

Abstract
CM2 is the second membrane protein of influenza C virus. Although its biochemical characteristics, coding strategy, and properties as an ion channel have been extensively studied, the role(s) of CM2 in the virus replication cycle remains to be clarified. In order to elucidate this role, in the present study we generated CM2-deficient influenza C virus-like particles (VLPs) and examined the VLP-producing 293T cells, VLPs, and VLP-infected HMV-II cells. Quantification of viral RNA (vRNA) in the VLPs by real-time PCR revealed that the CM2-deficient VLPs contain approximately one-third of the vRNA found in wild-type VLPs although no significant differences were detected in the expression levels of viral components in VLP-producing cells or in the number and morphology of the generated VLPs. This finding suggests that CM2 is involved in the genome packaging process into VLPs. Furthermore, HMV-II cells infected with CM2-deficient VLPs exhibited significantly reduced reporter gene expression. Although CM2-deficient VLPs could be internalized into HMV-II cells as efficiently as wild-type VLPs, a smaller amount of vRNA was detected in the nuclear fraction of CM2-deficient VLP-infected cells than in that of wild-type VLP-infected cells, suggesting that the uncoating process of the CM2-deficient VLPs in the infected cells did not proceed in an appropriate manner. Taken together, the data obtained in the present study indicate that CM2 has a potential role in the genome packaging and uncoating processes of the virus replication cycle.
Influenza C virus has seven RNA segments of negative polarity, which encode PB2, PB1, P3, hemagglutinin esterase-fusion (HEF) glycoprotein, nucleoprotein (NP), matrix (M1) protein and CM2, and the nonstructural proteins (NS1 and NS2) (34). PB2, PB1, and P3 are subunits of the RNA polymerase of the virus. HEF, which has receptor-binding, receptor-destroying, and fusion activities, forms a spike on the virion (10). NP participates in forming ribonucleoproteins (RNPs) with viral RNA (vRNA), PB2, PB1, and P3. M1 is abundantly present beneath the envelope, which gives rigidity to the virion. CM2 is the second membrane protein of the virus (11, 12). NS1 is involved in viral mRNA splicing (31), and NS2 is a nuclear export protein (35) and is incorporated into the virions (21).
RNA segment 6 (M gene) of influenza C virus is 1,180 or 1,181 nucleotides in length and encodes the M1 and CM2 proteins. The predominant mRNA detected in virus-infected cells lacks a region from nucleotides 755 to 982 (numbering is based on the sequence composed of 1,181 nucleotides) and encodes a 242-amino-acid matrix protein (M1) (46). Unspliced mRNA from the RNA segment, synthesized in small quantities, encodes the P42 protein, which contains an additional 132 amino acids on the C terminus of M1 (11, 13). P42 is cleaved by signal peptidase at an internal cleavage site to generate CM2 composed of the C-terminal 115 amino acids, in addition to the M1′ protein composed of the N-terminal 259 amino acids (14, 37).
The biochemical characteristics of CM2 have been extensively analyzed. CM2 is a type III membrane protein that is oriented in membranes with a 23-amino-acid N-terminal extracellular domain, a 23-amino-acid transmembrane domain, and a 69-amino-acid C-terminal cytoplasmic domain (8, 12, 36). It is abundantly expressed in virus-infected cells, and a small amount of CM2 was shown to be incorporated into the progeny virus particles (12). It forms disulfide-linked dimers and tetramers and is posttranslationally modified by palmitoylation and phosphorylation (12, 22, 36, 42). It is also modified by N-glycosylation at asparagine residue 11, and, as a result, three forms of CM2 with different electrophoretic mobilities (CM20, CM2a, and CM2b) are detected in infected cells (12, 36).
The function of CM2 has recently been reported by several groups. CM2 was found to form a Cl− channel when expressed in Xenopus laevis oocytes (15), and electrophysiological studies of CM2-expressing mouse erythroleukemia cells have identified Na+-activated proton permeability in addition to the low pH-activated Cl− permeability (Y. Muraki, I. V. Chizhmakov, D. C. Ogden, and A. J. Hay, unpublished data). When expressed together with a pH-sensitive hemagglutinin (HA) of influenza A virus, CM2 was demonstrated to modulate the pH of the exocytic pathway, suggesting that CM2 has proton permeability (3). Thus, the features of CM2 have been precisely characterized from both biochemical and functional perspectives.
Ion channel proteins of influenza A and B viruses have been shown to play critical roles in the virus life cycle. The M2 protein of influenza A virus functions as a proton channel during the entry of the virus into susceptible cells by allowing the acidification of the virion interior (34). In addition, M2 has been demonstrated to have a role in infectious virus production and virion morphology (5, 18, 25, 26, 38). Larger truncations of the M2 cytoplasmic tail resulted in a significant decrease in infectious virus production, indicating that M2 cytoplasmic region is required for efficient genome packaging. The influenza B virus BM2 protein has also been shown to have a proton channel activity (28) and is involved in the incorporation of the virus genome into virions (16, 17). It is likely, therefore, that CM2 is also crucial in influenza C virus replication because the biochemical characteristics of CM2 are closely similar to those of M2 (12, 36), and, like M2 and BM2, CM2 seems to have permeability to proton as described above. However, the precise role(s) of CM2 in the influenza C virus replication cycle remains to be elucidated.
Uncoating is the release of viral nucleic acid from its protective protein coat or envelope. In the case of influenza A virus, the HA spike glycoprotein on the virions binds to sialic acid-containing receptors, and the virion-receptor complex is endocytosed. Upon acidification within the endosome, the viral HA undergoes a conformational rearrangement that produces a fusogenic peptide, resulting in fusion of the viral and endosomal membrane (envelope fusion). To allow release of viral RNP (vRNP) into the cytoplasm, the H+ ions in the acidic endosome are introduced into the virion interior through the M2 ion channel. As a result, vRNP is primed to dissociate from M1 after envelope fusion. The released vRNP is then transported into the nucleus through the nuclear pore complex via a nuclear localization signal-dependent mechanism (45). The entry and uncoating processes of influenza C virus are considered to be similar to those of influenza A virus. However, in vitro analysis of virions showed that the M1 protein of influenza C virus readily disintegrates under neutral and alkaline conditions, a phenomenon that is contrary to the process in influenza A virus in which M1 dissociates under the acidic conditions (48). Thus, the uncoating process in influenza C virus remains to be clarified, particularly in relation to CM2 function.
