

Abstract
Myotonic dystrophy (DM1) is an autosomal dominant neuromuscular disorder associated with a (CTG)n expansion in the 3′-untranslated region of the DM1 protein kinase (DMPK) gene. To explain disease pathogenesis, the RNA dominance model proposes that the DM1 mutation produces a gain-of-function at the RNA level in which CUG repeats form RNA hairpins that sequester nuclear factors required for proper muscle development and maintenance. Here, we identify the triplet repeat expansion (EXP) RNA-binding proteins as candidate sequestered factors. As predicted by the RNA dominance model, binding of the EXP proteins is specific for dsCUG RNAs and proportional to the size of the triplet repeat expansion. Remarkably, the EXP proteins are homologous to the Drosophila muscleblind proteins required for terminal differentiation of muscle and photoreceptor cells. EXP expression is also activated during mammalian myoblast differentiation, but the EXP proteins accumulate in nuclear foci in DM1 cells. We propose that DM1 disease is caused by aberrant recruitment of the EXP proteins to the DMPK transcript (CUG)n expansion.
Keywords: DM1/EXP proteins/muscleblind/myotonic dystrophy/RNA dominance
Introduction
Myotonic dystrophy (dystrophia myotonica, DM1) is one of 13 identified triplet repeat expansion disorders that include Huntington’s disease, fragile X syndrome and several types of spinocerebellar ataxia (for reviews see Ashley and Warren, 1995; Groenen and Wieringa, 1998; Korade-Mirnics et al., 1998). These disorders are characterized by genetic anticipation in which disease severity is proportional, and age-of-onset inversely proportional, to the size of the expansion mutation. The DM1 expansion is a (CTG)n repeat in the 3′-untranslated region (3′-UTR) of the DM protein kinase (DMPK) gene. This CTG repeat ranges in size from 5–37 repeats in the normal population, 50–1000 repeats in mild to classical symptom adult-onset patients with premature cataract formation, myotonia and muscle weakness, and >1000 repeats in severely affected congenital individuals (CDM) with neonatal respiratory distress, hypotonia and mental retardation. Surprisingly, the DM1 inheritance pattern is autosomal dominant even though the expansion mutation occurs in a region of the DMPK gene that does not encode protein.
Three different models have been suggested to explain how a 3′-UTR triplet repeat expansion leads to a dominantly inherited disease. The DMPK haploinsufficiency model proposes that the DM1 expansion mutation reduces expression of the mutant allele. DM1 cells may thus express only half of the normal level of DMPK protein, and this may be sufficient for disease progression. Several observations argue against this model. For example, the correlation between loss of the DMPK protein and disease severity is not strong, and no loss-of-function mutations have been described in the DMPK coding region (for reviews see Groenen and Wieringa, 1998; Korade-Mirnics et al., 1998). Moreover, efforts to reproduce the eye and muscle defects characteristic of DM1 disease using Dmpk knockout mice have also been unsuccessful (Jansen et al., 1996; Reddy et al., 1996).
The second, or chromatin structure model speculates that the (CTG)n expansion creates an exceptionally stable nucleosomal region that inhibits adjacent gene expression (Wang et al., 1994; Otten and Tapscott, 1995). While several studies have shown that transcription of the mutant DMPK allele is not severely affected by CTG expansion (Krahe et al., 1995; Davis et al., 1997), expression of the adjacent upstream DMWD (formerly gene 59) and downstream SIX5 (formerly DMAHP) genes is depressed in some DM1 individuals (Klesert et al., 1997; Thornton et al., 1997; Korade-Mirnics et al., 1999). However, this loss of expression may be due to inhibition of mRNA nucleocytoplasmic export rather than a transcriptional defect (Alwazzan et al., 1999). Since DM1 is often associated with premature cataract formation, the SIX5 gene product is particularly interesting since it is homologous to the Drosophila sine oculis protein involved in photoreceptor development and both Six5+/– and Six5–/– mice develop cataracts (Winchester et al., 1999; Klesert et al., 2000; Sarkar et al., 2000).
Finally, the RNA dominant mutation model proposes that triplet repeat expansion causes a gain-of-function at the RNA level possibly by impairing nucleocytoplasmic mRNA export (Taneja et al., 1995; Wang et al., 1995; Caskey et al., 1996; Davis et al., 1997; Hamshere et al., 1997). Recent studies using C2C12 myoblast cultures and transgenic mice support this idea (Amack et al., 1999; Mankodi et al., 2000). Several DM1-associated phenotypes, including accumulation of (CUG)n expansion-bearing transcripts in the nucleus, are observed when (CTG)n expansions are introduced into the 3′-UTR of several different transgenes. To explain how RNA dominance might cause DM1 disease, the protein sequestration hypothesis has been proposed (Caskey et al., 1996; Timchenko et al., 1996). According to this hypothesis, the DM1 expansion leads to sequestration of (CUG)n-binding proteins on mutant DMPK RNAs and depletion from other transcripts that require these proteins for normal gene expression. Two types of (CUG)n expansion-binding proteins could exist. The first type would bind to the wild-type (non-expanded) DMPK pre-mRNA and mRNA, but binding might be influenced by (CUG)n expansion either directly, by acting as a binding site, or indirectly, by altered transcript folding. Although CUG-binding protein 1 (CUG-BP1) is a candidate for this type of factor since it has been implicated in DM1 and alternative pre-mRNA splicing of muscle gene transcripts (Timchenko et al., 1996; Philips et al., 1998), recent studies argue that it is unlikely to be directly involved in disease pathogenesis (Michalowski et al., 1999). The second type of protein may not associate with (CUG)n repeats within the normal range, but would specifically recognize the larger disease-associated expansions.
In this study, we identify triplet repeat expansion (EXP) double-stranded (ds) RNA-binding proteins, which selectively associate with (CUG)n expansions. Human EXP proteins are homologous to the Drosophila muscleblind (mbl) proteins that play essential roles in the terminal differentiation of embryonic pharyngeal, visceral and somatic muscles as well as ommatidial photoreceptors (Begemann et al., 1997; Artero et al., 1998). EXP expression, which is most prominent in blood, eye, cardiac muscle and skeletal muscle, is induced during mouse myoblast differentiation, suggesting an important role for these proteins during, and possibly following, terminal differentiation of DM1-affected tissues. Our results support the RNA dominance model for DM1 pathogenesis, and suggest that nuclear accumulation of mutant DMPK transcripts leads to aberrant recruitment of EXP proteins and subsequent deleterious effects on cellular differentiation.
