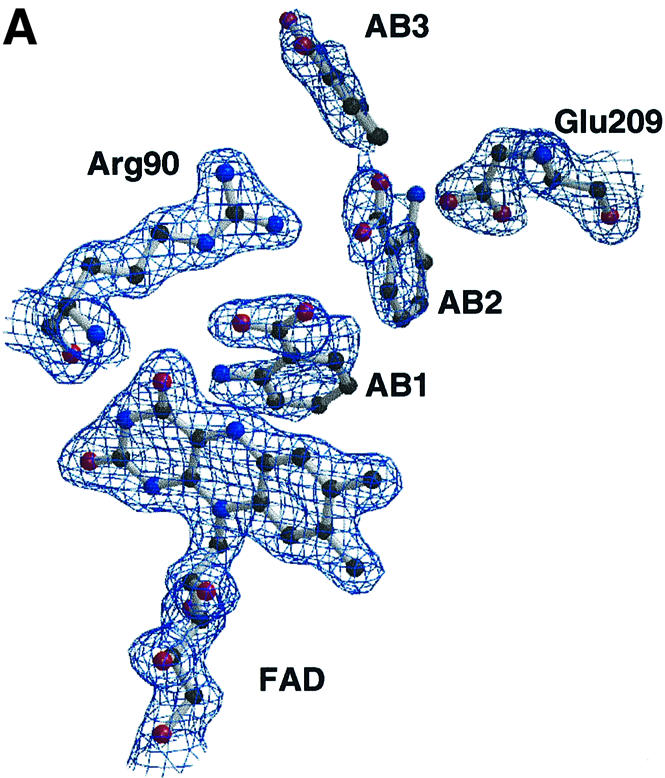


Fig. 3. (A) Electron density in the region of the active site of the LAAO–AB complex. Some protein side chains, the FAD and the three AB positions are shown. The electron density is from a 2Fo – Fc map, averaged over eight protomers in the asymmetric unit, and contoured at 1.0σ. (B) Molecular surface representation of the LAAO–AB complex with the electrostatic potential mapped between –15.0 kT (red) and +15.0 kT (blue). The protein main chain is shown as a colored tube with blue as the helical domain, green as the substrate-binding domain and red as the FAD-binding domain. The AB and FAD molecules are represented as capped sticks. A magnified view of the funnel and the active site containing the AB ligands is shown in the inset. (C) Stereo representation of the interactions between the protein and the three AB ligands in the LAAO–AB complex. The protein main chain regions are represented as a green coil. Amino acid residues, the FAD and the AB molecules are shown in yellow, gray and cyan bonds, respectively. The alternative conformation of His223 is colored magenta. Hydrogen bonds are depicted as black dashed lines.
In the LAAO–AB structure, the positions of the three bound AB molecules suggest the trajectory taken by the substrate from the surface of the molecule to the active site (Figure 3B). The orientation of the AB molecules appears to be determined by the electrostatics of the funnel. The surface closest to the carboxylates of AB1, AB2 and AB3 is uniformly electropositive, whereas the surface most proximal to the amino groups of the ligands is predominantly electronegative in character. The three AB molecules make contacts with a number of residues within the funnel and the active site (Figure 3C). The outermost ligand, AB3, lies ∼10 Å within the funnel between helices α5, α8 and α9 and makes van der Waals contacts with side chain residues within these three helices. Further stabilization at this position is due primarily to an ionic interaction between the amino group of AB3 and the carboxylate of Asp224. AB2 lies 5.5 Å closer to the active site than AB3. The AB2 carboxyl group is within 3.7 Å of the imidazole portion of His223, and the amino group makes contact with the carboxyl group of Glu209; the side chains of both of these residues protrude from the lining of the active site funnel. The most well defined of the three inhibitors, AB1, lies within the active site nearest to the isoalloxazine ring with C1 positioned 3.9 Å from N5 of FAD. A salt bridge exists between Arg90 and the carboxylate moiety of AB1. The AB1 carboxylate is also stabilized by a hydrogen bond with the side chain hydroxyl group of Tyr372. Finally, the AB1 amino group is within hydrogen bonding distance of the backbone carbonyl oxygen atom of Gly464. In addition, His223 in the LAAO–AB complex can adopt an alternative conformation to that seen in the LAAO–CIT complex (Figure 3C). This indicates that the active site of the enzyme has space to accommodate alternative positions for the imidazole side chain of His223, which may be required for catalysis.