In the present study, to clarify the role of CM2 in the influenza C virus replication cycle, we generated CM2-deficient influenza C VLPs and analyzed the VLP-producing cells, VLPs, and VLP-infected cells. Evidence was obtained that CM2 is involved in the packaging of the reporter gene into VLPs and in the uncoating process of the VLP, suggesting that CM2 is essential for virus replication.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
293T cells were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (29). The HMV-II line of human malignant melanoma cells was maintained in RPMI 1640 medium with 10% calf serum (32). The Ann Arbor/1/50 (AA/50) strain of influenza C virus was grown in the amniotic cavity of 9-day-old embryonated hens' eggs as previously described (47). Monoclonal antibodies (MAbs) against the HEF, NP, and M1 proteins of AA/50 and antisera against CM2, NS1, and NS2 were reported previously (2, 11, 31, 39, 40). Anti-lamin B and anti-α-tubulin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and Sigma (St. Louis, MO), respectively.
Plasmid DNAs.
Plasmid DNAs, pPolI/NP-AA.GFP(−) and pPolI/NP-AA.Luc(−), from which green fluorescent protein-vRNA (GFP-vRNA) and luciferase-vRNA are, respectively, transcribed under the control of the RNA polymerase I promoter, were described previously (29). The influenza C virus protein-expressing plasmid DNAs, pcDNA/PB2-AA, pcDNA/PB1-AA, pcDNA/P3-AA, pCAGGS.MCS/NP-AA, pME18S/HEF-AA, pCAGGS.MCS/M1-AA, pME18S/Met-CM2-YA, pME18S/NS1-YA, and pME18S/NS2-YA, were described previously (29).
Generation and purification of VLPs.
For the generation of wild-type (WT) VLPs (see Results section), 293T cells in a 35 mm-petri dish were transfected with the following 10 plasmids as described previously (29): 0.5 μg of pPolI/NP-AA.GFP(−) or pPolI/NP-AA.Luc(−) and 0.125 μg of pcDNA/PB2-AA, 0.25 μg of pcDNA/PB1-AA, 0.25 μg of pcDNA/P3-AA, 0.25 μg of pCAGGS.MCS/NP-AA, 1.25 μg of pME18S/HEF-AA, 0.3 μg of pCAGGS.MCS/M1-AA, 0.0875 μg of pME18S/Met-CM2-YA, 0.7 μg of pME18S/NS1-YA, and 1.0 μg of pME18S/NS2-YA. For the generation of VLPs lacking CM2 (ΔCM2 VLPs) (see Results section), 293T cells were transfected with a mixture containing same amounts of the following nine plasmids: pPolI/NP-AA.GFP(−) or pPolI/NP-AA.Luc(−), pcDNA/PB2-AA, pcDNA/PB1-AA, pcDNA/P3-AA, pCAGGS.MCS/NP-AA, pME18S/HEF-AA, pCAGGS.MCS/M1-AA, pME18S/NS1-YA, and pME18S/NS2-YA. In the latter mixture, 0.0875 μg of pME18S was included instead of pME18S/Met-CM2-YA to adjust the total amount of plasmid DNA. To prepare a large quantity of VLPs, 293T cells in a 100 mm-petri dish were transfected with a proportionally increased amount of the plasmids described above. The culture medium of the transfected 293T cells was collected at 48 h posttransfection (p.t.) and clarified by low-speed centrifugation. The supernatant was layered onto 30% (wt/vol) sucrose in NTE buffer (100 mM NaCl, 10 mM Tris [pH 7.4], 1 mM EDTA) and centrifuged at 200,000 × g for 2 h at 4°C in a Beckman SW40 Ti rotor (Beckman, Fullerton, CA). The resulting pellet was suspended in 10% glycerol in phosphate-buffered saline (PBS).
Electron microscopy.
The culture medium from 293T cells transfected with the mixtures of 9 and 10 plasmids was collected at 48 h p.t. and clarified by low-speed centrifugation. As described previously (18), the resultant supernatants were put onto a Formvar-coated copper grid, stained with 2% phosphotungstic acid solution, and examined with a JEM-1200EX electron microscope at 80 kV.
Immunoblotting of 293T cells and VLPs.
Transfected 293T cells, purified VLPs, and nuclear and cytoplasmic fractions of HMV-II cells (see below) were resolved by SDS-PAGE on 17.5% gels containing 4 M urea under reducing conditions (47). After SDS-PAGE, immunoblotting was carried out as described previously (29) by using the MAbs and antisera described above. The proteins were detected by an enhanced chemiluminescence Western blotting system (GE Healthcare, Piscataway, NJ) according to the manufacturer's instructions. Band intensities were measured by ImageJ software, version 1.38 (W. Rasband, National Institutes of Health [http://rsb.info.nih.gov/ij/]).
Infection of HMV-II cells with VLPs.
The VLPs suspended in PBS or the supernatant of the plasmid-transfected 293T cells were treated with tosylamide-phenylethyl chloromethyl ketone (TPCK)-treated trypsin (20 μg/ml) at 37°C for 10 min, followed by the addition of soybean trypsin inhibitor. The monolayered HMV-II cells were infected with the VLPs at 33°C for 60 min and subsequently infected with the helper virus (AA/50) at a multiplicity of infection (MOI) of 5 and incubated for up to 48 h. In the real-time PCR for the quantification of incoming GFP-vRNA (see below), the helper virus was not used for infection. GFP-positive HMV-II cells were observed under a fluorescent microscope (Leica Microsystems, Exton, PA) and photographed. Luciferase activity expressed in the HMV-II cells infected with VLPs was measured with Lumat 9507 (Berthold, Bad Wildungen, Germany) according to the manufacturer's instructions for the luciferase assay system (Promega, Madison, WI) (29).
RNA extraction, reverse transcription, and real-time PCR.
The RNAs were extracted from the plasmid-transfected 293T cells, purified VLPs, and nuclear and cytoplasmic fractions of VLP-infected HMV-II cells using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The RNA preparations were treated with Turbo DNA-free DNase (Ambion, Austin, TX) to degrade the residual plasmid DNAs and then cleaned using an RNeasy Mini Kit. The cDNA was synthesized by reverse transcription using a primer complementary to the 12 nucleotides of the 3′ end of the influenza C vRNA (19) and subjected to real-time PCR as follows.