Results
Identification of triplet repeat expansion RNA-binding proteins
Mutant DMPK allele transcripts carry expansions ranging from 50 to >2000 CUG repeats (Figure 1A), and chemical modification studies indicate that RNAs with ≤10 CUG repeats are predominantly single-stranded while larger repeat RNAs (CUG≥20) form stable RNA hairpins (Figure 1B) (Napierala and Krzyzosiak, 1997). Recently, this proposal has been corroborated for large RNAs by the direct visualization of (CUG)130 and DMPK-(CUG)90 RNA hairpins in the electron microscope (Michalowski et al., 1999), and by thermal melting and nuclease digestion studies (Tian et al., 2000). Although the structure of expanded CUG repeats in vivo is unknown, we postulated that factors which recognize (CUG)n expansions should be dsRNA-binding proteins and the extent of binding to DMPK mRNAs should be proportional to RNA hairpin length. To test these possibilities, 32P-labeled DMPK RNAs containing variable numbers of CUG repeats (6, 54, 90), or an antisense transcript containing six CAG repeats, were synthesized by in vitro transcription. Labeled transcripts were then incubated in HeLa cell nuclear extracts followed by UV-light induced crosslinking to covalently attach any proteins that were directly bound to the CUG and CAG repeat DMPK RNAs. Following RNase digestion and gel electrophoresis, the total proteins crosslinked to triplet repeat RNAs were visualized by label transfer. Many proteins crosslinked weakly to DMPK 3′-UTR RNAs with either six CAG (6as) or six CUG (6s) repeats (Figure 1C). The ∼41 kDa protein that more prominently crosslinked to DMPK 6as and 6s RNAs was identified as the hnRNP C1 protein by specific immunopurification (data not shown). However, three additional 40–45 kDa proteins crosslinked to DMPK RNAs with either 54 or 90 CUG repeats. Since binding appeared to be proportional to repeat size, these proteins were named the (CUG)n triplet repeat expansion (EXP) dsRNA-binding proteins.

Fig. 1. DM1 (CUG)n expansion mutation. (A) The structure of DMPK mRNA is illustrated with the positions of the DMPK coding region (stippled box), the 3′-UTR CUG repeat region (black box) and the poly(A) tail [(A)n] indicated. Also highlighted are CUG repeats corresponding to normal, pre-mutant and mutant DM1 RNAs. (B) (CUG)n RNAs showing that CUG repeats ≥20 spontaneously fold into dsRNA hairpins while (CUG)≤10 RNAs are primarily single stranded. (C) Multiple 38–45 kDa proteins crosslink preferentially to DMPK 3′-UTR RNAs with large CUG repeats. DMPK 3′-UTR RNAs containing either 6, 54 or 90 (6s, 54s, 90s) CUG repeats, or an antisense transcript containing six CAG (6as) repeats, were labeled with [α-32P]UTP, incubated in HeLa nuclear extract, photocrosslinked, digested with RNase and fractionated by SDS–PAGE. A bracket indicates the position of the EXP proteins. Sizes are indicated in kilodaltons. (D) Crosslinking of EXP proteins to dsCUG RNAs is proportional to the number of CUG repeats. RNAs composed of 11, 20, 35, 74 or 97 CUG repeats were labeled with [α-32P]UTP and used for photocrosslinking analysis. The relative amount of EXP crosslinking was quantified by PhosphorImager analysis and is illustrated together with a 35 kDa protein that binds preferentially to shorter CUG repeats.
These DMPK transcripts contained ∼400 nucleotides of DMPK 3′-UTR sequence in addition to the (CUG)n repeat. To ascertain whether the EXP proteins were binding directly to CUG repeats, and confirm that the extent of EXP protein binding was proportional to the number of CUG repeats, (CUG)n RNAs containing 11–97 repeats, but without DMPK sequence, were prepared and assayed by the photocrosslinking technique (Figure 1D). While the EXP proteins did not detectably crosslink to RNAs with ≤11 CUG repeats, association of these proteins with (CUG)≥20 RNAs was proportional to repeat number. In contrast, crosslinking of a 35 kDa protein, tentatively identified as hnRNP A2, and an unidentified 50 kDa protein was maximal with (CUG)11 RNA but diminished as CUG repeat number increased.
Novel dsRNA-binding proteins that preferentially recognize CUG hairpins
The RNA-binding specificity of the EXP proteins was examined using two different types of CG-rich dsRNAs. First, crosslinking of the EXP proteins to CUG versus CAG repeat RNAs was compared since large CAG repeat RNAs also form RNA hairpins (Michalowski et al., 1999). Several proteins of ∼35 and 50 kDa crosslinked to (CUG)10 RNAs while the EXP proteins did not, and very little protein binding to (CAG)10 or (CAG)54 RNAs was detectable with this assay (Figure 2A). In striking contrast, prominent binding of the EXP proteins to (CUG)54 RNAs was observed. Secondly, we determined whether the EXP proteins recognize the HIV-1 transactivation region (TAR) RNA hairpin, which has been demonstrated to bind the TAR dsRNA-binding protein (TRBP) (Gatignol et al., 1991). Surprisingly, the EXP proteins did not crosslink to either wild-type or mutant TAR RNAs (Figure 2B). To address the concern that the EXP proteins might bind, but not crosslink to TAR RNA, the ability of TAR to compete for EXP protein binding to (CUG)90 RNA was tested. EXP crosslinking to (CUG)90 was abolished by excess cold (CUG)90, but not by TAR RNA, demonstrating that the EXP proteins recognize preferentially dsCUG.
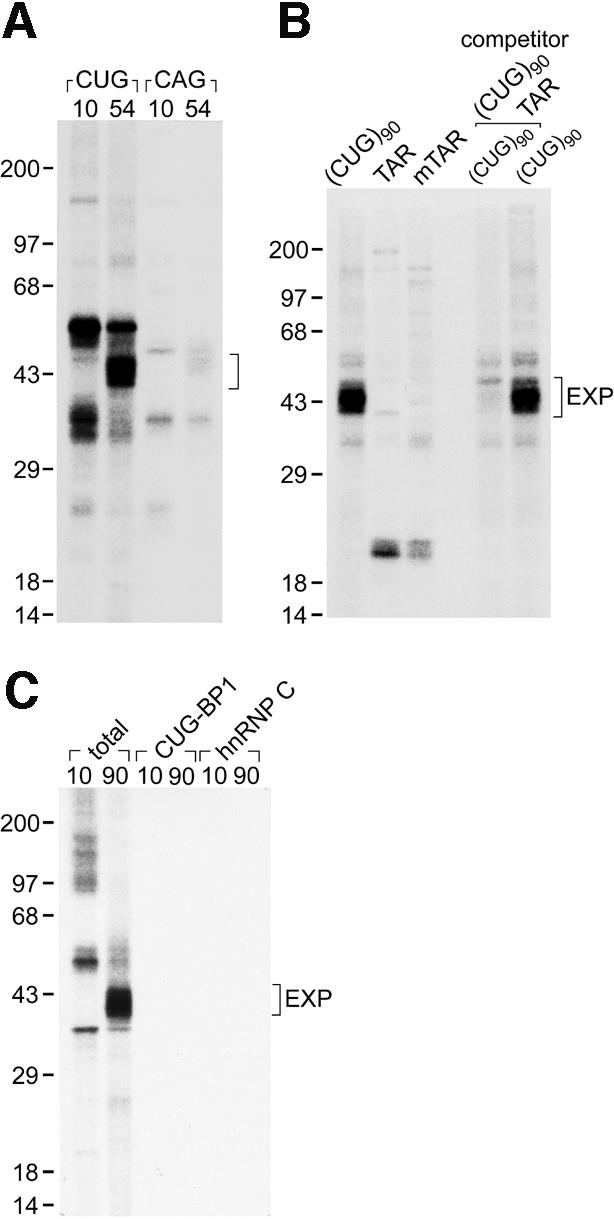
Fig. 2. EXP proteins are novel dsCUG-binding factors. (A) EXP proteins crosslink directly and preferentially to large CUG repeat RNAs. Photocrosslinking was performed as in Figure 1C except that RNAs were synthesized in the presence of [α-32P]GTP and transcribed from pCTG10, pCTG54, pCAG10 or pCAG54 which do not contain DMPK sequences. The bracket indicates EXP protein position. (B) EXP proteins do not crosslink to the HIV-1 TAR RNA. Plasmids containing either 90 CUG repeats [(CUG)90], the TAR RNA sequence (TAR) or a mutant TAR (mTAR) were transcribed in the presence of [α-32P]UTP and photocrosslinking performed as described in (A). The first three lanes indicate binding activity seen with each labeled RNA. In the last two lanes, labeled (CUG)90 was mixed with a 500-fold molar excess of unlabeled (CUG)90 or unlabeled TAR RNA prior to addition to HeLa nuclear extract. (C) EXP proteins are not immunologically related to CUG-BP1 or hnRNP C proteins. Immunopurifications of EXP crosslinked proteins were performed using mAbs 3B1 (anti-CUG-BP1) and 4F4 (anti-hnRNP C).