Comparison of the orientations of the three AB molecules reveals that the carboxylate group for each ligand points in the same direction, upward towards the funnel entrance to the active site, and the aromatic ring is directed downwards to the ribityl moiety of FAD. In AB1, the aromatic ring is sandwiched between the side chains of Ile430 and Ile374. The orientations of AB2 and AB3 are similar; however, that of AB1 is different, with the benzyl ring rotating by 108° relative to that of AB2 such that the amino group is positioned over the pyrimidine portion of the isoalloxazine ring. This orientation places the amino group within hydrogen bonding distance of the carbonyl oxygen of Gly464, at the N-terminus of the C-terminal helix in the structure. Interestingly, comparisons of the protein–ligand contacts seen in LAAO, DAAO and PAO all reveal a similar interaction of the ligand amino nitrogen atom with the carbonyl oxygen atom of a conserved glycine residue at the N-terminus of the final α-helix in the structures. Apart from the above-mentioned hydrogen bond interaction, the amino group makes no further contacts with the protein.
Structural comparisons with d-amino acid oxidase
LAAO specifically catalyzes the oxidative deamination of l-amino acids; the enzyme DAAO oxidizes d-amino acids. Two crystal structures of DAAO have been determined in the presence of AB (Mattevi et al., 1996; Umhau et al., 1999) as well as a reduced complex with the reaction product imino tryptophan (Todone et al., 1997). The structure of the LAAO–AB complex allows a detailed comparison of these two enzymes, revealing factors that determine their enantiomeric specificity. The overall structures of LAAO and porcine DAAO (pcDAAO) show some similarity in particular in the FAD-binding domain. Both enzymes also contain a large β-pleated sheet in the substrate-binding domain; however, the orientation of the sheet is significantly different between the two structures. Furthermore, pcDAAO lacks the helical domain observed in LAAO. Rather, the entrance to the active site of DAAO is formed by a loop termed the ‘active site lid’, which is present in a rudimentary form in the yDAAO structure (Umhau et al., 1999).
In the pcDAAO–AB complex structure, the aromatic ring of the inhibitor is sandwiched between the isoalloxazine ring of FAD and the side chain of Tyr224. The carboxylate moiety of the ligand is oriented towards the ribityl chain of FAD; it forms a salt bridge with the guanidinium group of Arg283 and makes a further hydrogen bond with Tyr228. A hydrophobic environment is observed around the aromatic ring of the ligand, with the side chains of Leu51, Ile215 and Ile230 forming van der Waals contacts. In the structure of the complex of LAAO–AB, the carboxylate group of the AB1 inhibitor points away from the ribityl moeity of FAD and forms a salt bridge with Arg90. The aromatic ring is surrounded by the hydrophobic side chains of Val374, Ile430 and Trp465. Superposition of the FAD cofactors of LAAO and pcDAAO indicates that the respective AB molecules are roughly co-localized without superposition of the C1 atoms of the ligand. In addition, the atoms from those residues possibly involved in abstraction of a proton from the amino group of the substrate during hydride transfer (see below) do not superimpose (Figure 4A). The carbonyl oxygens from Gly464 in LAAO and Gly313 in pcDAAO, which are involved in binding of the ligand amino group, co-localize, consistent with the approximate superposition of the AB amino groups.



Fig. 4. (A) Stereo view of the active sites of LAAO and porcine DAAO (pcDAAO) with the respective FAD cofactors superimposed along the isoalloxazine ring system. (B) Stereo view of the active sites of LAAO and the mirror-image pcDAAO with the AB ligands superimposed. Atoms and bonds are represented as capped sticks, with those corresponding to DAAO being shown in green and those from LAAO shown in red. The modeled His223 rotamer optimally situated to abstract a proton from the amino group from the substrate is shown in cyan. Superimposed AB molecules are shown in magenta. The mirror plane co-incident with the catalytic axis is shown in gray and those atoms involved in catalysis that lie along the mirror plane are enlarged. (C) Stereo representation of the modeled l-phenylalanine substrate within the active site. Amino acid residues, the FAD and l-phenylalanine molecules are shown in yellow, gray and cyan bonds, respectively. Hydrogen bonds are depicted as black dashed lines.