Quantification of GFP-vRNA by real-time PCR was carried out using a LightCycler (Roche, Mannheim, Germany). PCR was performed in a 20-μl mixture containing 2 μl of cDNA preparation, 10 μl of SYBR Green Real-time PCR Master Mix (Toyobo, Osaka, Japan), 0.8 μl of sense primer, 0.8 μl of antisense primer, and 6.4 μl of RNase-free water. The PCR protocol consisted of an initial denaturation step at 95°C for 30 s, followed by 40 cycles: denaturation at 95°C for 5 s, annealing at 55°C for 10 s, and extension at 72°C for 15 s. Primer sequences are as follows (27): GFP-forward, 5′-AGAAGAACGGCATCAAGGTG-3′; GFP-reverse, 5′-GAACTCCAGCAGGACCATGT-3′. The standard curve was calculated based on the results of real-time PCR using a series of 10-fold dilutions of pPolI/NP-AA.GFP(−) as a template. As a loading control of RNAs extracted from the cells, β-actin mRNA was quantified by real-time PCR using the following primers (1): β-actin-forward, 5′-CCACACTGTGCCCATCTACG-3′; β-actin-reverse, 5′-AGGATCTTCATGAGGTAGTCAGTCAG-3′. The standard curve for β-actin mRNA quantification was calculated using a series of 10-fold dilutions of the 459-bp product of β-actin cDNA that had been PCR amplified with a set of primers (primer sequences available on request).
Flow cytometry of HMV-II cells infected with VLPs.
Monolayered HMV-II cells were trypsinized using Accutase (Sigma-Aldrich, Munich, Germany) and then added to the supernatants of the transfected 293T cells containing VLPs so that the ratio of the number of the VLPs to that of HMV-II cells was maintained at approximately 3:1. After incubation of the cells on ice for 30 min, an aliquot of the cells was transferred to 33°C and incubated for 180 min. The cells were then washed twice with Hanks' balanced salt solution and incubated with anti-HEF MAb J14 (primary antibody) and Alexa Fluor 488-conjugated anti-mouse IgG antibody (secondary antibody) (Molecular Probes, Eugene, OR). The cells were subjected to flow cytometry using a FACSCalibur instrument (Becton Dickinson, San Jose, CA). The histogram was drawn using Cell Quest software (Beckton Dickinson).
VLP-mediated hemolysis.
VLP-mediated hemolysis was analyzed as described previously (20). Briefly, 100 μl of the VLP suspension in PBS (32 HA units/ml [HAU/ml]) was added to 0.5 ml of 2% chicken erythrocytes in PBS at pH 7.0 and incubated on ice for 30 min. The mixture was centrifuged at 500 × g, and the pellet was suspended in 0.5 ml of saline buffered with 10 mM morpholineethanesulfonic acid (MES) of various pH levels and incubated at 37°C for 60 min. The mixture was then centrifuged, and the supernatants were measured for the optical density at 540 nm.
Cell fractionation.
VLP-infected HMV-II cells were subjected to fractionation as described previously (41). Briefly, HMV-II cells mildly solubilized with RSB buffer (10 mM Tris-HCl, 100 mM NaCl, 1.5 mM MgCl2, pH 8.0) containing 0.3% NP-40 for 30 min at 0°C were divided into two fractions by centrifugation at 1,200 × g for 5 min at 4°C. The precipitate was washed twice with the RSB buffer containing 0.3% NP-40 and then used as the nuclear fraction. The supernatant was recentrifuged at 10,000 × g for 5 min at 4°C, and the resulting supernatant was used as the cytoplasmic fraction. Aliquots of the respective fractions were then used for immunoblotting and RNA extraction.
Statistical analysis.
All functional tests were carried out independently at least three times. Data between groups were analyzed using a Student's t test or paired t test. A P value of less than 0.05 was considered statistically significant. All statistical analyses were performed using SPSS software, version 17.
RESULTS
CM2 is not required for influenza C VLP generation.
We previously reported that the transfection of 10 plasmid DNAs [pPolI/NP-AA.GFP(−), pcDNA/PB2-AA, pcDNA/PB1-AA, pcDNA/P3-AA, pCAGGS.MCS/NP-AA, pME18S/HEF-AA, pCAGGS.MCS/M1-AA, pME18S/Met-CM2-YA, pME18S/NS1-YA and pME18S/NS2-YA] into 293T cells resulted in the generation of influenza C virus VLPs (referred to as WT VLPs in the present study) (29). The GFP-vRNA minigenome and nine virus proteins (PB2, PB1, P3, NP, HEF, M1, CM2, NS1, and NS2) were expressed from the respective plasmids in 293T cells, and the cells were incubated at 33°C. At 48 h p.t., the influenza C VLPs were detected in the culture medium by electron microscopy. HMV-II cells infected with the generated VLPs and a helper virus (AA/50) exhibited GFP expression at 48 h postinfection (p.i.), indicating that the GFP-vRNA in the VLPs was transferred to HMV-II cells. GFP expression in the VLP-infected HMV-II cells without superinfection was only background levels (29), indicating that GFP expression in the VLP-infected cells is dependent on superinfection with a helper virus.
In the present study, as a control experiment, the supernatant of the 293T cells transfected with the above 10 plasmids was examined by electron microscopy. We confirmed that the supernatant contained WT VLPs of filamentous morphology (Fig. 1A, left panel). Next, to investigate the effects of CM2 on VLP generation, we eliminated the CM2-expressing plasmid, pME18S/Met-CM2-YA, in which a methionine-CM2 open reading frame (ORF) of C/Yamagata/1/88 was inserted under the expression promoter (29). The mixture containing the nine plasmids, not including pME18S/Met-CM2-YA, was transfected into 293T cells, and the supernatant was also confirmed to contain filamentous VLPs (Fig. 1A, right panel). The packing of surface glycoproteins in regular hexagonal arrays, a striking feature of influenza C virus preparations reported previously (6, 7, 9, 33, 44), was clearly detected on the VLPs (Fig. 1A, right panel, triangle), indicating that the defect in CM2 expression in the 293T cells did not lead to any inhibition of VLP formation or change in VLP morphology. In the subsequent section, therefore, VLPs generated from the 293T cells transfected with the nine plasmids are referred to as ΔCM2 VLPs.
FIG. 1.