In an attempt to identify the EXP proteins, we screened previously characterized 35–50 kDa RNA-binding proteins for EXP activity. EXP activity was not immunopurified with antibodies to the 50 kDa CUG-BP1 or the 41–43 kDa hnRNP C proteins (Figure 2C). Several additional hnRNP proteins (A1, A2/B1, D), TRBP, the La autoimmune antigen, and the translational initiation factor eIF4A were also eliminated as EXP protein candidates by immunopurification analysis. Therefore, we concluded that the EXP proteins are novel dsCUG-binding proteins.
Homology to Drosophila muscleblind proteins
The strategy used to purify the EXP proteins from HeLa cell nuclear extract is illustrated in Figure 3A. The majority of EXP activity eluted from single-stranded (ss) DNA–agarose at 0.2 M NaCl (EXP1), but significant EXP activity was also detectable in the 0.2 M NaCl fraction (EXP2) from poly(U)–Sepharose (Figure 3B). Polypeptides in the 45 kDa region of the gel were excised, electroeluted, and the proteins re-fractionated by SDS–PAGE. The purified EXP1 fraction was composed primarily of several 38–45 kDa polypeptides while EXP2 migrated as a single 40 kDa band (Figure 3C). These EXP polypeptides were subjected to in-gel trypsin digestion followed by mass spectrometry protein identification analysis. For EXP1, several significant matches were found to the human 42 kDa MBNL and 40 kDa KIAA0428 proteins (see Materials and methods). No matches were found for the EXP2 protein fraction. To confirm that MBNL was related to the EXP1 proteins, two primers bordering the MBNL coding sequence were used to amplify a cDNA clone encoding the full-length protein from a HeLa S3 cell library. This cDNA clone (hEXP42) was subcloned into pET15b for subsequent production of a His-tagged protein, which was used to immunize mice for antibody production. Immunoblot analysis demonstrated that these anti-hEXP42 antibodies recognized weakly two 43/44 kDa polypeptides from HeLa cells and a single 43 kDa protein from normal and DM1 lymphoblastoid cell lines consistent with the sizes of the protein isoform predicted from the hEXP42 cDNA (Figure 3D).

Fig. 3. Purification of HeLa cell EXP proteins. (A) Purification scheme. The initial purification step included digestion of HeLa cell nuclear extract (HeLa NE) with micrococcal nuclease (MN) followed by DEAE Fast-Flow Sepharose chromatography and collection of the flow-through (FT). Final fractions are EXP1 eluate fraction 1 (eF1) and EXP2 eluate fraction 1 (eF1). (B) Label transfer activities of various fractions (top panel). Fractions were incubated with 32P-labeled (CUG)90 RNA followed by photocrosslinking analysis. The bottom panel is a silver-stained SDS–polyacrylamide gel showing the protein composition of each fraction. (C) Silver-stained SDS–polyacrylamide gel showing purified EXP1 and EXP2 fractions. Proteins from the EXP1 (bracket) and EXP2 (asterisk) regions indicated in (B) were excised and refractionated by SDS–PAGE. (D) Anti-hEXP42 polyclonal antibodies recognize two minor proteins in HeLa nuclear extract and a single protein in normal and DM1 lymphoblast nuclear extracts. An immunoblot using the anti-CUG-BP1 mAb 3B1 is also shown as a protein loading control. (E) EXP activity is immunopurified with anti-hEXP42 antibodies. Crosslinking reactions with (CUG)10 and (CUG)54 RNAs and HeLa nuclear extracts were performed as described in Figure 2A except that in vitro transcription was performed with [α-32P]UTP. Immunopurifications were performed using either the anti-hEXP42 antiserum, a non-immunized control antiserum, or mAb 3B1 (anti-CUG-BP1). The last lane is a crosslinking reaction using 32P-labeled (CUG)90 with 50 ng of purified His-tagged hEXP42 in the absence of HeLa nuclear extract.
Identification of the hEXP42/MBNL and KIAA04288 proteins in the EXP1 fraction suggested that these proteins might be responsible for EXP activity. To obtain direct evidence that hEXP42 possessed dsCUG-binding activity, HeLa proteins were crosslinked to dsCUG RNAs and immunopurifications performed. A significant fraction of the HeLa cell EXP 43–45 kDa activity was immunopurified with the anti-hEXP42 antiserum (Figure 3E). Although these antibodies did not recognize any of the proteins crosslinked to (CUG)10, they efficiently immunopurified (CUG)54 binding activity while control antibodies did not. In addition, purified His-tagged hEXP42 protein alone bound and crosslinked to all dsCUG RNAs tested, including (CUG)90 (Figure 3E). On the basis of these results, we concluded that hEXP42 is identical to MBNL.
Since three EXP proteins were detectable by dsCUG-binding activity (Figure 1C) and the anti-hEXP42 antibody did not immunopurify the 35–40 kDa EXP activity (Figure 3E), we speculated that smaller EXP isoforms exist. Therefore, the HeLa cDNA library was re-screened using hEXP42 as a hybridization probe. Three full-length cDNAs (hEXP42, hEXP40, hEXP35) were characterized (Figure 4A). The hEXP42 and hEXP40 cDNA sequences were identical to MBNL and KIAA04288, respectively, while the hEXP35 cDNA encoded a novel protein isoform. All three EXP proteins are presumably produced by alternative pre-mRNA splicing, and contain four repeats of a zinc-knuckle related motif, CCCH (C3H), which has been shown previously to bind RNA (Figure 4B) (Anderson et al., 1993). The MBNL and KIAA04288 cDNAs were identified during cDNA library screens as human homologs of the Drosophila muscleblind (mbl) proteins required for terminal differentiation of muscle and photoreceptor cells (Begemann et al., 1997; Artero et al., 1998). The N-terminal regions of the three human EXP and the largest Drosophila mbl protein (mblB) were very similar while the C-terminal regions were considerably more divergent (Figure 4A). Homology of the EXP proteins to the fly mbl protein family was an intriguing observation since DM1 is characterized by specific eye and muscle defects. These results suggested that the MBNL/EXP gene might be expressed differentially in human tissues. As anticipated, poly(A)+ RNA blot analysis demonstrated that the human MBNL/EXP gene was most highly expressed in both cardiac and skeletal muscle with considerably lower levels observed in all other tissues examined (Figure 4C). It is also interesting to note that hEXP42 was also present at relatively high levels in lymphoblastoid cell lines (Figure 3D) because immunoglobulin levels are reduced in DM1 patients (Meola and Moxley, 1999).

