

Abstract
The rapid induction of type I interferon (IFN) is essential for establishing innate antiviral responses. During infection, cytoplasmic viral RNA is sensed by two DExD/H box RNA helicases, RIG-I and MDA5, ultimately driving IFN production. Here, we demonstrate that purified genomic RNA from HIV-1 induces a RIG-I-dependent type I IFN response. Both the dimeric and monomeric forms of HIV-1 were sensed by RIG-I, but not MDA5, with monomeric RNA, usually found in defective HIV-1 particles, acting as a better inducer of IFN than dimeric RNA. However, despite the presence of HIV-1 RNA in the de novo infection of monocyte-derived macrophages, HIV-1 replication did not lead to a substantial induction of IFN signaling. We demonstrate the existence of an evasion mechanism based on the inhibition of the RIG-I sensor through the action of the HIV-1 protease (PR). Indeed, the ectopic expression of PR resulted in the inhibition of IFN regulatory factor 3 (IRF-3) phosphorylation and decreased expression of IFN and interferon-stimulated genes. A downregulation of cytoplasmic RIG-I levels occurred in cells undergoing a single-cycle infection with wild-type provirus BH10 but not in cells transfected with a protease-deficient provirus, BH10-PR−. Cellular fractionation and confocal microscopy studies revealed that RIG-I translocated from the cytosol to an insoluble fraction during the de novo HIV-1 infection of monocyte-derived macrophages, in the presence of PR. The loss of cytoplasmic RIG-I was prevented by the lysosomal inhibitor E64, suggesting that PR targets RIG-I to the lysosomes. This study reveals a novel PR-dependent mechanism employed by HIV-1 to counteract the early IFN response to viral RNA in infected cells.
Human immunodeficiency virus type 1 (HIV-1) causes profound immune deregulation that results in immune hyperactivation, CD4+ T-cell depletion, and progression to AIDS (reviewed in reference 5). HIV-1 has also evolved numerous mechanisms to evade various aspects of the innate and adaptive immune response (reviewed in reference 36), including the ability to subvert several host innate immune factors that limit retroviral replication (reviewed in reference 14). Since the immune system fails to eradicate HIV-1 in infected cells, further studies are required to evaluate the strategies utilized by HIV-1 to counteract the innate immune response.
The induction of the innate immune response by viral pathogens is characterized by a rapid production of type I interferons (IFNs) (IFN-β/α), which play a major role in the inhibition of virus replication. Upon recognition of viral products, antiviral responses are initiated by either Toll-like receptors (TLRs) or retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), which elicit multiple intracellular signaling cascades culminating in the production of immunoregulatory molecules, including type I IFN, proinflammatory cytokines, and IFN-stimulated genes (ISGs). The activation of these pathways disrupts virus replication and initiates adaptive immune responses (23, 24, 34).
The recognition of viral RNA differs between TLRs and RLRs due to their subcellular localizations and capacities to recognize specific nucleic acid sequences and structures. TLRs sense incoming viral nucleic acids present in the extracellular environment or in endosomes (reviewed in reference 48). Several studies have highlighted a critical role for TLRs in the regulation of HIV-1 replication. Indeed, uridine-rich oligonucleotides derived from HIV-1 RNA have been shown to induce the production of IFN-α and proinflammatory cytokines via TLR7 and TLR8 in dendritic cells and macrophages (20, 43). Furthermore, in human plasmacytoid dendritic cells (pDCs), TLR7 is crucial for the detection of HIV-1 infection (3). Recently, TLR8 and DC-SIGN have been shown to promote HIV-1 replication by enhancing the transcription of full-length viral transcripts (19). Although TLR7 and TLR8 play a major role in HIV-1 recognition, the persistent activation of TLRs by HIV-1 can often lead to chronic immune activation that enhances CD4+ T-cell loss (1, 7, 43).
In contrast to the TLR-dependent sensing of HIV-1 RNA, the RLRs RIG-I and melanoma differentiation-associated gene 5 (MDA5) are DExD/H box RNA helicases that detect viral RNA in the cytoplasm of infected cells. RLRs are pivotal for the recognition of viral infection in almost all cell types, including epithelial, fibroblastic, and conventional dendritic cells (cDCs), as well as macrophages (31, 74). Upon RNA binding through the helicase domain, RIG-I interacts with the downstream CARD-containing adapter molecule MAVS (mitochondrial antiviral signaling protein [also called IPS-1, VISA, or CARDIF]) (35, 45, 64, 78). MAVS in turn activates the IKK-related kinases TBK1 and IKKɛ, which results in the phosphorylation and activation of interferon regulatory factor 3 (IRF-3) and IRF-7. The coordinated activation of these factors as well as NF-κB and AP-1 results in the induction of the IFN response (24, 41, 56).
RIG-I distinguishes viral from cellular RNA in part by recognizing 5′-triphosphorylated (5′-PPP) structures, a modification that is not found on capped or processed cellular RNA (2, 26, 54, 55, 57, 58). RIG-I is required for the detection of single-stranded RNA viruses, including negative-stranded viruses like influenza A virus, measles virus, vesicular stomatitis virus (VSV), and Sendai virus (SeV), and positive-stranded viruses, such as hepatitis C virus (HCV) or Japanese encephalitis virus (JEV). RIG-I was also shown previously to be activated by the complex double-stranded DNA (dsRNA) structures found in the untranslated regions (UTRs) of the HCV genome (60).