A mirror-symmetrical relationship has been shown to exist between pcDAAO and flavocytochrome B2 such that a mirror plane coincides with the superimposed isoalloxazine ring systems of the enzymes. This results in postioning of the active site residues on the respective Re and Si faces of FAD (Mattevi et al., 1996). Given that LAAO and DAAO catalyze identical reactions but with mirror-symmetrical substrates, we were interested in comparing the arrangement of catalytic atoms between these two enzymes. Two possibilities exist to reverse enantiomeric specificity. First, the orientation of the substrate and functional groups can be mirrored through the plane of the isoalloxazine ring. This has been real ized upon comparison of DAAO with the subclass of α-hydroxy acid-oxidizing flavoenzymes (Mattevi et al., 1996) such as flavocytochrome b2 (Xia and Mathews, 1990) and glycolate oxidase (Lindqvist, 1989). Secondly, substrate binding can occur on the same face of the isoalloxazine ring. In this case, the groups involved in substrate binding are mirrored through a plane perpendicular to the isoalloxazine ring. As discussed below, the second case is observed upon comparison of LAAO and DAAO. In order to superimpose the tetrahedral geometry of the enantiomeric substrates, we generated a mirror-image pcDAAO–AB structure (henceforth referred to as pcDAAO′/AB′) through the application of an inversion matrix. Superposition of the ABLAAO and AB′pcDAAO′ molecules results in overlap of the residues involved in binding the carboyxlate (Arg90 and Arg283 in LAAO and DAAO′, respectively) and those involved in hydrophobic contacts with the aromatic portion of AB (Ile430 and Leu51 in LAAO and DAAO′, respectively) (Figure 4B). More importantly, the superposition of AB ligands in LAAO and DAAO′ clearly reveals the existence of a mirror plane coincident with the catalytic axis (substrate Cα–FAD N5) and perpendicular with the isoalloxazine ring system of FAD. This suggests that the active sites of LAAO and DAAO have evolved with a high degree of enantiomeric conservation.
In the structure of pcDAAO, no functional group has been identified that might assist in base-catalyzed hydride transfer, and Mattevi et al. (1996) have suggested that the reductive half reaction involves a direct transfer of the substrate α-hydrogen to the flavin. However, the superposition of LAAO and pcDAAO′ reveals that the hydroxyl group of Tyr224 (in pcDAAO′) and NE2 of a His223 (in LAAO) rotamer, positioned between the two observed conformations, lie in close proximity to one another. Furthermore, these atoms also lie along the catalytic mirror plane (Figure 4B). This suggests the possibility that pcDAAO may utilize Tyr224 to assist hydride transfer similarly to what we propose for the role of His223. Indeed, all of the atoms which appear to be involved in the catalytic reaction lie on the mirror plane and those which are involved in binding the different enantiomeric substrates lie off of the plane. We performed the identical superposition using the structure of yDAAO (not shown). A similar mirror-plane relationship between yDAAO′ and LAAO is observed; however, a suitable base positioned along the mirror plane is not present.
Model for substrate entry and catalysis
The presence of three occupied AB subsites in the structure of the LAAO–AB complex provides us with a rare opportunity to postulate a model for the trajectory of the l-amino acid substrate as it traverses the tunnel towards the active site. Upon entrance into the funnel, the substrate will orient itself according to the electrostatic characteristics of the funnel walls; specifically an electropositive surface guides the carboxyl group whereas an electronegative surface, including residues Asp224 and Glu209 which contact AB3 and AB2, respectively (Figure 3C), guides the amino group. The carboxylate group of AB1 probably binds in a similar orientation to that of the natural amino acid substrate, held in position by Arg90. Furthermore, the C1 atom of AB1 is proximal to N5 of the isoalloxazine ring and mimics Cα of the substrate. This is consistent with the proposed hydride transfer mechanism from the αC-H to N5 of FAD. The amino group of AB1 is not expected to mimic exactly the position of the amino group of the substrate. Finally, the position of the benzoate ring between the side chains of Ile430 and Trp465 correlates well with the preference of the enzyme for hydrophobic amino acid substrates.
Using the AB inhibitor as a template for the substrate, we modeled l-phenylalanine into the active site of the enzyme (Figure 4C). In this model, the hydrogen atom on the α carbon of the substrate is positioned such that it points toward the isoalloxazine ring enabling reduction of FAD via either a hydride transfer mechanism or a radical-based mechanism. The modeled substrate with a tetrahedral center at the α carbon atom places the carboxylate group in an ideal position for a strong salt bridge interaction with the guanidinium group of Arg90. In fact, the hydrogen bond distances are shorter in the model than we observe in the actual structure of the AB complex, probably due to the fact that the inhibitor is a planar molecule and therefore does not exactly mimic the tetrahedral substrate. This orientation of the substrate places the amino group near to the position of C2 of the benzoate ring in AB1. Due to differences in hybridization states between inhibitor and the natural substrate, we predict the substrate amino group to lie between C2 and NH2 of AB1. This position of the substrate/product amino/imino groups supports its role in stabilizing the negative charge developing on the pyrimidine moiety of the reduced FAD during hydride transfer (Umhau et al., 1999). Furthermore, the phenylalanine is positioned between the side chains of Ile374 and Ile430, as suggested by the binding mode for the benzoate ring in the AB complex (Figure 4C).