Fig. 4. EXP proteins are homologous to the Drosophila muscleblind family. (A) Alignment of the human hEXP42/MBNL, hEXP40/KIAA0428 and hEXP35 proteins with the largest of the four muscleblind proteins, mblB. Identical amino acid positions are highlighted by black boxes while similarities are shaded. (B) Illustration showing the organization of the four C3H motifs (C3H I–IV) and a potential transmembrane domain (TM) in the EXP proteins. (C) Poly(A)+ RNA (2 µg/lane) blot analysis of human tissues showing high EXP expression in cardiac and skeletal muscle. Lanes were standardized for β-actin levels, and sizes are indicated in kilobases.
Activation of EXP expression during mammalian myoblast differentiation
Because RNA levels might not reflect the amount of protein present in various cell types, we also examined protein levels in several mouse tissues using immunoblot analysis. Anti-hEXP42 polyclonal antibodies detected high levels of mEXP42 in skeletal muscle, lower levels in heart and eye, and no mEXP42 in brain or testes (Figure 5A). In contrast, the 43 kDa TRBP protein was prominently expressed only in testes, as shown previously (Lee et al., 1996). Thus, these EXP expression patterns correlated with pathological effects seen in eye and muscle tissues of affected DM1 individuals.

Fig. 5. Characterization of EXP proteins in mouse tissues and during myoblast differentiation. (A) Immunoblot analysis using anti-hEXP42 antibodies showing that the EXP proteins are expressed in muscle and eye. Equal protein loads (15 µg/lane) were confirmed by Coomassie Blue staining (data not shown). (B) EXP expression is induced during myoblast differentiation. Immunoblot showing myosin heavy chain (αmyosin HC), mEXP42 (αhEXP42) and CUG-BP1 (αCUG-BP1) protein levels prior to (Day 0) and during (Days 1–5) myoblast differentiation. (C) Phase and immunofluorescence microscopy showing two examples of multinucleated myotubes, present on Day 4 following induction of myoblast differentiation, as well as undifferentiated myoblasts.
The discovery that the EXP proteins were structurally related to the Drosophila mbl proteins and expressed at a high level in skeletal muscle suggested that EXP proteins might play key roles during the terminal differentiation of mammalian cell precursors. To examine this possibility, murine C2C12 myoblasts were grown to >90% confluency and myoblast differentiation was induced. Under these conditions, myoblasts fuse to form multinucleated syncytia followed by cell elongation and myotube formation. Initial cell fusion resulted in the appearance of multinucleated cells within 48 h following induction, and cells subsequently elongated to form multinucleated myotubes by about Day 3. During this myoblast fusion process, the expression of mEXP42 was followed by immunoblot analysis. As a marker for differentiation, myosin heavy chain (myosin HC) was detectable within 48 h following the induction of differentiation and increased up to Day 5 (Figure 5B). In agreement with the proposal that mammalian EXP proteins are important during terminal differentiation of muscle cells, mEXP42 protein was not detectable until Day 2 and increased to a maximum level by Day 4. The level of CUG-BP1, which has been implicated in DM1 pathogenesis and as a troponin T pre-mRNA intronic enhancer binding protein (Philips et al., 1998), remained constant during the transition from myoblasts to multinucleated myotubes.
During myoblast differentiation, the subcellular distributions of CUG-BP1 and mEXP42 were also monitored by cell immunofluorescence. In both myoblasts and myotubes, CUG-BP1 localized primarily to the nucleus (Figure 5C). As anticipated from the immunoblot analysis, mEXP42 was not detectable until Day 2 following the induction of myoblast differentiation. Surprisingly, this protein localized primarily to the cytoplasm of multinucleated myotubes. This result was unexpected since the (CUG)n-binding activity of the EXP proteins was predominantly nuclear in HeLa and lymphoblastoid cell lines. These results suggested that the EXP proteins might also function in the cytoplasm during this early stage of C2C12 myoblast differentiation.
Nuclear accumulation of EXP proteins in DM1 cells
Previous studies have shown that DMPK and unrelated chimeric transcripts carrying (CUG)n expansions accumulate in nuclear foci. These RNAs have been detected using fluorescence in situ hybridization (FISH) employing either fluorochrome-conjugated DMPK or (CAG)n oligonucleotides (Taneja et al., 1995; Davis et al., 1997; Amack et al., 1999). On the basis of these observations, several studies have suggested that the nucleocytoplasmic export of mutant DMPK, and possibly other mRNAs may be compromised in DM1 cells. We, thus, employed FISH analysis to confirm that the DM1 myoblasts used in this study showed nuclear accumulation of DMPK mutant transcripts. In contrast to normal myoblasts, the (CAG)10 probe detected multiple (CUG)n foci per DM1 nucleus (data not shown).
If the EXP/muscleblind proteins are recruited by CUG expansions in vivo, the protein sequestration hypothesis predicts that the subcellular distribution of these proteins should also be altered in cells expressing mutant DMPK RNAs. To test this idea, we examined EXP protein localization in normal and DM1 myoblasts by cell immunofluorescence using anti-hEXP42 polyclonal antibodies. Significantly more hEXP42 protein was detected in human myoblasts as opposed to mouse C2C12 myoblasts prior to differentiation. In normal human myoblasts, this EXP protein was distributed in both the cytoplasm and nucleus (Figure 6A). However, multiple nuclear foci were enriched in hEXP42 in DM1 cells. DM1 myoblasts exhibited an average of 15 EXP-enriched foci per nucleus with a range of 6–30 foci per nucleus. Efforts to co-localize this EXP protein with (CUG)n-enriched foci failed because the in situ hybridization procedure inhibited specific binding of the anti-EXP polyclonal antibody.

Fig. 6. EXP proteins accumulate in the nucleus of DM1 cells. (A) Cell immunofluorescence of normal and DM1 myoblasts using anti-EXP polyclonal antibodies. Phase contrast microscopy highlights cell position while DNA was detected with DAPI. The arrow indicates one of the EXP-enriched foci. (B) FISH analysis with a Cy3-labeled (CAG)10 probe (red), which detects DMPK (CUG)n expansions in DM1 cell nuclei (shown by DAPI staining). (C) Cell immunofluorescence, using anti-EXP antibodies, of normal fibroblasts infected with a MyoD adenovirus (normal fibroblast + MyoD) or DM1 fibroblasts either uninfected (DM1 fibroblast – MyoD) or infected (DM1 fibroblast + MyoD). Arrows indicate several sizes of foci enriched in hEXP42.
Previous work has shown that expression of the myogenic determination factor MyoD in DM1 fibroblasts increases DMPK expression as well as the number of nuclear foci enriched in mutant allele transcripts (Davis et al., 1997). As additional evidence for sequestration of the EXP proteins in vivo, we expressed MyoD in normal and DM1 fibroblasts. DM1 fibroblasts infected with a MyoD adenovirus exhibited a marked increase in the number of DMPK mutant transcript foci when compared with uninfected DM1 or infected normal fibroblasts (Figure 6B). Strikingly, this increase in DMPK foci is concomitant with an increase in the number and size of hEXP42-enriched nuclear foci (Figure 6C). AdMyoD-infected fibroblasts generally showed more foci enriched in DMPK transcripts than hEXP42. This discrepancy may be due to the difference in sensitivity between the FISH and immunofluorescence procedures or to the fact that the anti-hEXP42 antibody recognizes only one of the EXP proteins. In summary, DMPK mutant transcripts and hEXP42 accumulate in nuclear foci in both DM1 myoblasts and AdMyoD-infected DM1 fibroblasts, suggesting that DMPK mutant RNAs recruit and sequester these dsRNA-binding proteins in vivo.