Retroviral genomic RNAs (gRNAs) are capped and polyadenylated, as are host mRNAs, yet they contain complex secondary structures in their 5′- and 3′-untranslated regions, which may be recognized by the cellular innate immune system. Mature HIV-1 viral particles contain two copies of the full-length positive-sense viral genomic RNA that are found tightly associated as a compact dimer. The conformation of the stable dimeric RNA structure appears to be important for the production of infectious HIV-1 particles (8, 9; reviewed in reference 47). Indeed, RNA from infectious mature virions is mainly dimeric, while RNA isolated from immature, noninfectious viral particles obtained from protease-defective (PR−) mutant viruses is predominantly monomeric (18). Although the significance of HIV-1 genomic RNA dimerization is still not fully understood, it appears that HIV-1 protease plays a key role in this event.
Despite numerous reports describing the inhibitory effects of type I IFN production on retroviral replication (22, 25, 73), the ability of lentiviral RNA to trigger RLR signaling has not been described. In the present study, we provide evidence that RIG-I, but not MDA5, detects both dimeric and monomeric forms of HIV-1 viral RNA, resulting in the activation of the RIG-I pathway. Furthermore, we describe a novel evasion mechanism in which HIV-1 uses the viral protease to deplete RIG-I from the cytoplasm, thus inhibiting the initiation of the RIG-I signaling cascade.
MATERIALS AND METHODS
Cell culture, transfections, and luciferase assays.
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors at the Royal Victoria Hospital, Montreal, Quebec, Canada, with informed consent, in agreement with Royal Victoria Hospital, Jewish General Hospital, and McGill University Research Ethics Committees. PBMCs were isolated by density centrifugation on Ficoll-Paque Plus (Amersham Biosciences, Uppsala, Sweden) from fresh apheresis specimens obtained with informed consent from healthy donors. Monocytes were isolated from PBMCs by magnetic cell sorting using anti-CD14-conjugated microbeads and an Automacs instrument (Miltenyi Biotec, Auburn, CA) and cultured in Iscove medium (Wisent Technologies, Rocklin, CA) supplemented with 2% human serum A/B (Wisent Technologies), 700 U/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (a generous gift from Cangene Corporation, Mississauga, Canada), 100 U/ml penicillin G, and 100 μg/ml streptomycin in gas-permeable thermoplastic nonadherent culture bags (Origen Biomedical). On day 7, monocyte-derived macrophages (MDMs) were harvested and resuspended in complete McCoy's 5A medium (supplemented with 10% fetal bovine serum [FBS] and antibiotics) (Wisent Technologies). Monocyte-derived macrophage differentiation and purity were analyzed by flow cytometry as described previously (59).
Human hepatoma Huh7.0 cells, HEK293 cells, HeLa cells, and RIG-I knockout (RIG-I−/−) and MDA5−/− mouse embryonic fibroblasts (MEFs) as well as the corresponding wild-type (WT) MEFs were used for transient transfections and were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum and antibiotics. Both RIG-I−/− and MDA5−/− MEFs were obtained from Robert H. Silverman, Lerner Research Institute, Cleveland Clinic, with the permission of Shizuo Akira, Osaka University, who generated the original RIG-I and MDA5 knockout mice (31, 33). For luciferase assays, subconfluent HeLa cells and WT and MDA5−/− MEFs were transfected by using TransIT-LT1 transfection reagent (Mirus) with 100 ng of the pRL-TK reporter (Renilla luciferase [Luc] for an internal control) and 100 ng of the respective Luc or pGL3 reporter (firefly luciferase, experimental reporter). RNA transfection was performed 24 h later with 2 μg of HIV-1 RNA dimers and RNA monomers, respectively, and 500 ng of RNA bearing 5′-triphosphate by using Transmessenger transfection reagent (Qiagen). At 24 h post-RNA transfection, reporter gene activity was measured by a dual-luciferase reporter assay, according to the manufacturer's instructions (Promega). Luciferase activity was measured as the fold activation (relative to the basal level of the reporter gene of the nontransfected sample after normalization with the cotransfected Renilla luciferase activity). In RIG-I−/− and RIG-I WT MEFs, HIV-1 gRNA transfections were performed as described above. For dose-response experiments, subconfluent HEK293 cells were transfected by the calcium phosphate coprecipitation method with 100 ng of the pRL-TK reporter (Renilla luciferase for an internal control); 100 ng of the IFNB pGL3 reporter (firefly luciferase, experimental reporter); 200 ng of the ΔRIG-I, TBK1, and pEGFP-C1 expression plasmids; and 50 to 500 ng of the green fluorescent protein (GFP)-protease expression plasmid as indicated. IFNB promoter activity was measured at 24 h posttransfection by use of the dual-luciferase reporter assay as described above. Single-cycle infections were performed with subconfluent HEK293 cells, where cells were transfected by the calcium phosphate coprecipitation method with 1 μg of Myc-RIG-I expression plasmid and 1 μg of either WT BH10 or the BH10-PR− proviral clone.
Plasmid constructs.