The catalytic mechanism for LAAO may involve a base for abstraction of a proton from the amino group of the substrate and transfer of a hydride from the α carbon to N5 of the isoalloxazine ring of FAD to form the imino intermediate. In order for such a base-catalyzed mechanism to occur, the Michaelis complex must contain the zwitterionic form of the substrate with the amino group situated proximally to a potential base. Alternatively, if the Michaelis complex contains the anionic substrate, hydride transfer would probably occur without the assistance of a base. This scenario has been proposed by Porter and Bright at high pH (>7) where the substrate would be anionic (Porter and Bright, 1980). In the structure of yDAAO, a potential basic residue is not present proximal to the AB-binding site (Umhau et al., 1999), suggesting that hydride transfer occurs without the assistance of a base. In this case, the pH of the active site environment must be sufficiently high to enable the substrate to bind in the anionic form. In contrast, the structures of LAAO and pcDAAO suggest possible basic residues (His223 and Tyr224, respectively) that lie along the catalytic mirror plane (Figure 4B). This observation supports the presence of a zwitterionic substrate in the Michaelis complex as has been suggested by Fitzpatrick and Massey for pcDAAO (Fitzpatrick and Massey, 1982).
The second step in amino acid conversion (Scheme 1) involves attack of a water molecule on the α carbon center of the imino acid intermediate. For DAAO, there is no indication that this step is enzyme mediated; however, as discussed above, both PAO and LAAO show a structurally conserved water molecule held in position near N5 of the FAD and within hydrogen bonding distance of a lysine residue (Lys300 in LAAO and Lys326 in PAO). This structural conservation of the water molecule, the lysine residue and the hydrophobic environment surrounding the lysine side chain suggests a possible active role for the protein in promoting the hydrolysis reaction resulting in deamination in PAO and LAAO. Further studies will be required to confirm the role of the protein in this stage of the reaction in LAAO.
In conclusion, these observations suggest that although the structural frameworks of members of the PHBH family (Mattevi, 1998) are similar, they can be subclassified on the basis of modes of substrate entry and arrangement of active site residues. In the cases of LAAO and DAAO, divergent evolution of the active site has occurred such that the positions of essential residues needed for substrate binding and orientation have evolved to select one enantiomer over the other while the spatial arrangement of atoms needed for catalysis is conserved.
Materials and methods
Protein purification and crystallization
LAAO from C.rhodostoma was purified from isolated snake venom as outlined by Ponnudurai et al. (1994). The protein was dialyzed into 50 mM Tris–HCl pH 7.5 and concentrated to 8 mg/ml using a Centricon 10 (Amicon Inc.). Protein concentrations were determined by Bradford assay using bovine serum albumin as the protein standard.
Crystallization conditions for the native enzyme were screened by the hanging drop vapor diffusion technique. Initial trials were carried out using a sparse-matrix screen, described by Jancarik and Kim (1991). Spherulite-type crystals were obtained from polyethylene glycol (PEG) Mr 4000, ammonium acetate and sodium citrate pH 4.1. Further refinement of these initial conditions resulted in small single crystals from 18.5% PEG4K, 75 mM ammonium sulfate, 100 mM sodium citrate pH 4.5 and 10% glycerol. These small crystals, which appeared within 2–7 days, were used for macroseeding by transferring to a solution containing a mixture of equal volumes of fresh protein at 8 mg/ml and a dissolving mother liquor, 16% PEG4K, 75 mM ammonium sulfate, 100 mM citrate pH 4.5 and 10% glycerol. After 3 days of equilibration against the initial precipitant solution, large rectangular crystals grew to an average size of 0.3 mm×0.16 mm×0.1 mm. The native crystals belong to space group P21 with unit cell dimensions of a = 79.49 Å, b = 159.50 Å, c = 102.95 Å and β = 109.5°. The solvent content, assuming two homodimers of 110 kDa in the asymmetric unit, was calculated to be 45%.