Discussion
RNA dominance: role for triplet repeat expansion RNA-binding proteins
Previous studies have led to the prevailing view that DM1 is an RNA dominant disease (Groenen and Wieringa, 1998; Korade-Mirnics et al., 1998). How does a gain-of-function mutation at the RNA level cause disease? Although the (CUG)n expansion mutation might lead to altered activity of the RNA by itself, another possibility is that the triplet repeat expansion results in the formation of a novel RNA structure that serves as a binding site for an RNA-binding protein. Here, we provide evidence for this latter possibility by identifying the EXP proteins. Several characteristics of these proteins support the RNA dominance model. First, large (CUG)n expansions form stable RNA hairpins (Napierala and Krzyzosiak, 1997; Michalowski et al., 1999; Tian et al., 2000), and the EXP proteins bind preferentially to dsCUG RNAs. More importantly, binding is proportional to the length of the RNA hairpin. Secondly, the EXP proteins are homologous to the Drosophila mbl proteins that are essential for terminal differentiation of both muscle and photoreceptor cells (Begemann et al., 1997; Artero et al., 1998). Enhanced expression of the EXP proteins during myoblast differentiation supports a role for these dsRNA-binding factors in mammalian development and DM1 disease pathogenesis. Disruption of the mbl gene, which was originally identified as a dominant suppressor of the sev-svp2 (Sevenless-Seven-up) eye defect, is embryonic lethal. In wild-type flies, mbl is expressed in the ventral nerve cord and the photoreceptor system as well as pharyngeal, visceral and somatic muscles. We demonstrate that EXP expression is low in undifferentiated mouse myoblasts, but is activated upon induction of differentiation during the period when DMPK gene expression also increases (Davis et al., 1997). Thirdly, both DMPK mutant transcripts and the EXP proteins accumulate in nuclear foci in DM1 myoblasts. Although additional work is required to determine whether the EXP proteins are directly bound to these (CUG)n RNAs in vivo, our results suggest that the characteristic muscle wasting and weakness seen in DM1 patients may be due to recruitment and sequestration of the EXP proteins by (CUG)n expansion RNAs during myoblast differentiation.
The other dominantly inherited myotonic dystrophies, DM2 and proximal myotonic myopathy (PROMM), are clinically related to DM1, but the CTG repeat in the DMPK gene 3′-UTR expansion is not expanded. Since the DM2 and PROMM loci have been localized to chromosome 3q21, these disorders may be caused by different mutations in the same, or closely linked, genes (Ranum et al., 1998; Ricker et al., 1999). Previous work has suggested that genetic anticipation may be associated with PROMM disease (Ricker et al., 1999) so the DM2/PROMM loci may contain undetected, and possibly different, expansion mutations. Alternatively, these related myotonic dystrophies might be caused by mutations in the MBNL/EXP gene, encoding hEXP35, hEXP40 and hEXP42, or in genes encoding factors that regulate EXP activity. Recent mapping studies, which have confirmed previous work, do not support the first alternative since the MBNL/EXP gene maps distal to the DM2 locus at chromosome 3q25 (performed in collaboration with L.Ranum, University of Minnesota, MN). Nevertheless, database searches have revealed several additional EXP-related genes so it is possible that another EXP gene will be mapped to the DM2/PROMM interval on chromosome 3q21. If further analysis confirms that EXP functions are affected in DM2 and PROMM, then a unifying feature of DM1 and other autosomal dominant muscular dystrophies may be that disease is caused by alterations of EXP activity.
Molecular mechanisms involved in DM1 disease pathogenesis
What is the cellular pathway that is adversely affected by RNA dominance? Current evidence suggests that DM1 cells are not consistently defective in DMPK pre-mRNA splicing, 3′-end formation or mRNA stability (for reviews see Groenen and Wieringa, 1998; Korade-Mirnics et al., 1998). In contrast, the export of DMPK mutant transcripts from the nucleus is inhibited (Taneja et al., 1995; Davis et al., 1997; Hamshere et al., 1997). An important suggestion that has emerged from these studies is that retention of DMPK mutant transcripts might lead to a dominant-negative effect on the export of other transcripts. Recent evidence supports the idea of an RNA dominant effect that is specific for large CUG repeats, but independent of flanking sequences in the DMPK mRNA. Mouse C2C12 myoblasts have been transfected with reporter genes that contain various DMPK 3′-UTR segments with variable numbers of CUG repeats (Amack et al., 1999). Expression of these constructs with large CUG repeats (≥57) results in decreased translation of the reporter and nuclear accumulation of the transcript. More importantly, stable C2C12 lines expressing a β-galactosidase/DMPK 3′-UTR (CUG)>200 transgene show significant defects in myoblast fusion and differentiation, and revertants that possess normal fusion activity delete the CUG repeat region and downstream sequences. Thus, (CUG)n expansions might compromise the nuclear export of mutant RNAs, and this may lead to a dominant-negative effect on the export of other gene transcripts. However, the discovery that the EXP proteins are distributed in both the nucleus and cytoplasm during myoblast differentiation introduces the possibility that other cellular process may be directly affected by the DM1 expansion mutation. We propose a model in which the DM1 mutation produces a competing dsRNA-binding substrate that recruits the EXP proteins and sequesters them away from their normal RNA-binding sites during a critical period of cell differentiation (Figure 7). Current work is focused on identifying the RNAs that are normally associated with the EXP proteins in vivo and characterizing EXP functions in the nucleus and cytoplasm.

Fig. 7. RNA dominance model for DM1 pathogenesis. The processing and nucleocytoplasmic export of DMPK transcripts from the normal and mutant alleles are illustrated. This model proposes that the normal RNA-binding sites for the EXP proteins are distributed in both the nucleus and cytoplasm.