HIV-1 protease cDNA was amplified from the pNL4.3 HIV-1 expression plasmid and cloned into the pEGFP C1 expression plasmid at the EcoRV and BamHI cloning sites using the following set of primers: 5′-CCGCGAATTCGATGCCTCAGATCACTCTTTGG-3′ (forward) and 5′-CCGCTCTAGACTAAAAATTTAAAGTGCAGCCAATCTG-3′ (reverse). Plasmids encoding ΔRIG-I, RIG-I, RIG-IC, TBK1, IFN-β-pGL3, interferon-stimulated response element (ISRE)-Luc, NF-κB-pGL3, and pRL-TK were previously described (35, 38, 65, 80).
Isolation of HIV-1 viral RNA.
HIV-1 genomic RNA dimers and monomers were produced by the transfection of HeLa cells with BH10 WT, BH10-PR− (protease defective), and FL proviral clones for 48 h (29). In the BH10 FL transfection, HeLa cells were incubated with the protease inhibitor saquinavir (1 μM) (28). Virus-containing supernatants were collected and centrifuged at 40,000 rpm for 1 h onto a 20% (wt/vol) sucrose cushion in phosphate-buffered saline (PBS) by using a Beckman SW40 rotor. The virus pellet was dissolved in sterile lysis buffer (50 mM Tris [pH 7.4], 50 mM NaCl, 10 mM EDTA, 1% [wt/vol] SDS, 50 μg of tRNA/ml, and 100 μg proteinase K/ml) and extracted twice at 4°C with an equal volume of buffer-saturated phenol-chloroform-isoamyl alcohol (25:24:1) (Invitrogen). The aqueous phase containing the viral RNA was precipitated overnight at −80°C with 0.1 volumes of 3 M sodium acetate (pH 5.2) and 2.5 volumes of 95% (vol/vol) ethanol and centrifuged at 4°C for 30 min. The gRNA pellet was rinsed in 70% (vol/vol) ethanol and dissolved in 20 μl of buffer S (10 mM Tris [pH 7.5], 100 mM NaCl, 10 mM EDTA, 1% SDS). The genomic viral RNA concentration was determined by absorption at 260 nm using a Nanodrop ND-1000 spectrophotometer, while the purity of RNA dimers and monomers was verified by electrophoresis on a nondenaturing 1% (wt/vol) agarose gel and subsequently analyzed by Northern blotting as described previously (29, 68). Densitometric analysis was performed by using NIH 1.6.3 software to evaluate the amount of dimers and monomers in each RNA preparation prior to RNA transfection (further described in reference 28). The percentage of monomeric RNA in the FL mutant was approximately 93% ± 8%. A total of 2 μg of HIV-1 RNA dimers and RNA monomers was used for transfections.
Viral stocks and infections.
The R5-tropic HIV-1 B primary isolate was provided by the McGill AIDS Center at the Lady Davis Institute (Montreal, Canada) and was isolated from a treatment-naïve HIV-1-infected patient from Montreal. MDMs and THP-1 cells were infected in a pellet for 2 h at a multiplicity of infection (MOI) of 1 and washed twice with PBS to remove unbound viral particles. Cells were then plated onto RPMI 1640 medium supplemented as described above. Cells were harvested at 6, 24, and 72 h after infection for RNA extraction. MDMs and THP-1 cells were infected with SeV at 40 hemagglutinating units (HAU) per 10E6 cells in serum-free medium for the first 2 h, and cells were harvested for RNA or protein extraction at 6 h. SeV strain Cantell was obtained from Charles River Laboratories (North Franklin, CT). The HIV-1 proviral clones WT BH10, BH10-PR− (protease defective), and pNL4.3 were generously provided by Mark Wainberg (McGill AIDS Center, Montreal, Canada).
Reagents.
Cells were treated with 5 μM MG132 (Boston Biochem), 50 μM Z-VAD-fmk (R&D systems), 10 μM E64 (Roche Applied Science), 5 μM saquinavir (NIH AIDS Research and Reference Reagent Program), and 10 μg/ml of TLR7 ligand ssRNA40/LyoVec (Invivogen). 5′-PPP RNA was synthesized by using the T7 Megascript kit (Ambion).
Immunoblot analysis.
Whole-cell extracts (WCEs) were prepared in Nonidet P-40 (NP-40) lysis buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 1% NP-40, 2 mM EDTA, 10% glycerol, 5 mM NaF, 0.1 mM Na3VO4, 10 mM β-glycerophosphate [pH 7.4], 10 mM p-nitrophenyl phosphate disodium salt hexahydrate [PNPP], 0.1 mM phenylmethylsulfonyl fluoride [PMSF], and 5 μg/ml each of leupeptin, pepstatin, and aprotinin). Cell debris was removed by centrifugation at 15,000 rpm for 30 min. For subcellular fractionation, cell pellets were washed with PBS and further lysed in a solution containing 1% NP-40, 1% SDS, and 0.5% deoxycholate (DOC), followed by sonication. WCEs (50 μg) were subjected to electrophoresis on 7.5 to 12% acrylamide gels by SDS-PAGE and were transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Mississauga, Canada). The membranes were blocked in 5% dried milk in PBS plus 0.1% Tween 20 and then probed with primary antibodies. Anti-Flag (M2) and anti-Myc (9E10), each used at a concentration of 1 μg/ml, were purchased from Sigma-Aldrich (Oakville, Canada), and anti-GFP (Roche) was prepared in blocking solution. Anti-IRF-3 (1:1,000; IBL, Japan) and the phospho-specific antibodies directed against IRF-3 Ser-396 (1:1,000) (63), anti-p24 (1:1,000; in-house) (10), anti-RIG-I (1:1,000; in-house) (42), and β-actin (MAb1501; Chemicon) were prepared in 5% bovine serum albumin (BSA)-PBS-Tween. After washing in PBS-0.1% Tween 20, the membranes were incubated for 1 h with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (1:8,000). Immunoreactive bands were visualized with an enhanced chemiluminescence detection system (ECL, Amersham Biosciences).