VLPs generated from 293T cells transfected with plasmid DNAs. VLPs generated from 293T cells transfected with the 10 (WT VLP) or nine (ΔCM2 VLP) plasmid DNAs were analyzed (see Materials and Methods for plasmid composition). (A) The supernatant of the 293T cells was negatively stained and observed using an electron microscope. Black triangles indicate hexagonal arrays of HEF (right panel). Bar, 200 nm. (B) Purified VLPs were lysed, electrophoresed by SDS-PAGE, and subjected to immunoblotting using a mixture of MAbs against HEF, NP, and M1 (left) and with antiserum against CM2 (right). Band intensities of HEF, NP, and M1 in the respective lanes were measured, and the ratios of HEF/M1, NP/M1, and NP/HEF are shown. (C) RNA was extracted from the purified VLPs, treated with DNase, reverse transcribed, and then subjected to real-time PCR for quantification of GFP-vRNA. Because the initial data were not normally distributed, they were log transformed for statistical analysis. The copy number of the GFP-vRNA in the WT VLPs was used for normalization. Each bar represents the mean ± standard errors of the means.
CM2 is involved in the packaging of the reporter gene into VLPs.
In order to compare the number of WT VLPs and ΔCM2 VLPs generated, a given amount of the supernatant from a constant number of the transfected 293T cells was clarified through 30% sucrose in NTE buffer, and the pellet containing the VLPs was subjected to hemagglutination and determination of protein concentration (23). In three independent experiments, the hemagglutinin titers of the clarified WT VLPs and ΔCM2 VLPs were reproducibly 128 HAU/ml, and the protein concentrations of WT VLPs and ΔCM2 VLPs were 1.1 ± 0.2 and 1.1 ± 0.3 mg/ml, respectively. This observation indicates that there was no significant difference in the number of the VLPs generated. Thus, the absence of CM2 did not appear to affect the generation of VLPs.
Next, we investigated virus proteins in the VLPs (Fig. 1B). A given amount of the clarified VLPs was analyzed by immunoblotting using a mixture of MAbs against HEF (S16), NP (H27), and M1 (L2) and antisera against CM2. No significant differences were observed in the amounts of HEF and M1 between the WT VLPs and ΔCM2 VLPs, and, as expected, CM2 was detected only in the WT VLPs.
It should be noted that the amount of NP present in the ΔCM2 VLPs was slightly less than that in the WT VLPs (Fig. 1B). The ratios of both NP/M1 and NP/HEF were lower in the ΔCM2 VLPs than in the WT VLPs although the ratio of HEF/M1 exhibited no significant difference (Fig. 1B), suggesting that the amount of vRNP in the ΔCM2 VLPs is less than that in the WT VLPs. We therefore examined the amount of GFP-vRNA in the VLPs. RNA was extracted from an identical amount of WT VLPs and ΔCM2 VLPs, and reverse-transcribed cDNAs were subjected to real-time PCR using a pair of primers specific for the GFP sequence. As shown in Fig. 1C, four independent experiments showed that the amount of GFP-vRNA in the ΔCM2 VLPs was approximately 37% of that in the WT VLPs (P < 0.01).
To demonstrate the specificity in terms of the generation of ΔCM2 VLPs, an experiment was carried out in which the M1-expressing plasmid was eliminated from the system. The mixture containing the nine plasmid DNAs, not including pCAGGS.MCS/M1-AA, was transfected into 293T cells, and the supernatant was subjected to Western blotting and reverse transcription-PCR. As a result, the HEF, NP, and M1 proteins as well as GFP-vRNA were not detected in the supernatant of 293T cells transfected with the nine plasmids (data not shown). As M1 is considered to be involved in virion morphogenesis, we concluded that the detected proteins and GFP-vRNA shown in Fig. 1B and C are specific to the assembly and budding process in VLP generation and do not result from the nonspecific disintegration of the transfected 293T cells.
To rule out the possibility that the elimination of pME18S/Met-CM2-YA affects the expression of other components in the VLP-producing cells, 293T cells transfected with the 9 or 10 plasmids were analyzed at 24 h p.t. (Fig. 2). Immunoblotting using a mixture of MAbs against HEF (S16), NP (H27), and M1 (L2) and antisera against CM2, NS1, and NS2 revealed that, except for CM2 expression, no significant differences were observed in the expression levels of viral proteins between WT VLP- and ΔCM2 VLP-producing cells (Fig. 2A). Fluorescent microscopy showed that GFP expression in the ΔCM2 VLP-producing cells was comparable to that in the WT VLP-producing cells (Fig. 2B). Furthermore, real-time PCR revealed no significant difference in the amounts of GFP-vRNA in the two cell groups (Fig. 2C). Thus, based on the comparison of GFP-vRNA and viral proteins in the VLPs and 293T cells, we were able to conclude that CM2 is involved in GFP-vRNA packaging into the VLPs.
FIG. 2.

Expression of viral components and GFP in the 293T cells transfected with plasmid DNAs. 293T cells transfected with the 10 (WT) or 9 (ΔCM2) plasmid DNAs were analyzed (see Materials and Methods for plasmid composition). (A) The lysates of the 293T cells at 24 h p.t. were electrophoresed and subjected to immunoblotting using a mixture of MAbs against HEF, NP, and M1 (left) and with antisera against CM2, NS1, and NS2 (right). (B) The plasmid-transfected 293T cells were observed using a fluorescent microscope. The mock-transfected cells are shown in the left panel (magnification, ×50). (C) RNA was extracted from the 293T cells, treated with DNase, reverse transcribed, and then subjected to real-time PCR for quantification of GFP-vRNA. The relative copy number of the GFP-vRNA from 293T cells transfected with the 10 plasmids (WT) is expressed as 1.0. The result from mock-transfected cells was shown as the control. A representative result is shown.
CM2 is indispensable to efficient gene transfer by VLPs.
We previously reported that WT VLPs are capable of transferring GFP-vRNA to HMV-II cells (29), indicating that transcription and translation of GFP-vRNA occurred in the HMV-II cells infected with WT VLPs. In the present study, in order to investigate the ability of ΔCM2 VLPs to transfer the reporter gene, an equal amount of WT VLPs or ΔCM2 VLPs, based on hemagglutinin units and protein concentrations, was added to the monolayered HMV-II cells, followed by superinfection with the helper virus. As shown in Fig. 3A, GFP expression was detected in the HMV-II cells infected with WT VLPs, whereas only a few HMV-II cells infected with ΔCM2 VLPs expressed GFP. To quantify the result, VLPs containing luciferase-vRNA were generated by transfecting pPolI/NP-AA.Luc(−) (29) and the nine (or eight) virus protein-expressing plasmids into 293T cells and tested. Five independent experiments revealed that the relative luciferase unit value in the HMV-II cells infected with ΔCM2 VLPs was 3.5% of that in the HMV-II cells infected with WT VLPs (P < 0.05) (Fig. 3B). Taken together, the results showed that the transcription and translation of GFP-vRNA were significantly reduced in the HMV-II cells infected with ΔCM2 VLPs.