Materials and methods
Cells
HeLa S3 cells were grown on plates at 37°C in Dulbecco’s modified Eagle medium (DMEM; Gibco-BRL, Rockville, MD) containing 5% calf serum (CS) and 1% penicillin/streptomycin (P/S). For isolation of the EXP proteins, HeLa S3 cells were grown in spinner flasks in Joklik-modified MEM (Gibco-BRL), 5% CS and 1% P/S, or obtained from the Cell Culture Center (Minneapolis, MN). Normal (gift of T.Cooper, Baylor University, Houston, TX) and DM1 (gift of L.Timchenko, Baylor University) myoblasts were maintained in F10 (Ham’s) nutrient mixture (Gibco-BRL), 15% fetal bovine serum (FBS), 5% defined supplemented CS (Hyclone, Logan, UT), 0.5% chick embryo extract and 1% P/S. Mouse C2C12 myoblasts (ATCC, Rockville, MD), were maintained in DMEM, 20% FBS and 1% P/S, and then transferred to fusion medium (DMEM, 2% horse serum, 1% P/S) for differentiation. For cell immunofluorescence, myoblasts were grown on glass slides coated with rat tail collagen (Type 1, Sigma). Normal (GM03928) and DM1 (GM03986) human lymphoblasts were obtained from the NIGMS Human Genetic Cell Repository (HGCR; Coriell Institute for Medical Research, Camden, NJ), and grown in RPMI 1640 (Gibco-BRL) supplemented with 20% FBS and 1% P/S. Normal (GM03523) and DM1 (GM03132) fibroblasts were also obtained from HGCR and grown in DMEM supplemented with 20% FBS and 1% P/S. For fibroblast differentiation, cells were treated with a MyoD adenovirus, prepared as described previously (Murry et al., 1996). The AdMyoD virus stock (8 × 1010 particles/ml) was diluted 1:100 in DMEM supplemented with 20% FBS and 1% P/S, and applied to normal (GM03523) and DM1 (GM03132) fibroblasts grown to near confluency on collagen-coated slides. Following incubation for 4 h at 37°C, cells were washed with phosphate-buffered saline (PBS) and transferred to differentiation media (DMEM, 2% horse serum, 1% P/S), and FISH/cell immunofluorescence was performed following 3 days of differentiation.
DMPK, triplet repeat and TAR clones
DMPK clones with variable numbers of repeats were created using PCR mutagenesis of DMPK cDNA (DDBJ/EMBL/GenBank accession No. M87312) as described previously (Michalowski et al., 1999). Antisense transcript was created by cutting pDMPK.8-4 with EcoRV and transcribing using SP6 polymerase. This resulted in the introduction of 15 nucleotides of polylinker 5′, and 22 nucleotides of polylinker 3′, to the DMPK sequence.
Plasmids pCTG54 and pCTG90 were constructed by subcloning the CTG repeats from pDMPK.8-16 and pDMPK.8-6 into pSP72 at EcoRI and BamHI. Plasmid pCTG10.2 was created by subcloning a PCR-derived fragment with an identical surrounding sequence and 10 CTG repeats into pSP72. RNA transcripts were prepared by digesting pCTG10.2, pCTG54 and pCTG90 with BamHI and transcribing with T7 polymerase. The resulting transcripts contain 29 nucleotides of polylinker from pSP72 5′ to the CTG repeats. To create CAG transcripts, pCTG10.2 and pCTG54 were digested with EcoRI and transcribed using SP6 polymerase. The resulting RNAs contained 45 nucleotides of polylinker from pSP72 5′ to the CAG repeat. Clones containing 11, 20, 35, 74 and 97 CTG repeats were cut with SspI and XhoI. RNA transcripts containing CUG repeats with no polylinker sequence were created by in vitro transcription using T7 RNA polymerase. TAR and mTAR clones were created by PCR using overlapping oligonucleotides, cut with XhoI and HindIII and cloned into pSP72. The TAR and mutant TAR sequences used were identical to published sequences (wild type and mutant BL234, respectively) (Gatignol et al., 1991). pTAR (gaacucgagGGGUCUCUCUGGUUAGACCAGAUCUGAGCCUGGGAGCUCUCUGGCUAACUAGGGAACCCAaagcuu) and pmTAR (gaacucgagGGGUCUCUCUGGUUAGACCAGAUCUGUCGCUGGGAGCUCUCUGGCUAACUAGGGAACCCAaagcuu) transcripts were generated by digesting the plasmid with HindIII and transcribing using SP6 polymerase. The resulting RNAs contained three nucleotides of polylinker 5′ to the XhoI site.
EXP activity and immunopurification assays
EXP activity was determined using a photocrosslinking approach. Labeled RNA was transcribed from the DNA templates described above in the presence of either [α-32P]UTP or [α-32P]GTP using T7 or SP6 RNA polymerases (Gibco-BRL), and then purified on 8% polyacrylamide–7 M urea gels. Photocrosslinking assays were performed as described previously (Timchenko et al., 1996) using either HeLa nuclear extract or 50 ng of recombinant hEXP42. Quantification of the relative amount of EXP crosslinking, illustrated in Figure 1D, was obtained using a Storm PhosphorImager (Molecular Dynamics) to scan SDS–polyacrylamide gels in which each lane was loaded with equal radioactive counts. Crosslinked proteins were identified by specific immunopurification using antibody-bound protein A–Sepharose beads as described previously (Timchenko et al., 1996). Antibodies tested in this immunopurification assay included mouse anti-hEXP42 antiserum, monoclonal antibody (mAb) 3B1 against CUG-BP1 (Timchenko et al., 1996), mAbs 4B10 (anti-human hnRNPA1) and 4F4 (anti-human hnRNP C1/C2) (gifts of G.Dreyfuss, University of Pennsylvania, Philadelphia, PA), rabbit anti-human hnRNP D (gift of G.Brewer, Wake Forest University, Winston-Salem, NC), rabbit anti-mouse PRBP (gift of R.Braun, University of Washington, Seattle, WA), rabbit anti-human TRBP (gift of K.-T.Jeang, NIAID, Bethesda, MD) and rabbit anti-human eIF4A (gift of N.Sonenberg, McGill University, Montreal, Canada).
EXP protein purification and identification
HeLa cell nuclear extract was prepared as described (Swanson and Dreyfuss, 1988) from 40 l of S3 cells, dialyzed against buffer P [20 mM HEPES–KOH pH 7.9, 10% glycerol, 0.1 mM EDTA, 1.5 mM MgCl2, 1 mM dithiothreitol (DTT)], and treated with 20 U/ml micrococcal nuclease (Roche, Indianapolis, IN) in the presence of 1 mM CaCl2 for 10 min at 30°C. Digestion was stopped by addition of EGTA to 5 mM followed by centrifugation at 3000 g for 10 min. The supernatant was collected and loaded onto a DEAE–Sepharose Fast-Flow column (Pharmacia, 2.5 × 7.2 cm) that had been equilibrated in buffer P. EXP activity in the flow-through and wash fractions was collected, and 1 M NaCl in buffer P was added to a final concentration of 50 mM. This sample was then applied to a heparin–Sepharose CL6B column (Pharmacia, 2.5 × 8.1 cm) pre-equilibrated with 50 mM NaCl in buffer P. Following washing, EXP activity was eluted with 0.15 M NaCl in buffer P, and the eluate was concentrated (Centricon Plus-80 Biomax-30, Millipore, Bedford, MA) and then diluted >3-fold in 0.1% Tween-20 in buffer P. The heparin–Sepharose eluate was applied to a poly(U)–Sepharose 4B column (Sigma) pre-equilibrated with buffer P. The EXP2 activity, which bound to poly(U)–Sepharose, was eluted with 0.2 M NaCl in buffer P. The flow-through and wash fractions from poly(U)–Sepharose were applied to a ssDNA–agarose column equilibrated with buffer P. After washing with buffer P, EXP1 activity was eluted with 0.2 M NaCl in buffer P. The eluates from ssDNA–agarose (EXP1) and poly(U)–Sepharose (EXP2) were concentrated (Centricon-30, Millipore) to a volume of 100 µl. Samples were initially fractionated on a 15% SDS–polyacrylamide gel which was stained with 0.1% Coomassie Blue R-250 in water for 30 min. Proteins in the 45 kDa region corresponding to EXP activity were excised and the gel slices incubated in elution buffer (0.05% SDS, 4 mM Tris–acetate pH 8.4, 0.2 mM EDTA) for 15 min. The EXP proteins were eluted from the gel by electrophoresis (Little Blue Tank, ISCO, Lincoln, NE).