Real-time PCR.
DNase-treated total RNA was prepared by using the RNeasy kit (Qiagen Inc.). Total RNA was reverse transcribed with 100 U of Superscript II Plus RNase H− reverse transcriptase (RT) using oligonucleotide AnCT primers (Gibco BRL Life Technologies). Quantitative PCR (Q-PCR) assays were performed in triplicates using FastStart Universal SYBR green master mix (Rox) (Roche) on the AB 7500 Fast real-time PCR system (Applied Biosystems, Foster City, CA). The human primer pairs used were as follows: 5′-TTGTGCTTCTCCACTACAGC-3′ (forward) and 5′-CTGTAAGTCTGTTAATGAAG-3′ (reverse) for IFN-β, 5′-CCTGATGAAGGAGGACTCCATT-3′ (forward) and 5′-AAAAAGGTGAGCTGGCATACG-3′ (reverse) for IFN-α2, 5′-ACCCGCAGATGTCCATGAG-3′ (forward) and 5′-GTGGCATCATGTAGTTGTGAACCT-3′ (reverse) for IRF-7, 5′-AGCTCCATGTCGGTGTCAG-3′ (forward) and 5′-GAAGGTCAGCCAGAACAGGT-3′ (reverse) for ISG15, 5′-CAACCAAGCAAATGTGAGGA-3′ (forward) and 5′-AGGGGAAGCAAAGAAAATGG-3′ (reverse) for ISG56, 5′-TTCCTGCAAGCCAATTTTGTC-3′ (forward) and 5′-TCTTCTCACCCTTCTTTTTCATTGT-3′ (reverse) for CXCL10, 5′-ATGCTCCAGAAGGCCAGACA-3′ (forward) and 5′-CCTCCACTGTGCTGGTTTTATCT-3′ (reverse) for interleukin-12α (IL-12α), 5′-GGAGACTTGCCTGGTGAAAA-3′ (forward) and 5′-ATCTGAGGTGCCCATGCTAC-3′ (reverse) for IL-6, 5′-GGTCAGAGGACGGCATGAGA-3′ (forward) and 5′-GCAGGACCCAGGTGTCATTG-3′ (reverse) for APOBEC3G, and 5′-CCTTCCTGGGCATGGAGTCCT-3′ (forward) and 5′-AATCTCATCATCTTGTTTTCTGCG-3′ (reverse) for β-actin. The HIV-1 Gag primer set was 5′-GGAGCTAGAACGATTCGCAGTTA-3′ (forward) and 5′-GGTTGTAGCTGTCCCAGTATTTGTC-3′ (reverse). The mouse primers set used were as follows: 5′-CACAGCCCTCTCCATCAACT-3′(forward) and 5′-TCCCACGTCAATCTTTCCTC-3′ (reverse) for mouse IFN-β (mIFN-β), 5′-GAAGGACAGGAAGGATTTTGGA-3′ (forward) and 5′-TGAGCCTTCTGGATCTGTTGGT-3′ (reverse) for mIFN-α4, 5′-GGGCCACAGCAACATCTATGA-3′ (forward) and 5′-CCGCTGGGACACCTTCTTC-3′ (reverse) for mISG15, 5′-CAACCAAGCAAATGTGAGGA-3′ (forward) and 5′-AGGGGAAGCAAAGAAAATGG-3′ (reverse) for mISG56, and 5′-CACCAGTTCGCCATGGAT-3′ (forward) and 5′-CCTCGTCACCCACATAGGAG-3′ (reverse) for mβ-actin. All data are presented as relative quantification (RQ) using the comparative threshold cycle (CT) method as the expression of the target gene versus the reference gene β-actin. The absence of genomic DNA contamination was demonstrated routinely by analyses of PCRs performed with total RNA using each of the primer sets.
RT-PCR.
DNase-treated total RNA was prepared by using the RNeasy kit (Qiagen Inc.). Total RNA was reverse transcribed with 100 U of Superscript II Plus RNase H− reverse transcriptase using oligonucleotide AnCT primers (Gibco BRL Life Technologies). The cDNAs were amplified in 50 μl of PCR buffer containing deoxynucleoside triphosphate (dNTP), MgCl2, and Taq polymerase (Amersham Biosciences, England). A total of 20, 25, 30, and 35 cycles were carried out, and PCR products were analyzed by electrophoresis on a 2% agarose gel. Pictures show ethidium bromide fluorescence of PCR products obtained at a given number of PCR cycles, i.e., before reaching saturation levels. The primer pairs used were 5′-TGCAAGCTGTGTGCCTCT-3′ (forward) and 5-CATCTTTGTCTGGCATCTGG-3′ (reverse) for RIG-I and 5′-CCTTCCTGGGCATGGAGTCCT-3′ (forward) and 5′-AATCTCATCATCTTGTTTTCTGCG-3′ (reverse) for β-actin.