FIG. 3.

Reporter gene expression in HMV-II cells infected with VLPs. (A) HMV-II cells infected with mock (Control), WT VLPs, or ΔCM2 VLPs, followed by superinfection with AA/50, were incubated for 48 h and observed by fluorescence microscopy (magnification, ×100). (B) WT VLPs or ΔCM2 VLPs containing luciferase-vRNA were used for infection, and luciferase activity detected in the infected HMV-II cells was measured at 24 h p.i. and is expressed as relative light units (RLU). The RLU value from the HMV-II cells infected with WT VLPs was expressed as 100 and used for normalization. Each bar represents the mean ± standard errors of the means. The supernatant from mock-transfected 293T cells was used as a control.
Attachment, entry, and fusion activities of VLPs.
The amount of GFP-vRNA in the ΔCM2 VLPs was approximately one-third of that in the WT VLPs (Fig. 1C). To examine if the difference in reporter gene expression in VLP-infected HMV-II cells is actually observed when the same amount of GFP-vRNA is added to the HMV-II cells, we conducted a gene transfer experiment in which the amount of GFP-vRNA contained in the ΔCM2 VLPs used for infection was comparable to that in the WT VLPs. HMV-II cells were infected with ΔCM2 VLPs or WT VLPs at a ratio of 3:1, followed by superinfection with the helper virus, and reporter gene (luciferase) expression was measured and compared. The results showed that there was also a significant difference in reporter gene expression (WT to ΔCM2, 27:1) (data not shown). Taken together, these data have led us to conclude that the CM2 protein in VLPs plays a critical role in efficient gene transfer. Therefore, we analyzed the VLP-infected cells by the following steps.
First, we examined the attachment and entry of the VLPs. VLP-infected HMV-II cells were analyzed by flow cytometry as described in Materials and Methods. WT VLPs attached to HMV-II cell surfaces were shown to be internalized into the cells as a left-shift of the histogram was observed after the cells were incubated at 33°C for 180 min (Fig. 4, left panel). There was no difference in the histograms from the WT VLP- and ΔCM2 VLP-infected cells (Fig. 4, right panel), indicating that attachment and entry of the ΔCM2 VLPs occurred as efficiently as that of the WT VLPs.
FIG. 4.

Flow cytometry of HMV-II cells infected with VLPs. HMV-II cells infected with WT VLPs or ΔCM2 VLPs were analyzed by flow cytometry using anti-HEF MAb J14. The VLP-infected cells were incubated at 4°C for 30 min and then incubated at 33°C for a further 180 min. The histogram from mock-infected cells is shown as a shaded area. Vertical and horizontal lines indicate the number of cells and fluorescence intensities, respectively.
Next, we examined whether the envelope fusion of VLPs occurs appropriately since differences in the envelope fusion of internalized VLPs should result in a difference in reporter gene expression. As envelope fusion within infected cells is difficult to detect, we adopted a method published previously (20) in which the fusion activity of the virus particles was expressed as hemolytic activity. As a result, no significant differences were observed in the obtained values for optical density between WT VLPs and ΔCM2 VLPs (Fig. 5). This finding suggests that the significant difference in the reporter gene transfer between the WT VLPs and ΔCM2 VLPs was not due to any difference in the fusion activity of the VLPs.
FIG. 5.
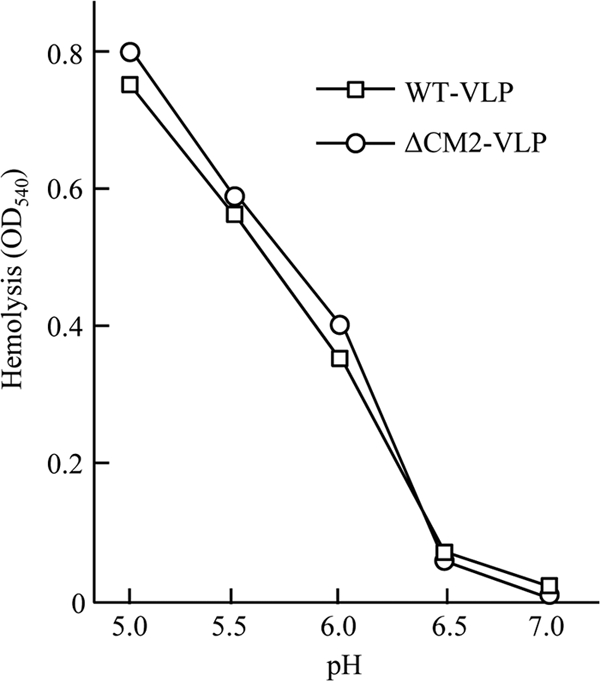
VLP-mediated hemolysis. Hemolysis mediated by WT VLPs or ΔCM2 VLPs was measured using chicken erythrocytes. A total of 100 μl of VLP suspension in PBS was added to 0.5 ml of 2% (vol/vol) chicken erythrocytes in PBS at pH 7.0 and incubated on ice for 30 min. The mixture was centrifuged at 500 × g, and the pellet was suspended in 0.5 ml of saline buffered with 10 mM MES at various pH levels and incubated at 37°C for 60 min. The mixture was then centrifuged, and the supernatants were measured for the optical density at 540 nm (OD540).
Quantification of GFP-vRNA in the VLP-infected HMV-II cells.
The fact that the attachment, entry, and fusion activities of ΔCM2 VLPs were comparable to those of WT VLPs indicates that the significant difference observed in reporter gene expression (Fig. 3) is derived from a step(s) that occurs after envelope fusion. In the present study, we focused on the uncoating process in the infected cells. Since the majority of influenza A virus particles showed a half-time for penetration of about 25 min after adsorption and since 10 min later vRNP is found in the nucleus (24), we attempted to detect the incoming GFP-vRNA at 60 min p.i. in the nucleus of VLP-infected cells. As described in Materials and Methods, HMV-II cells were divided into nuclear and cytoplasmic fractions, each of which was analyzed for immunoblotting using specific antibodies to the nucleus (lamin B) and cytoplasm (α-tublin). As a result, the cells were confirmed to have been successfully fractionated (Fig. 6A).
FIG. 6.