Following re-electrophoresis, the gel was stained and gel slices containing the EXP proteins were subjected to trypsin in-gel digestion and protein identification analysis at the HHMI Biopolymer Laboratory and W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University. Selected peptides were subjected to nanospray MS/MS analysis on a quadrupole time-of-flight (Q-Tof) mass spectrometer, and the resulting spectra were searched using the Sequest program (Thermoquest, San Jose, CA) combined with manual analysis. Significant matches were made to the MBNL (DDBJ/EMBL/GenBank accession No. CAA74155) and KIAA04288 (DDBJ/EMBL/GenBank accession No. BAA24858) proteins, including the acetylated N-terminal peptide (residues 1–12) and an internal peptide (residues 249–275). A third mass-matched peptide was also identified (residues 236–246). The EXP2 protein was not identified. Sequence data for hEXP35 have been submitted to the DDBJ/EMBL/GenBank database under accession No. AF255334.
cDNA clone isolation and transcript characterization
A HeLa S3 cell cDNA library (Stratagene) was screened as described previously (Timchenko et al., 1996) using a 32P-labeled PCR-generated DNA fragment prepared using oligonucleotide primers MSS789 (5′-GACCGACCATATGGCTGTTAGTGTCACACCAATTCGGG-3′) and MSS790 (5′-CGGGATCCCTACATCTGGGTAACATACTTGTGGCTAGTC-3′) derived from the MBNL cDNA sequence. Filters were hybridized overnight with 5 × 105 c.p.m./ml of labeled DNA and washed as described (Michalowski et al., 1999). Positive plaques were purified, the corresponding plasmids isolated following in vivo excision, and the cDNA inserts characterized by DNA sequencing. EXP mRNA expression in human tissues was analyzed using a human multi-tissue RNA blot (MTN blot, Clontech). Hybridization was performed for 14 h using a 32P-labeled hEXP42 cDNA NdeI–BamHI fragment as a probe. The blot was washed twice at room temperature for 15 min in 2× SSC and once for 30 min at 68°C in 0.2× SSC.
Recombinant protein isolation, antibody preparation and immunoblotting
To produce a His-tagged hEXP42 protein, a DNA fragment corresponding to full-length hEXP42 was synthesized by PCR using MSS789 and MSS790 as primers and a HeLa cDNA λZAP library as template. This hEXP42 cDNA was digested with NdeI and BamHI and subcloned into NdeI–BamHI digested pET15b (Novagen). Recombinant protein was prepared as described previously (Michalowski et al., 1999), and used to immunize mice for the production of anti-EXP polyclonal antibodies (Timchenko et al., 1996).
Mouse tissues for immunoblotting were prepared by disrupting the dissected tissue (Polytron cell disrupter) in lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 2 mM phenylmethylsulfonyl fluoride, 6 µg/ml aprotinin, 1 µg/ml leupeptin), followed by repeated passage through a 21G needle and addition of NP-40 to 1%. After incubation on ice (15 min), protein concentrations were determined (Coomassie Plus-200 Protein Assay Reagent, Pierce, Rockford, IL) and 15 µg of protein loaded per lane. For immunoblotting of mouse C2C12 myoblasts, whole cell extracts were prepared by washing confluent plates twice with PBS followed by addition of 1 ml of buffer E (50 mM Tris–HCl, pH 8.0, 0.1 mM EDTA, 1 mM DTT, 12.5 mM MgCl2, 20% glycerol, 0.1 M KCl, 1% Triton X-100, 1 µg/ml leupeptin/pepstatin) and scraping with a rubber policeman. After incubating on ice for 5 min, samples were spun at 4300 g for 4 min, and the supernatant was collected and stored at –80°C. Immunoblotting was performed as described previously using a 1:100 dilution of mouse anti-hEXP42 antiserum, a 1:250 dilution of mouse anti-human TRBP antiserum, a 1:500 dilution of mAb 3B1 (anti-CUG-BP1), a 1:400 dilution of mAb MY-32 (anti-myosin HC, Sigma) or a 1:1000 dilution of mAb 4F4 (anti-hnRNP C proteins) (Timchenko et al., 1996; Michalowski et al., 1999).
Fluorescence in situ hybridization and cell immunofluorescence
FISH was performed essentially as described previously (Taneja et al., 1995) using Cy3-conjugated (CAG)10 (Operon Technologies, Alameda, CA) except that salmon sperm DNA was omitted from the hybridization solution. The subcellular distribution of the EXP proteins was determined by immunofluorescence microscopy (Zeiss Axioplan 2 fluorescence microscope) using a 1:100 dilution of the anti-hEXP 42 antiserum and a 1:20 dilution of a fluorescein isothiocyanate-conjugated IgG1-specific goat anti-mouse secondary antibody (Southern Biotechnology, Birmingham, AL).
Acknowledgments
Acknowledgements
We thank P.Non for technical assistance, D.Duke and L.Green of the University of Florida Hybridoma Core Facility for antibody preparation, the HHMI Biopolymer Laboratory & W.M.Keck Foundation Biotechnology Resource Laboratory at Yale University for protein analysis, and R.Braun, G.Brewer, T.Cooper, G.Dreyfuss, K.-T.Jeang, N.Sonenberg and L.Timchenko for reagents. We also thank C.Brannan, W.Hauswirth, A.Lewin and J.Resnick for comments on the manuscript. This work was supported by grants from the NIH (GM46272 and AR46799 to M.S.S., T32 AI07110-18 to C.R.U.), and an American Heart Association Established Investigator Grant (M.S.S.).