Confocal microscopy.
A549 cells were seeded onto Ibidi (München, Germany) μ-Slides VI0.4 and grown overnight to 50% confluence. A549 cells were transfected by using Lipofectamine LTX and Plus reagent (Invitrogen) with GFP or GFP-protease with or without saquinavir (5 μM), respectively. Cells were maintained in F-12K nutrient mixture (Kaighn's modified) supplemented with 10% heat-inactivated fetal bovine serum, antibiotics, and 25 mM HEPES to help maintain cell viability during the experiment (17). At 15 h posttransfection, A549 cells were set up for live imaging at 37°C in a humidified 5% CO2 chamber using a Chamlide environmental control system (LCI, Seoul, South Korea) installed on a Leica DM16000B microscope. The microscope was equipped with a WaveFX spinning disk confocal head (Quorum Technologies, Ontario, Canada), and images were acquired with a Hamamatsu ImageEM EM-charge-coupled-device (CCD) camera. After confirming the expression of GFP-positive live cells, samples were fixed with 4% paraformaldehyde for 15 min and washed in PBS. Cells were permeabilized and blocked in 3% BSA and 0.25% Triton X-100 in PBS. Primary anti-LAMP-1 antibody (DSHB) and anti-RIG-I (1:1,000; in-house) (42) were incubated for 1 h at room temperature. Secondary antibodies, anti-rabbit IgG(H+L)-Alexa Fluor546 (1:1,000) (catalog number A11071; Molecular Probes) and anti-rat IgG(H+L)-Alexa Fluor647 (1:200) (catalog number A21472; Molecular Probes) conjugates, were applied for 1 h at room temperature. Controls were prepared by immunostaining without the primary antibody. Images were acquired by using a 63×/1.4-plan-apochromat oil immersion objective. The images were acquired by using Volocity Imaging software (version 4.3.2; Improvision, Perkin-Elmer, Waltham, MA) and then processed by using the Fiji package of ImageJA (version 1.44f).
Statistical analysis.
Data were analyzed as means ± standard errors of the means (SEM). Statistical significance was assessed by a paired Student's t test. Analyses were performed by using Prism 5 software (GraphPad). Statistical significance was evaluated by using the following P values: a P values of <0.05, a P value of <0.01, or a P value of <0.001.
RESULTS
HIV-1 genomic RNA induces IFN signaling.
HIV-1 genomic RNA (gRNA) was obtained from an infectious HIV-1 molecular clone, BH10, that generates predominantly HIV-1 RNA dimers. RNA monomers were virion isolated from a previously characterized noninfectious BH10 HIV-1 strain (29). This mutant strain also contains a mutation in the amino-terminal zinc finger and linker region of the nucleocapsid, resulting in the formation of monomeric RNA (29). The mutant virus was grown in the presence of the HIV-1 protease inhibitor saquinavir for 48 h to ensure the complete formation of monomeric viral RNA (28). RNA extracted from the respective virions was electrophoresed in a nondenaturing agarose gel and visualized by autoradiography (Fig. 1A). Only two types of migrating bands were observed: a slower-migrating band corresponding to RNA dimers and a faster-migrating band corresponding to HIV-1 monomers.
FIG. 1.

HIV-1 genomic RNA induces IFN signaling. (A) HIV-1 viral RNA dimers were obtained from WT BH10 virions, whereas HIV-1 RNA monomers were isolated from BH10 virions grown in the presence of the protease inhibitor saquinavir (S) (1 μM) for 48 h. Also, a joint mutation in the amino-terminal finger and linker region of the nucleocapsid was included in these virions. Genomic RNAs extracted from the respective virions were electrophoresed on a nondenaturing 1% (wt/vol) agarose gel and analyzed by Northern blotting. (B) Activity of type I IFN promoters by HIV-1 RNA. HeLa cells were transfected with the respective reporter plasmids for IFNB-pGL3, ISRE-Luc, and NF-κB-pGL3. At 24 h posttransfection, HIV-1 gRNA dimers, monomers, or control 5′-PPP RNA was transfected. Luciferase activity was analyzed at 24 h posttransfection. Values are as means ± SEM. Statistical significance was evaluated by using P values of <0.05 (*), <0.01 (**), and <0.001 (***). NT, not treated.
While RIG-I is an important cytoplasmic sensor of RNA viruses (reviewed in reference 48), no reports have examined RIG-I activation during HIV-1 infection. To explore the role of RLR cytoplasmic sensors in the recognition of HIV-1, the ability of transfected monomeric and dimeric HIV-1 gRNA to activate the IFN response was initially evaluated in HeLa cells by a reporter gene assay (Fig. 1B). A luciferase reporter under the control of either the IFNB promoter or consensus ISRE or NF-κB elements was introduced together with HIV-1 dimeric or monomeric RNA into HeLa cells, and promoter activity was assessed at 24 h. Both forms of HIV-1 RNA exhibited significant IFN-inducing activity, although the monomeric form was consistently a more potent ligand. Indeed, the IFNB promoter was stimulated 7.5-fold with HIV-1 dimers (P < 0.001) and 15-fold with HIV-1 monomers (P < 0.05), while the ISRE was activated 3.5-fold (P < 0.001) and 7-fold (P < 0.05), respectively. Similarly, NF-κB-responsive elements were stimulated 6-fold (P < 0.001) by the HIV-1 RNA dimers and 9-fold (P < 0.05) with the RNA monomers.