The LAAO–AB complex was crystallized using conditions identical to those for the native enzyme except that 50 mM AB was added to the protein solution prior to setting up the crystallization experiments. The crystals obtained from these conditions are of space group P21 with cell dimensions a = 77.55 Å, b = 137.18 Å, c = 212.63 Å and β = 105.63°. This change in cell dimensions results in an asymmetric unit with four dimers present and a solvent content of 43%.
Data collection and structure determination
Crystals were transferred briefly into a drop of the cryoprotectant, Paratone 8277 (Exon Oil), mounted in a rayon loop (Hampton Research Co.) and transferred to a stream of nitrogen at 83 K. The data were collected on a MAR image plate detector with a double focusing mirror system (Supper, Ltd) mounted on a Rigaku RU-200 rotating anode X-ray generator (CuKα radiation). The structure was solved by the method of multiple isomorphous replacement using two heavy atom derivatives (K2HgCl2 and KAuCl4) at pH 4.5, a double derivative (K2HgCl2 and KAuCl4) at pH 4.5 and one heavy atom derivative (K2HgCl2) at pH 6.0. The altered pH resulted in different number of heavy atom sites as well as different occupancies. High-resolution native data were collected at beamline X8-C (NSLS, Brookhaven National Laboratory, New York). The data collection statistics are shown in Table III. The X-ray images were processed using the HKL suite of software (Minor, 1993; Otwinowski, 1993). Heavy atom sites were identified from Patterson and difference Fourier techniques using the CCP4 suite of software (Collaborative Computational Project, 1994). The program SHARP (de la Fortelle and Bricogne, 1997) was used for heavy atom refinement. Table II gives the phasing refinement statistics. The overall Figure of Merit after heavy atom refinement and phasing was 0.35 (to 2.8 Å). Solvent flipping density modification was carried out using the program SOLOMON (Abrahams and Leslie, 1996) with a solvent content of 40%. Four-fold averaging using the RAVE suite of software (Jones, 1992; Kleywegt and Read, 1997) was carried out using the phases after solvent flipping, and the non-crystallographic symmetry matrices determined using the heavy atom positions. The resulting electron density map was clearly interpretable with most of the secondary structure elements visible. An initial model for a single protomer was built using the program O (Jones et al., 1991). Since the amino acid sequence for the C.rhodostoma enzyme was not yet available, the sequence of the C.adamanteus enzyme was incorporated into the initial model. The initial model for the native structure consisted of 481 amino acid residues (387 amino acid residues included side chains from the C.adamanteus sequence) as well as the FAD cofactor for a single monomer.
Table III. Data collection statistics.
| Data set | pH | Resolution (Å) | Completeness (%) | Total reflections | Independent reflections | I/σ | Rmergea |
|---|---|---|---|---|---|---|---|
| Native (1) | 4.5 | 2.6 | 87.4 | 110 458 | 49 460 | 14.7 | 0.045 |
| Native (2) | 4.5 | 2.0 | 88.1 | 391 000 | 139 242 | 18.9 | 0.046 |
| K2HgCl2 | 4.5 | 2.8 | 82.9 | 76 227 | 47 166 | 15.4 | 0.054 |
| K2HgCl2 | 6.0 | 2.8 | 88.8 | 104 515 | 49 741 | 12.0 | 0.066 |
| KAuCl4 | 4.5 | 2.8 | 88.0 | 96 712 | 50 503 | 16.8 | 0.042 |
| K2HgCl2/KAuCl4 | 4.5 | 2.8 | 87.3 | 105 067 | 50 092 | 24.1 | 0.048 |
aRmerge = ΣΣ|Ih,i – I| /ΣΣIh,i (summed over all intensities).
Table II. Heavy atom phasing statistics.
| Data set | pH | Resolution (Å) | Rderiva | No. of heavy atom sites | RCullisb | Phasing power |
|---|---|---|---|---|---|---|
| K2HgCl2 | 4.5 | 2.8 | 0.16 | 4 | 0.75 | 2.0 |
| K2HgCl2 | 6.0 | 2.8 | 0.32 | 10 | 0.78 | 1.8 |
| KAuCl4 | 4.5 | 2.8 | 0.12 | 10 | 0.83 | 1.4 |
| K2HgCl2/KAuCl4 | 4.5 | 2.8 | 0.24 | 8 | 0.78 | 2.2 |
aRderiv = Σ|FPH – FP|/ΣFP
bRCullis = Σ||FPH ± FP| – FH (calc)/Σ|FPH ± FP| (for centric reflections only).