References
- Alwazzan M., Newman,E., Hamshere,M.G. and Brook,J.D. (1999) Myotonic dystrophy is associated with a reduced level of RNA from the DMWD allele adjacent to the expanded repeat. Hum. Mol. Genet., 8, 1491–1497. [DOI] [PubMed] [Google Scholar]
- Amack J.D., Paguio,A.P. and Mahadevan,M.S. (1999) Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum. Mol. Genet., 8, 1975–1984. [DOI] [PubMed] [Google Scholar]
- Anderson J.T., Wilson,S.M., Datar,K.V. and Swanson,M.S. (1993) NAB2: a yeast nuclear polyadenylated RNA-binding protein essential for cell viability. Mol. Cell. Biol., 13, 2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artero R., Prokop,A., Paricio,N., Begemann,G., Pueyo,I., Mlodzik,M., Perez-Alonso,M. and Baylies,M.K. (1998) The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev. Biol., 195, 131–143. [DOI] [PubMed] [Google Scholar]
- Ashley C.T. Jr and Warren,S.T. (1995) Trinucleotide repeat expansion and human disease. Annu. Rev. Genet., 29, 703–728. [DOI] [PubMed] [Google Scholar]
- Begemann G., Paricio,N., Artero,R., Kiss,I., Perez-Alonso,M. and Mlodzik,M. (1997) muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development, 124, 4321–4331. [DOI] [PubMed] [Google Scholar]
- Caskey C.T., Swanson,M.S. and Timchenko,L.T. (1996) Myotonic dystrophy: discussion of molecular mechanism. Cold Spring Harb. Symp. Quant. Biol., 61, 607–614. [PubMed] [Google Scholar]
- Davis B.M., McCurrach,M.E., Taneja,K.L., Singer,R.H. and Housman,D.E. (1997) Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl Acad. Sci. USA, 94, 7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatignol A., Buckler-White,A., Berkhout,B. and Jeang,K.-T. (1991) Characterization of a human TAR RNA-binding protein that activates the HIV-1 LTR. Science, 251, 1597–1600. [DOI] [PubMed] [Google Scholar]
- Groenen P. and Wieringa,B. (1998) Expanding complexity in myotonic dystrophy. BioEssays, 20, 901–912. [DOI] [PubMed] [Google Scholar]
- Hamshere M.G., Newman,E.E., Alwazzan,M., Athwal,B.S. and Brook,J.D. (1997) Transcriptional abnormality in myotonic dystrophy affects DMPK but not neighboring genes. Proc. Natl Acad. Sci. USA, 94, 7394–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G., Bächner,D., Coerwinkel,M., Wormskamp,N., Hameister,H. and Wieringa,B. (1995) Structural organization and developmental expression pattern of the mouse WD-repeat gene DMR-N9 immediately upstream of the myotonic dystrophy locus. Hum. Mol. Genet., 4, 843–852. [DOI] [PubMed] [Google Scholar]
- Jansen G. et al. (1996) Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nature Genet., 13, 316–324. [DOI] [PubMed] [Google Scholar]
- Klesert T.R., Otten,A.D., Bird,T.D. and Tapscott,S.J. (1997) Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nature Genet., 16, 402–406. [DOI] [PubMed] [Google Scholar]
- Klesert T.R., Cho,D.H., Clark,J.I., Maylie,J., Adelman,J., Snider,L., Yuen,E.C., Soriano,P. and Tapscott,S.J. (2000) Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nature Genet., 25, 105–109. [DOI] [PubMed] [Google Scholar]
- Korade-Mirnics Z., Babitzke,P. and Hoffman,E. (1998) Myotonic dystrophy: molecular windows on a complex etiology. Nucleic Acids Res., 26, 1363–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korade-Mirnics Z., Tarleton,J., Servidei,S., Casey,R.R., Gennarelli,M., Pegoraro,E., Angelini,C. and Hoffman,E.P. (1999) Myotonic dystrophy: tissue-specific effect of somatic CTG expansions on allele-specific DMAHP/SIX5 expression. Hum. Mol. Genet., 8, 1017–1023. [DOI] [PubMed] [Google Scholar]
- Krahe R., Ashizawa,T., Abbruzzese,C., Roeder,E., Carango,P., Giacanelli,M., Funanage,V.L. and Siciliano,M.J. (1995) Effect of myotonic dystrophy trinucleotide repeat expansion on DMPK transcription and processing. Genomics, 28, 1–14. [DOI] [PubMed] [Google Scholar]
- Lee K., Fajardo,M.A. and Braun,R.E. (1996) A testis cytoplasmic RNA-binding protein that has properties of a translational repressor. Mol. Cell. Biol., 16, 3023–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A., Logigian,E., Callahan,L., McClain,C., Henderson,D., Krym,M. and Thornton,C.A. (2000) Myotonic dystrophy in mice expressing an expanded CUG repeat. Science, in press. [DOI] [PubMed] [Google Scholar]
- Meola G. and Moxley,R.T.,III (1999) Myotonic disorders: myotonic dystrophy and proximal myotonic myopathy. In Schapira,A.H.V. and Griggs,R.C. (eds), Muscle Diseases. Butterworth-Heinemann, Boston, MA, pp. 115–134. [Google Scholar]
- Michalowski S., Miller,J.W., Urbinati,C.R., Paliouras,M., Swanson,M.S. and Griffith,J. (1999) Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucleic Acids Res., 27, 3534–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry C.E., Kay,M.A., Bartosek,T., Hauschka,S.D. and Schwartz,S.M. (1996) Muscle differentiation during repair of myocardial necrosis in rats via gene transfer with MyoD. J. Clin. Invest., 98, 2209–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napierala M. and Krzyzosiak,W.J. (1997) CUG repeats present in myotonin kinase RNA form metastable ‘slippery’ hairpins. J. Biol. Chem., 272, 31079–31085. [DOI] [PubMed] [Google Scholar]
- Otten A.D. and Tapscott,S.J. (1995) Triplet repeat expansion in myotonic dystrophy alters the adjacent chromatin structure. Proc. Natl Acad. Sci. USA, 92, 5465–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips A.V., Timchenko,L.T. and Cooper,T.A. (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science, 280, 737–741. [DOI] [PubMed] [Google Scholar]
- Ranum L.P.W., Rasmussen,P.F., Benzow,K.A., Koob,M.D. and Day,J.W. (1998) Genetic mapping of a second myotonic dystrophy locus. Nature Genet., 19, 196–198. [DOI] [PubMed] [Google Scholar]
- Reddy S. et al. (1996) Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nature Genet., 13, 325–335. [DOI] [PubMed] [Google Scholar]
- Ricker K. et al. (1999) Linkage of proximal myotonic myopathy to chromosome 3q. Neurology, 52, 170–171. [DOI] [PubMed] [Google Scholar]
- Sarkar P.S., Appukuttan,B., Han,J., Ito,Y., Ai,C., Tsai,W., Chai,Y., Stout,J.T. and Reddy,S. (2000) Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nature Genet., 25, 110–114. [DOI] [PubMed] [Google Scholar]
- Swanson M.S. and Dreyfuss,G. (1988) RNA binding specificity of hnRNP proteins: a subset bind to the 3′ end of introns. EMBO J., 11, 3519–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja K.L., McCurrach,M., Schalling,M., Housman,D. and Singer,R.H. (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol., 128, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton C.A., Wymer,J.P., Simmons,Z., McClain,C. and Moxley,R.T.,III (1997) Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nature Genet., 16, 407–409. [DOI] [PubMed] [Google Scholar]
- Tian B., White,R.J., Xia,T., Welle,S., Turner,D.H., Mathews,M.B. and Thornton,C.A. (2000) Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA, 6, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko L.T., Miller,J.W., Timchenko,N.A., DeVore,D.R., Datar, K.V., Lin,L., Roberts,R., Caskey,C.T. and Swanson,M.S. (1996) Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res., 24, 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Pegoraro,E., Menegazzo,E., Gennarelli,M., Hoop,R.C., Angelini,C. and Hoffman,E.P. (1995) Myotonic dystrophy: evidence for a possible dominant-negative RNA mutation. Hum. Mol. Genet., 4, 599–606. [DOI] [PubMed] [Google Scholar]
- Wang Y.H., Amirhaeri,S., Kang,S., Wells,R.D. and Griffith,J.D. (1994) Preferential nucleosome assembly at DNA triplet repeats from the myotonic dystrophy gene. Science, 265, 669–671. [DOI] [PubMed] [Google Scholar]
- Winchester C.L., Ferrier,R.K., Sermoni,A., Clark,B.J. and Johnson,K.J. (1999) Characterization of the expression of DMPK and SIX5 in the human eye and implications for pathogenesis in myotonic dystrophy. Hum. Mol. Genet., 8, 481–492. [DOI] [PubMed] [Google Scholar]