Fractionation and real-time PCR of HMV-II cells infected with VLPs. (A) HMV-II cells were fractionated as described in Materials and Methods, and the whole-cell lysates, the nuclear fraction, and the cytoplasmic fraction of the cells were analyzed by immunoblotting using anti-lamin B and anti-α-tubulin antibodies. (B) The HMV-II cells infected with WT VLPs or ΔCM2 VLPs were incubated at 4°C for 30 min and then transferred to 33°C and incubated for 60 min. The cells were divided into cytoplasmic and nuclear fractions, and the GFP-vRNA contained in the respective fractions was quantified by real-time PCR. The vertical line indicates the copy number of GFP-vRNA from 1.0 × 106 HMV-II cells infected with VLPs. The data obtained from three independent experiments are shown as the means ± standard deviations. All comparisons between groups were statistically evaluated by using a paired t test. NS, not significant.
HMV-II cells were infected with VLPs so that the MOI was maintained at less than 1.0 and the copy number of GFP-vRNA included in WT VLPs was equal to that in ΔCM2 VLPs; i.e., preparations containing approximately three times as many ΔCM2 VLPs as WT VLPs were used for infection. In this experiment, to avoid the replication of GFP-vRNA in the VLP-infected cells, HMV-II cells were not superinfected with the helper virus. The VLP-infected cells were kept at 4°C for 30 min, transferred to 33°C, and then incubated for up to 60 min. The cells were then divided into nuclear and cytoplasmic fractions, and RNA was extracted from the respective fractions and subjected to real-time PCR for the quantification of GFP-vRNA. As shown in Fig. 6B, there was no difference in the total copy number of GFP-vRNA in the cells just after the incubation for 30 min at 4°C, indicating that an equal number of VLPs was attached onto the HMV-II cells. At this point, only a trace amount of GFP-vRNA was detected in the nuclear fractions of WT VLP- and ΔCM2 VLP-infected cells, indicating that a small proportion of GFP-vRNA is recovered in the nuclear fraction in this analysis. On the other hand, more importantly, after incubation at 33°C for a further 60 min, the copy number in the nuclear fraction of WT VLP-infected cells significantly increased (P < 0.05), whereas that of ΔCM2 VLP-infected cells did not change. Furthermore after the 60 min-incubation, a significant difference in the copy numbers in the nuclear fraction was observed between WT VLP- and ΔCM2 VLP-infected cells (P < 0.05). These findings are consistent with the notion that GFP-vRNA uncoated from the WT VLPs was efficiently transported to the nucleus and that the gene from the ΔCM2 VLPs was not.
DISCUSSION
Based on the results of a VLP generation experiment, we previously reported that the expression of major virus proteins, such as HEF and M1, in 293T cells is indispensable to efficient gene transfer (29), presumably because HEF and M1 are essential for the formation of VLPs. When a plasmid mixture without HEF- or M1-expressing plasmid was transfected into 293T cells, the culture medium did not exhibit any hemagglutination activity, and the supernatant was not capable of transferring the GFP gene to HMV-II cells (29). To make use of this method, in the present study, we first attempted to eliminate CM2 expression in transfected 293T cells and found that CM2 is not required for VLP formation (Fig. 1A). These findings indicate that CM2 is not involved in VLP morphogenesis and suggest that it is the major virus proteins, such as HEF and M1, that mainly participate in VLP morphogenesis. The NP/M1 and NP/HEF ratios in the ΔCM2 VLPs were lower than those in the WT VLPs (Fig. 1B), and the amount of GFP-vRNA in the ΔCM2 VLPs was approximately one-third that of in the WT VLPs (Fig. 1C). From these observations we concluded that CM2 is involved in the packaging of GFP-vRNA into VLPs because there were no significant differences in the expression levels of viral components between WT VLP- and ΔCM2 VLP-producing 293T cells (Fig. 2).
The involvement of CM2 in the genome packaging process should be further studied. For influenza A virus M2 and influenza B virus BM2, the cytoplasmic regions of the proteins are involved in the incorporation of the respective virus genome (5, 16, 17, 18, 25, 26). Therefore, it is possible that the cytoplasmic region of CM2 participates in the genome packaging through interaction with vRNP. Furthermore, the specific amino acid(s) in the cytoplasmic region responsible for packaging remains to be determined. Influenza C VLPs harboring CM2 with mutations in the cytoplasmic region may be used to answer the questions.
No significant differences in adsorption, internalization, or fusion activities of VLPs were observed between WT VLPs and ΔCM2 VLPs (Fig. 4 and 5), indicating that the infection process of ΔCM2 VLPs up to envelope fusion occurred in an appropriate manner. Therefore, we focused on the uncoating step of the VLPs and attempted to quantify GFP-vRNA in the nucleus, to which the vRNA released from the VLP is transported. In the real-time PCR, the copy number of GFP-vRNA in the nuclear fraction of WT VLP-infected cells significantly increased after incubation at 33°C for 60 min (P < 0.05), whereas no increase was observed in the ΔCM2 VLP-infected cells (Fig. 6B). This finding suggests that the uncoating step of WT VLPs occurred properly, whereas that of the ΔCM2 VLPs did not. Taken together, the results suggest that CM2 plays a role in the uncoating process of VLPs.
The total (cytoplasm plus nucleus) amount of GFP-vRNA in the WT VLP-infected cells decreased after incubation for 60 min (Fig. 6B). The amount of GFP-vRNA in WT VLP-infected cells without fractionation became 40% of that in the cells before the incubation at 33°C (data not shown), indicating that the decrease shown in Fig. 6B was not due to an artificial degradation of GFP-vRNA through the cell fractionation procedure. Brabec-Zaruba et al. examined the amount of viral protein 1 (VP1) and viral RNA in human rhinovirus type 2-infected HeLa cells up to 210 min postinfection and reported that at 120 min postinfection VP1 fell to below the detectable level; they found that viral RNA also decreased and that the lowest RNA level attained was roughly 30% of the input (4). The authors concluded (i) that empty capsids and virus that failed to uncoat are further transported to late endosomes and are finally degraded in lysosomes, and (ii) that intact viral RNA uncoated to the cytoplasm was used for replication and transcription. Considering the results reported in this article, it is likely that the amount of GFP-vRNA in the WT VLP-infected HMV-II cells decreased after incubation at 33°C under our experimental conditions. Based on our observations (Fig. 6B), we hypothesize that the decrease in GFP-vRNA in the WT VLP-infected cells is unavoidable and that a proportion of the GFP-vRNA successfully uncoated and transported to the nucleus is utilized for replication and transcription.