The HIV-1 viral RNA structure is well known to be sensed by TLR7 (3, 20, 43). To investigate if HIV-1 monomers and dimers have differential stimulatory activities through TLR7, an overlay of HIV-1 RNA dimers and monomers was also performed with Huh7.0 cells, which uniquely express TLR7 but not TLR3 (37). IFN-β, IFN-α2, and CXCL10 mRNA levels were induced by both HIV-1 RNA dimers and monomers at 24 h post-RNA overlay, but again, HIV-1 RNA monomers strongly stimulated IFN-α2 mRNA compared to HIV-1 RNA dimers (22-fold versus 2.5-fold, respectively) (data not shown). These results indicate that HIV-1 RNA dimers and monomers are bona fide ligands of TLR7.
RIG-I recognizes both dimeric and monomeric forms of HIV-1 gRNA.
To determine which RLR mediated HIV-1 RNA recognition, RIG-I knockout (RIG-I−/−) and the corresponding wild-type mouse embryo fibroblasts (MEFs) were transfected with either HIV-1 RNA dimers, monomers, or 5′-PPP control RNA, and the induction of IFN and ISG gene expression was assessed by Q-PCR. mIFN-β, mIFN-α4, mISG56, and mISG15 mRNAs were induced by HIV-1 RNA in RIG-I wild-type but not in RIG-I-deficient MEFs (Fig. 2A). As expected, the positive control 5′-PPP RNA also failed to induce detectable IFN levels in RIG-I knockout MEFs. HIV-1 RNA monomers elicited a 20- to 60-fold increase in mIFN-β, mIFN-α4, mISG56, and mISG15 gene expression levels, while a 7- to 20-fold induction was detected with HIV-1 dimers. A positive-feedback induction of the RIG-I pathway was also evident, as both forms of HIV-1 RNA induced RIG-I protein levels, a known ISG (30) (Fig. 2B). In contrast, in MDA5+/+ and MDA5−/− MEFs expressing the IFNB promoter, no significant difference in activation was observed with either dimeric or monomeric HIV-1 RNA (Fig. 2C). Thus, HIV-1 genomic RNA induces a strong IFN response through the specific activation of the RIG-I but not the MDA5 pathway. Furthermore, monomeric gRNA possessed a higher level of stimulatory activity than did dimeric gRNA, consistent with the premise that RNA conformation is important for RIG-I recognition (61, 62).
FIG. 2.

RIG-I recognizes both dimeric and monomeric forms of full-length HIV-1 viral RNA. (A) RIG-I+/+ and RIG-I−/− mouse embryo fibroblasts (MEFs) were transfected with HIV-1 gRNA dimers or monomers along with control 5′-PPP RNA, respectively. Total RNA was isolated at 24 h posttransfection and subjected to real-time PCR analysis for the quantification of type I IFN gene expression, such as mIFN-β, mIFN-α4, mISG15, mISG56, and mβ-actin. Results are presented as relative quantification (RQs). Values are means ± SEM. (B) Whole-cell extracts from RIG-I+/+ (WT) and RIG-I−/− (KO) MEFs transfected with HIV-1 gRNA dimers or monomers along with control 5′-PPP RNA, respectively, were subjected to Western blot analysis and probed with anti-RIG-I and anti-β-actin antibodies. (C) The IFNB-pGL3 promoter construct was transfected into MDA5+/+ and MDA5−/− MEFs. Subsequently, HIV-1 gRNA dimers, monomers, or control 5′-PPP RNA was transfected. IFNB promoter activity was analyzed at 24 h posttransfection. Values represent means ± SEM. IB, immunoblot.
RIG-I expression inhibits HIV-1 replication.
HIV-1 replication was inhibited during a single-cycle infection in the presence of different RIG-I expression constructs (Fig. 3). The active form of RIG-I (ΔRIG-I) (amino acids [aa] 1 to 229) or the adaptor protein MAVS (data not shown) blocked HIV-1 BH10 proviral DNA replication in the presence of both inducers of antiviral signaling (35, 80). The expression of full-length RIG-I or RIG-I C (helicase domain alone), which does not trigger significant IFN production (80), resulted in 2-fold (P < 0.05) and 10-fold (P < 0.05) decreases in HIV-1 replication, respectively, suggesting that the helicase domain alone possesses direct antiretroviral activity. When the proviral clone BH10 was transfected prior to RIG-I, an inhibition of HIV-1 replication was no longer noted. This suggests that HIV-1 products generated during viral replication target RIG-I activity.
FIG. 3.