The refined atomic coordinates of the LAAO–CIT complex were used as a molecular replacement search model for the LAAO–AB complex. The program AmoRe (Navaza, 1991) from the CCP4 suite of software (Collaborative Computational Project, 1994) was used to solve the structure by molecular replacement. The search model used consisted of the dimer of the LAAO–CIT complex with the FAD, citrate and water molecules removed. A rotation function was calculated using data from 20 to 3.0 Å. Eight solutions with peak heights of 18.7–25.6 σ were found, where σ is the standard deviation of the rotation function map. These corresponded to the four orientations of the dimer plus the 2-fold relationship between the monomers within each dimer. The translation function clearly identified the positions of the four dimers. Rigid body minimization of the correctly oriented dimers resulted in an R-factor of 33.7%. The FAD, AB and water molecules were built into the difference electron density using strict non-crystallographic symmetry.
Crystallographic refinement
For the LAAO–CIT structure, constrained crystallographic refinement using the program CNS (Brünger et al., 1998) was carried out initially to 2.4 Å resolution. As the resolution was increased to 2.0 Å, bulk solvent corrections were included and restrained non-crystallographic symmetry refinement carried out. Each cycle of refinement was followed by a manual rebuild using the program O. SIGMAA-weighted maps calculated with coefficients 2Fo – Fc and Fo – Fc were used for the model rebuilds. After the second cycle of refinement, the full amino acid sequence of the C.rhodostoma enzyme was completed and incorporated into the model. As the refinement progressed, water molecules were included in the model as well as carbohydrate chains attached to residues Asn172 and Asn361. In addition, difference density in the active site indicated that a molecule of citrate was bound. The final model consists of residues 4–486 for each of the four protomers. Each protomer contains one FAD, a citrate molecule and four sugar moieties: three at Asn172 and one at Asn361. Finally, the model includes 1791 water molecules.
The LAAO–AB complex was refined to 2.0 Å resolution. Since eight molecules were present in the asymmetric unit of this crystal form, restrained non-crystallographic symmetry refinement was performed throughout. The weights used in the refinement were higher than those used for the refinement of the LAAO–CIT model. The final model includes residues 5–486 for all eight protomers in the asymmetric unit. Each protomer contains one FAD, three AB molecules and a single sugar moiety at Asn172. The number of water molecules included in the final LAAO–AB model was 2378.
After the final round of refinement, the stereochemical parameters of the structures were evaluated with the program PROCHECK (Laskowski et al., 1993). The final refinement statistics for both structures are shown in Table I.
Enzyme assays
LAAO from C.rhodostoma was assayed according to a protocol adapted from Wellner (1966). Briefly, reactions were initiated at 37°C upon the addition of 1 µg of LAAO to an 800 µl reaction mixture containing 2.5 U of catalase, 100 mM acetate (pH 4.6), and varying concentrations of l-phenylalanine and AB. After an incubation period of 15 min, reactions were stopped by the addition of 200 µl of 25% trichloroacetic acid. An aliquot (100 µl) of this stopped reaction mixture was added to 1 ml of borate-arsenate solution (1 M, pH 6.5) and incubated at room temperature for 60 min. The absorbance of the reaction product complexed with borate-arsenate was then measured with a Cary 1 BIO UV-Visible Spectrophotometer at a wavelength of 300 nm. Blanks (reaction minus LAAO) were prepared in parallel to, and measured concurrently with, enzyme reactions. The rate of increase of OD300 over the 15 min time course was determined to be linear, and the coupled catalase activity was observed not to be inhibited by AB.
Accession numbers
The atomic coordinates and structure factors for the LAAO–CIT complex and the LAAO–AB complex have been deposited in the Brookhaven Protein Data Bank accession codes IF8R and IF8S, respectively (Bernstein et al., 1977).
Acknowledgments
Acknowledgements
We thank N.Croteau and B.Ahvazi for technical assistance. This work was supported by a grant from the Medical Research Council of Canada (MT13341 to A.V.) and a Chercheur-boursier salary support award from the Fonds de la recherche en santé du Québec (to A.V.).