The amount of the GFP-vRNA in the cytoplasmic fraction of VLP-infected cells after incubation for 60 min tended to be lower in ΔCM2 VLPs than in WT VLPs although the difference did not reach statistical significance (Fig. 6B). As shown in Fig. 5, no significant difference was observed in the hemolytic activities of WT VLPs and ΔCM2 VLPs, suggesting that envelope fusion of ΔCM2 VLPs in the endosome occurs as efficiently as does that of WT VLPs. We, therefore, offer the following speculations: (i) that uncoating of the ΔCM2 VLP is made difficult due to the absence of CM2, and, as the result, in the ΔCM2 VLP-infected cells the vRNP enclosed by an M1 shell left in the endosome progresses to lysosomes, where it may readily be degraded by lysosomal enzymes; and (ii) that, in contrast, in the WT VLP-infected cells, the vRNP that is in the process of uncoating and is being transported to the nucleus is not readily degraded.
Zhirnov and Grigoriev reported in vitro analysis of influenza C virus uncoating (48). They treated influenza C virions with nonionic detergent in neutral or alkaline medium and showed a release of RNP free of the M1 protein. Since we wished to demonstrate the difference in the uncoating process between WT VLPs and ΔCM2 VLPs by another approach, egg-grown influenza C virions were subjected to the in vitro analysis as an initial control experiment. However, their result could not be reproduced in our several attempts in that the HEF, NP, and M1 proteins were recovered in the pellet (RNP) fraction even in the neutral condition (data not shown). This observation may suggest that the in vitro analysis needs to be further studied under more critical conditions.
The roles of CM2 identified in the present study should be discussed in relation to the ion channel function of CM2. Hongo et al. postulated that the Cl− channel activity of CM2 may facilitate the interaction of M1 with vRNP by reducing the ionic strength beneath the viral budding site of the plasma membrane (15). If this is the case, the reduced packaging efficiency of GFP-vRNA into ΔCM2 VLPs is due to the loss of the Cl− channel function of CM2. In the present study, we observed that the gene transfer to HMV-II cells by WT VLPs did not occur in the presence of NH4Cl, a compound known to cause a rise in the pH value of endosomes (data not shown). This observation may suggest that, like the influenza A virus M2 protein, the proton permeability of CM2 reported by Betáková and Hay (3) is required for the uncoating process of influenza C virus although the notion has to be taken into account that the conformational change of the HEF protein to promote envelope fusion does not occur in the presence of NH4Cl. Thus, the role of the proton permeability of CM2 in virus uncoating remains to be studied.
A recombinant influenza A virus defect in M2 was not rescued using MDCK cells but was rescued using MDCK cells expressing M2 constitutively (5, 18, 25, 26, 43). An influenza B virus defect in BM2 was also rescued using MDCK cells expressing BM2 constitutively (16, 17). In the present study, using the established reverse-genetics system (30), we have attempted to generate recombinant influenza C viruses without CM2. However, no infectious recombinants have been rescued to date (data not shown). This finding suggests that CM2 is indispensable to the replication of influenza C virus, a notion consistent with evidence obtained from the present study.
Acknowledgments
We gratefully thank Kaoru Goto (Department of Anatomy and Cell Biology, Yamagata University Faculty of Medicine) and Ai Ito, Shoko Obata, Satoshi Kume, and Yu Shibahara (students of Yamagata University Faculty of Medicine) for technical assistance.
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, by the Takeda Science Foundation, by the Terumo Life Science Foundation, and by a Grant-in-Aid from the Global COE program of the Japan Society for the Promotion of Science.
Footnotes
Published ahead of print on 24 November 2010.
REFERENCES
- 1.Abrahamsen, H. N., T. Steiniche, E. Nexo, S. J. Hamilton-Dutoit, and B. S. Sorensen. 2003. Towards quantitative mRNA analysis in paraffin-embedded tissues using real-time reverse transcriptase-polymerase chain reaction: a methodological study on lymph nodes from melanoma patients. J. Mol. Diagn. 5:34-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alamgir, A. S. M., et al. 2000. Phylogenetic analysis of influenza C virus nonstructural (NS) protein genes and identification of the NS2 protein. J. Gen. Virol. 81:1933-1940. [DOI] [PubMed] [Google Scholar]
- 3.Betáková, T., and A. J. Hay. 2007. Evidence that the CM2 protein of influenza C virus can modify the pH of the exocytic pathway of transfected cells. J. Gen. Virol. 88:2291-2296. [DOI] [PubMed] [Google Scholar]
- 4.Brabec-Zaruba, M., B. Pfanzagl, D. Blaas, and R. Fuchs. 2009. Site of human rhinovirus RNA uncoating revealed by fluorescent in situ hybridization. J. Virol. 83:3770-3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, B. J., G. P. Laser, D. Jackson, and R. A. Lamb. 2008. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 82:10059-10070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Compans, R. W., D. H. Bishop, and H. Meier-Ewert. 1977. Structural components of influenza C virions. J. Virol. 21:658-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flewett, T. H., and K. Apostolov. 1967. A reticular structure in the wall of influenza C virus. J. Gen. Virol. 1:297-304. [PubMed] [Google Scholar]
- 8.Hay, A. J. 1998. Functional properties of the virus ion channels, p. 74-81. In K. G. Nicholson, R. G. Webster, and A. J. Hay (ed.), Textbook of influenza. Blackwell Science, Oxford, United Kingdom.