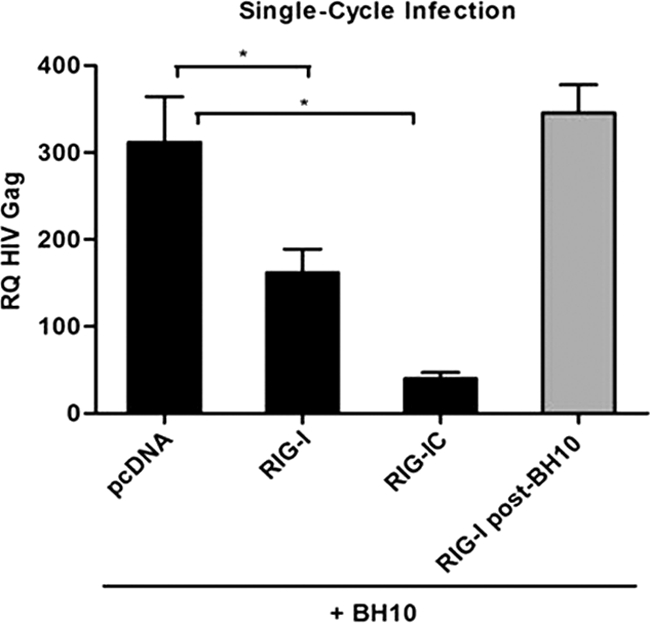
RIG-I overexpression inhibits HIV-1 replication in single-cycle infections. Total RNA was extracted from HEK293 cells 24 h after cotransfection of pcDNA3, Myc-RIG-I, and Myc-RIG-I C along with the BH10 proviral clone. BH10 transfection prior to Myc-RIG-I transfection is shown by the gray bar. cDNA samples were subjected to real-time PCR analysis with primers specific for the HIV-1 Gag gene or β-actin. HIV-1 replication is represented as an RQ based on the relative expression of the HIV-1 Gag gene versus β-actin as a reference gene. Values are means ± SEM. Statistical relevance was evaluated using a P value of <0.05 (*).
HIV-1 infection inhibits the type I IFN response.
Several reports, including our own, demonstrate that the induction of the type I IFN response is not detected during the early stages of HIV-1 infection in CD4+ T cells, macrophages, and immature dendritic cells (51, 67, 70, 72, 77). To demonstrate that HIV-1 monomeric and dimeric gRNAs act as functional RIG-I ligands in natural cellular targets of HIV-1 infection, the profiles of type I IFN and IFN-stimulated gene expression were determined in phorbol myristate acetate (PMA)-differentiated monocytic THP-1 cells (Fig. 4A). HIV-1 RNA dimers and monomers led to 10- to 24-fold increases in IFN-β gene expression, respectively. The classic antiviral ISGs IRF-7, ISG15, and ISG56 were induced 17-, 38-, and 34-fold, respectively, by the monomeric gRNA and 11-, 20-, and 25-fold, respectively, by the dimeric gRNA. The antiviral ISG APOBEC3G was induced 17-fold and 7-fold by the monomers and dimers, respectively. Levels of CXCL10 mRNA, a gene dually regulated by NF-κB and IRFs, were induced 500- to 800-fold in PMA-differentiated THP-1 cells. These findings demonstrate that HIV-1 viral RNA can induce a type I IFN response in macrophages.
FIG. 4.

HIV-1 viral RNA induces type I IFNs and interferon-stimulated genes in macrophages. (A) PMA-differentiated THP-1 cells were transfected with HIV-1 RNA dimers, RNA monomers, or RNA bearing 5′-triphosphate for 24 h, respectively. Total RNA was isolated at 24 h posttransfection and subjected to real-time PCR analysis for the quantification of IFN-stimulated gene expression, such as IFN-β, IRF-7, ISG15, ISG56, APOBEC3G, and β-actin. (B) Kinetics of HIV-1 replication in monocyte-derived macrophages (MDMs). MDMs were infected de novo (or not) with HIV-1 at an MOI of 1 for 6, 24, and 72 h. Total RNA was isolated at the indicated times and subjected to real-time PCR analysis with primers specific for the HIV-1 Gag gene or β-actin. (C and D) De novo HIV-1 infection of MDMs does not induce a type I IFN response but triggers a proinflammatory response. MDMs were infected de novo (or not) with HIV-1 at an MOI of 1 for 6, 24, and 72 h. MDMs infected with Sendai virus (40 hemagglutinating units/ml) for 6 h were used as a control for the induction of type I IFNs. Expression levels of IFN-β, IFN-α2, IRF-7, CXCL10, IL-12α, IL-6, and β-actin were analyzed by real-time PCR. Results are presented as RQs. Values are means ± SEM. NI, not infected.
However, de novo HIV-1 infection of primary monocyte-derived macrophages (MDMs) did not lead to a noticeable type I IFN response (Fig. 4C). MDMs were permissive to HIV-1 replication, as demonstrated by the detection of HIV-1 Gag transcripts as early as 6 h postinfection; a sharp increase in Gag mRNA levels at 72 h corresponded to the production of newly synthesized HIV-1 Gag mRNA (Fig. 4B). mRNA levels of IFN-β, IFN-α2, and the IFN-stimulated gene IRF-7 were unchanged after infection, whereas Sendai virus infection induced a strong antiviral response in MDMs (Fig. 4C). In contrast, an upregulation of the proinflammatory NF-κB-stimulated genes CXCL10, IL-6, and IL-12α was detected at 6 h but decreased to near-basal levels by 72 h (Fig. 4D). These results strongly suggest the existence of an evasion mechanism that targets the RIG-I signaling pathway during HIV-1 infection.
HIV-1 protease targets RIG-I.