References
- Abrahams J.P. and Leslie,A.G.W. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D, 52, 30–42. [DOI] [PubMed] [Google Scholar]
- Bernstein F.C., Koetzle,T.F., Williams,G.J., Meyer,E.E.J., Brice,M.D., Rodgers,J.R., Kennard,O., Shimanouchi,R. and Tasumi,M. (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol., 112, 535–542. [DOI] [PubMed] [Google Scholar]
- Binda C., Coda,A., Angelini,R., Rodolfo,F., Ascenzi,P. and Mattevi,A. (1999) A 30 Å long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure, 7, 265–276. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Chu C.C. and Paul,W.E. (1997) Fig1, an interleukin 4-induced mouse B cell gene isolated by cDNA representational difference analysis. Proc. Natl Acad. Sci. USA, 94, 2507–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles C.J., Edmondson,D.E. and Singer,T.P. (1977) Reversible inactivation of l-amino acid oxidase. Properties of the three conformational forms. J. Biol. Chem., 252, 8035–8039. [PubMed] [Google Scholar]
- Collaborative Computational Project, No 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Curti B., Massey,V. and Zmudka,M. (1968) Inactivation of snake venom l-amino acid oxidase by freezing. J. Biol. Chem., 243, 2306–2314. [PubMed] [Google Scholar]
- Curti B., Ronchi,S. and Simonetta,P.M. (1992) d- and l-amino acid oxidases. In Müller,F. (ed.), Chemistry and Biochemistry of Flavoenzyme. CRC Press, Boca Raton, FL, Vol. III, pp. 69–94. [Google Scholar]
- de Kok A. and Rawitch,A.B. (1969) Studies on l-amino acid oxidase. II. Dissociation and characterization of its subunits. Biochemistry, 8, 1405–1411. [DOI] [PubMed] [Google Scholar]
- de Kok A. and Veeger,C. (1968) Studies on l-amino acid oxidase. I. Effects of pH and competitive inhibitors. Biochim. Biophys. Acta, 167, 35–47. [DOI] [PubMed] [Google Scholar]
- de la Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement in the MIR and MAD methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick P.F. and Massey,V. (1982) Proton release during the reductive half reaction of d-amino acid oxidase. J. Biol. Chem., 257, 9958–9962. [PubMed] [Google Scholar]
- Hecht H.J., Kalisz,J.M., Hendle,J., Schmid,R.D. and Schomburg,D. (1993) Crystal structure of glucose oxidase from Aspergillus niger refined at 2.3 Å resolution. J. Mol. Biol., 229, 153–172. [DOI] [PubMed] [Google Scholar]
- Jancarik J. and Kim,S.H. (1991) Sparse-matrix sampling—a screening method for crystallization of proteins. J. Appl. Crystallogr., 24, 409–411. [Google Scholar]
- Jones T.A. (1992) A yaap asap @#*? A set of averaging programs. In Dodson,E.J., Gover,S. and Wolf,W. (eds), Molecular Replacement. SERC Daresbury Laboratory, Warrington, UK, pp. 91–105. [Google Scholar]
- Jones T.A., Zhou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. D., 49, 18–23. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J. and Read,R.J. (1997) Not your average density. Structure, 5, 1557–1569. [DOI] [PubMed] [Google Scholar]
- Kraulis P. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Li Z.Y., Yu,R.F. and Lian,E.C. (1994) Purification and characterization of l-amino acid oxidase from king cobra (Ophiophagus hannah) venom and its effect on human platelet aggregation. Toxicon, 32, 1349–1358. [DOI] [PubMed] [Google Scholar]
- Lindqvist Y. (1989) Refined structure of spinach glycolate oxidase at 2 Å resolution. J. Mol. Biol., 209, 151–166. [DOI] [PubMed] [Google Scholar]
- Macheroux P., Kitzing,K., Vetsch,M., Sappelt,M., Ghisla,S., Schwarz,M. and Kurfürst,M. (1999) Studies on the glycosylation of l-amino acid oxidase from the Malayan pit viper Calloselasma rhodostoma. In Ghisla,S., Kroneck,P., Macheroux,P. and Sund,H. (eds), Flavins and Flavoproteins. Ruldolf Weber, Berlin, pp. 583–586. [Google Scholar]
- Mattevi A. (1998) The PHBH fold: not only flavoenzymes. Biophys. Chem., 70, 217–222. [DOI] [PubMed] [Google Scholar]