- 9.Herrler, G., A. Nagele, H. Meier-Ewert, A. S. Bhown, and R. W. Compans. 1981. Isolation and structural analysis of influenza C virion glycoproteins. Virology 113:439-451. [DOI] [PubMed] [Google Scholar]
- 10.Herrler, G., and H. D. Klenk. 1991. Structure and function of the HEF glycoprotein of influenza C virus. Adv. Virus Res. 40:213-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hongo, S., et al. 1994. Identification of a second protein encoded by influenza C virus RNA segment 6. J. Gen. Virol. 75:3503-3510. [DOI] [PubMed] [Google Scholar]
- 12.Hongo, S., K. Sugawara, Y. Muraki, F. Kitame, and K. Nakamura. 1997. Characterization of a second protein (CM2) encoded by RNA segment 6 of influenza C virus. J. Virol. 71:2786-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hongo, S., et al. 1998. Identification of a 374 amino acid protein encoded by RNA segment 6 of influenza C virus. J. Gen. Virol. 79:2207-2213. [DOI] [PubMed] [Google Scholar]
- 14.Hongo, S., et al. 1999. Influenza C virus CM2 protein is produced from a 374-amino-acid protein (P42) by signal peptidase cleavage. J. Virol. 73:46-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hongo, S., et al. 2004. Detection of ion channel activity in Xenopus laevis oocytes expressing Influenza C virus CM2 protein. Arch. Virol. 149:35-50. [DOI] [PubMed] [Google Scholar]
- 16.Imai, M., S. Watanabe, A. Ninomiya, M. Obuchi, and T. Odagiri. 2004. Influenza B virus BM2 protein is a crucial component for incorporation of viral ribonucleoprotein complex into virions during virus assembly. J. Virol. 78:10007-10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai, M., K. Kawasaki, and T. Odagiri. 2008. Cytoplasmic domain of influenza B virus BM2 protein plays critical roles in production of infectious virus. J. Virol. 82:728-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwatsuki-Horimoto, K., et al. 2006. The cytoplasmic tail of the influenza A virus M2 protein plays a role in viral assembly. J. Virol. 80:5233-5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura, H., et al. 1997. Interspecies transmission of influenza C virus between humans and pigs. Virus Res. 48:71-79. [DOI] [PubMed] [Google Scholar]
- 20.Kitame, F., K. Sugawara, K. Ohwada, and M. Homma. 1982. Proteolytic activation of hemolysis and fusion by influenza C virus. Arch. Virol. 73:357-361. [DOI] [PubMed] [Google Scholar]
- 21.Kohno, Y., et al. 2009. Intracellular localization of influenza C virus NS2 protein (NEP) in infected cells and its incorporation into virions. Arch. Virol. 154:235-243. [DOI] [PubMed] [Google Scholar]
- 22.Li, Z. N., et al. 2001. The sites for fatty acylation, phosphorylation and intermolecular disulphide bond formation of influenza C virus CM2 protein. J. Gen. Virol. 82:1085-1093. [DOI] [PubMed] [Google Scholar]
- 23.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 24.Martin, K., and A. Helenius. 1991. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 65:232-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCown, M. F., and A. Pekosz. 2005. The influenza A virus M2 cytoplasmic tail is required for infectious virus production and efficient genome packaging. J. Virol. 79:3595-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCown, M. F., and A. Pekosz. 2006. Distinct domains of the influenza A virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J. Virol. 80:8178-8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moeller, F., F. C. Nielsen, and L. B. Nielsen. 2003. New tools for quantifying and visualizing adoptively transferred cells in recipient mice. J. Immunol. Methods 282:73-82. [DOI] [PubMed] [Google Scholar]
- 28.Mould, J. A., et al. 2003. Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes. Dev. Cell 5:175-184. [DOI] [PubMed] [Google Scholar]
- 29.Muraki, Y., et al. 2004. Identification of an amino acid residue on influenza C virus M1 protein responsible for formation of the cord-like structures of the virus. J. Gen. Virol. 85:1885-1893. [DOI] [PubMed] [Google Scholar]
- 30.Muraki, Y., et al. 2007. A mutation on influenza C virus M1 protein affects virion morphology by altering the membrane affinity of the protein. J. Virol. 81:8766-8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muraki, Y., et al. 2010. Influenza C virus NS1 protein up-regulates the splicing of viral mRNAs. J. Virol. 84:1957-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishimura, H., et al. 1989. A human melanoma cell line highly susceptible to influenza C virus. J. Gen. Virol. 70:1653-1661. [DOI] [PubMed] [Google Scholar]
- 33.Nishimura, H., et al. 1990. Characterization of the cord-like structures emerging from the surface of influenza C virus-infected cells. Virology 179:179-188. [DOI] [PubMed] [Google Scholar]
- 34.Palese, P., and M. L. Shaw. 2007. Orthomyxoviridae: the viruses and their replication, p. 1647-1689. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 35.Paragas, J., et al. 2001. Influenza B and C virus NEP (NS2) proteins possess nuclear export activities. J. Virol. 75:7375-7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pekosz, A., and R. A. Lamb. 1997. The CM2 protein of influenza C virus is an oligomeric integral membrane glycoprotein structurally analogous to influenza A virus M2 and influenza B virus NB proteins. Virology 237:439-451. [DOI] [PubMed] [Google Scholar]
- 37.Pekosz, A., and R. A. Lamb. 1998. Influenza C virus CM2 integral membrane glycoprotein is produced from a polypeptide precursor by cleavage of an internal signal sequence. Proc. Natl. Acad. Sci. U. S. A. 95:13233-13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rossman, J. S., et al. 2010. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J. Virol. 84:5078-5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugawara, K., H. Nishimura, S. Hongo, F. Kitame, and K. Nakamura. 1991. Antigenic characterization of the nucleoprotein and matrix protein of influenza C virus with monoclonal antibodies. J. Gen. Virol. 72:103-109. [DOI] [PubMed] [Google Scholar]
- 40.Sugawara, K., et al. 1993. Construction of an antigenic map of the haemagglutinin-esterase protein of influenza C virus. J. Gen. Virol. 74:1661-1666. [DOI] [PubMed] [Google Scholar]
- 41.Sugawara, K., Y. Muraki, E. Takashita, Y. Matsuzaki, and S. Hongo. 2006. Conformational maturation of the nucleoprotein synthesized in influenza C virus-infected cells. Virus Res. 122:45-52. [DOI] [PubMed] [Google Scholar]
- 42.Tada, Y., et al. 1998. Phosphorylation of influenza C virus CM2 protein. Virus Res. 58:65-72. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe, S., T. Watanabe, and Y. Kawaoka. 2009. Influenza A virus lacking M2 protein as a live attenuated vaccine. J. Virol. 83:5947-5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waterson, A. P., J. M. Hurrell, and K. E. Jensen. 1963. The fine structure of influenza A, B and C viruses. Arch. Gesamte Virusforsch 12:487-495. [DOI] [PubMed] [Google Scholar]
- 45.Whittaker, G. R., and A. Helenius. 1998. Nuclear import and export of viruses and virus genomes. Virology 246:1-23. [DOI] [PubMed] [Google Scholar]
- 46.Yamashita, M., M. Krystal, and P. Palese. 1988. Evidence that the matrix protein of influenza C virus is coded for by a spliced mRNA. J. Virol. 62:3348-3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokota, M., K. Nakamura, K. Sugawara, and M. Homma. 1983. The synthesis of polypeptides in influenza C virus-infected cells. Virology 130:105-117. [DOI] [PubMed] [Google Scholar]
- 48.Zhirnov, O. P., and V. B. Grigoriev. 1994. Disassembly of influenza C viruses, distinct from that of influenza A and B viruses requires neutral-alkaline pH. Virology 200:284-291. [DOI] [PubMed] [Google Scholar]