- Mattevi A., Vanoni,M.A., Todone,F., Rizzi,M., Teplyakov,A., Coda,A., Bolognesi,M. and Curti,B. (1996) Crystal structure of d-amino acid oxidase: a case of active site mirror-image convergent evolution with flavocytochrome b2. Proc. Natl Acad. Sci. USA, 93, 7496–7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W. (1993) XDISPLAYF Program. Purdue University. [Google Scholar]
- Miura R., Setoyama,C., Nishina,Y., Shiga,K., Mizutani,H., Miyahara,I. and Hirotsu,K. (1997) Structural and mechanistic studies on d-amino acid oxidase×substrate complex: implications of the crystal structure of enzyme×substrate analog complex. J. Biochem. (Tokyo), 122, 825–833. [DOI] [PubMed] [Google Scholar]
- Mizutani H., Miyahara,I., Hirotsu,K., Nishina,Y., Shiga,K., Setoyama,C. and Miura,R. (1996) Three-dimensional structure of porcine kidney d-amino acid oxidase at 3.0 Å resolution. J. Biochem. (Tokyo), 120, 14–17. [DOI] [PubMed] [Google Scholar]
- Navaza J. (1991) AMoRe: an automated package for molecular replacement. Acta Crystallogr. A, 50, 157–163. [Google Scholar]
- Otwinowski Z. (1993) Oscillation data reduction program. In Sawyer,L., Isaacs,N. and Bailey,S. (eds), Proceedings of the CCP4 Suite Study Weekend: Data Collection and Processing. SERC Daresbury Laboratory, Warington, UK, pp. 56–62. [Google Scholar]
- Ponnudurai G., Chung,M.C.M. and Tan,N.-H. (1994) Purification and properties of the l-amino acid oxidase from Malayan pit viper (Calloselasma rhodostoma) venom. Arch. Biochem. Biophys., 313, 373–378. [DOI] [PubMed] [Google Scholar]
- Porter D.J.T. and Bright,H.J. (1980) Interpretationof the pH dependence of flavin reduction in the l-amino acid oxidase reaction. J. Biol. Chem., 255, 2969–2975. [PubMed] [Google Scholar]
- Raibekas A.A. and Massey,V. (1998) Primary structure of the snake venom l-amino acid oxidase shows high homology with the mouse B cell interleukin 4-induced Fig1 protein. Biochem. Biophys. Res. Commun., 248, 476–478. [DOI] [PubMed] [Google Scholar]
- Suhr S.-M. and Kim,D.-K. (1996) Identification of the snake venom substance that induces apoptosis. Biochem. Biophys. Res. Commun., 224, 134–139. [DOI] [PubMed] [Google Scholar]
- Suhr S.-M. and Kim,D.S. (1999) Comparison of the apoptotic pathways induced by l-amino acid oxidase and hydrogen peroxide. J. Biochem., 125, 305–309. [DOI] [PubMed] [Google Scholar]
- Todone F., Vanoni,M.A., Mozzarelli,A., Bolognesi,M., Coda,A., Curti,B. and Mattevi,A. (1997) Active site plasticity in d-amino acid oxidase: a crystallographic analysis. Biochemistry, 36, 5853–5860. [DOI] [PubMed] [Google Scholar]
- Torii S., Naito,M. and Tsuruo,T. (1997) Apoxin I, a novel apoptosis-inducing factor with l-amino acid oxidase activity purified from western diamonback rattlesnake venom. J. Biol. Chem., 272, 9539–9542. [DOI] [PubMed] [Google Scholar]
- Umhau S., Diederichs,K., Welte,W., Ghisla,S., Pollegioni,L., Molla,G., Porrini,D. and Pilone,M.S. (1999) Very high resoution crystal structure of d-amino acid oxidase. Insights into the reaction mechanisms and mode of ligand binding. In Ghisla,S., Kroneck,P., Macheroux,P. and Sund,H. (eds), Flavins and Flavoproteins. Ruldolf Weber, Berlin, pp. 567–570. [Google Scholar]
- Vrielink A., Lloyd,L.F. and Blow,D.M. (1991) Crystal structure of cholesterol oxidase from Brevibacterium sterolicum refined at 1.8 Å resolution. J. Mol. Biol., 219, 533–554. [DOI] [PubMed] [Google Scholar]
- Wellner D. (1966) Evidence for conformational changes in l-amino acid oxidase associated with reversible inactivation. Biochemistry, 5, 1585–1591. [DOI] [PubMed] [Google Scholar]
- Wierenga R.K., DeJong,R.J., Kalk,K.H., Hol,W.G.J. and Drenth,J. (1979) Crystal structure of p-hydroxybenzoate hydroxylase. J. Mol. Biol., 131, 55–73. [DOI] [PubMed] [Google Scholar]
- Xia Z.X. and Mathews,F.S. (1990) Molecular structure of flavocytochrome b2 at 2.4 Å resolution. J. Mol. Biol., 212, 837–863. [DOI] [PubMed] [Google Scholar]
- Zeller A. and Maritz,A. (1944) Über eine neue l-Aminosäure-oxydase. Helv. Chim. Acta, 27, 1888–1903. [PubMed] [Google Scholar]