Many viruses circumvent the innate antiviral response by using viral proteins to antagonize the RIG-I/MAVS pathway at different levels (6). Viral proteases, in particular, are known to abrogate the RIG-I/MAVS pathway through the direct degradation of RIG-I (52) or the cleavage of the adaptor MAVS (38, 45). To investigate a potential role for the HIV-1 protease (PR) in the inhibition of RIG-I signaling, a sequential transfection of RIG-I followed 24 h later by the WT BH10 proviral clone or the BH10-protease-defective proviral clone was performed (Fig. 5). The transduction of BH10 decreased RIG-I protein levels by ∼3-fold, whereas RIG-I protein expression was unchanged in the presence of a PR-defective BH10 provirus (Fig. 5, lanes 3 and 4). Analysis of endogenous IRF-3 confirmed that IRF-3 degradation was not protease dependent, consistent with previous reports demonstrating that Vif and Vpr target IRF-3 for degradation (13, 51). This finding thus argues for a specific role for HIV-1 PR in the disruption of RIG-I signaling.
FIG. 5.
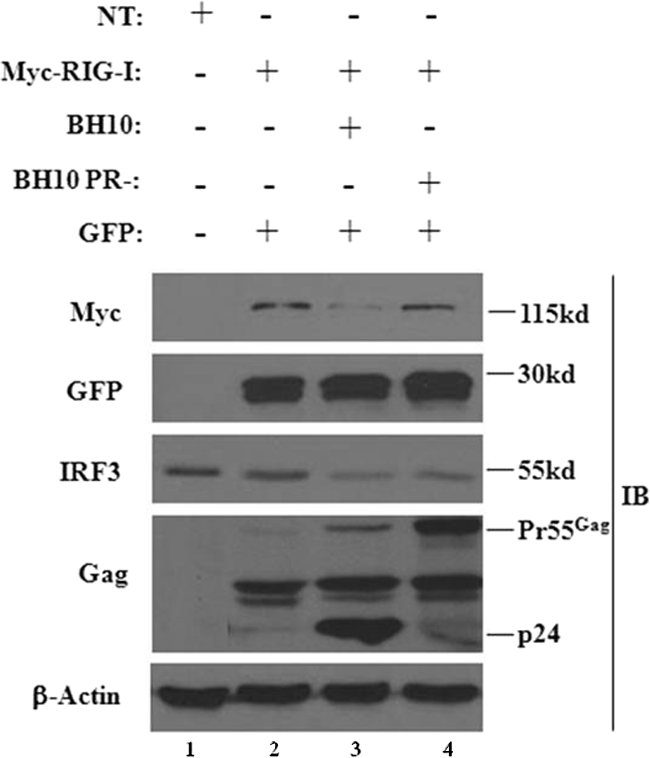
An HIV-1 protease-deficient provirus restores the expression of RIG-I. HEK293 cells were transfected with a plasmid expressing Myc-RIG-I. At 24 h posttransfection cells were transfected with either the WT BH10 proviral clone or the BH10-protease-defective proviral clone. A GFP-expressing plasmid alone was used as an internal control. Whole-cell extracts were subjected to Western blot analysis using anti-Myc, anti-GFP, anti-IRF-3, anti-p24 Gag, and anti-β-actin antibodies. The 55-kDa band represents the immature Gag polyprotein, whereas the 24-kDa band represents the mature Gag protein.
To further examine the role of PR in the inhibition of RIG-I signaling, increasing amounts of HIV-1 PR were coexpressed with constitutively active RIG-I (ΔRIG-I; aa 1 to 229); the activation of the IFNB promoter was dramatically decreased by PR in a dose-dependent manner (Fig. 6A). In contrast, the activation of the downstream kinase TBK1 was not affected, arguing that the protease targets RIG-I signaling upstream of TBK1. Concomitant with the inhibition of the RIG-I-mediated activation of the IFNB promoter, the level of RIG-I protein expression was also reduced in the presence of increasing amounts of the HIV-1 PR (Fig. 6B). To verify that RIG-I is specifically targeted by PR, the downregulation of other signaling proteins by PR was also examined (Fig. 6C). Regardless of the presence of PR, no changes in the expression of MDA5 or IRF-3 were observed. Also, RIG-I transcript levels were not affected by PR expression, indicating that RIG-I is specifically targeted by the protease at the protein level (data not shown).
FIG. 6.

HIV-1 PR targets RIG-I and interferes with IFN-β activation. (A) HIV-1 PR downregulates the IFNB promoter. HEK293 cells were transfected with an IFNB-pGL3 reporter plasmid, the pEGFP-C1 vector, or expression plasmids encoding ΔRIG-I and TBK1 along with increasing amounts (50 ng, 100 ng, 200 ng, and 500 ng) of the GFP-protease expression construct. IFNB promoter activity was measured at 24 h posttransfection. Values represent means ± SEM. (B) RIG-I is downregulated in the presence of HIV-I PR. HEK293 cells were cotransfected with expression vectors for Myc-RIG-I (2 μg) and increasing amounts (2 μg, 5 μg, and 10 μg) of GFP-protease as indicated. Cell lysates were subjected to Western blot analysis, and expression levels of RIG-I, protease, and β-actin were analyzed by immunoblotting with antibodies against Myc, GFP, and β-actin, respectively. (C) HIV-1 PR specifically targets RIG-I. HEK293 cells were cotransfected with plasmids expressing Myc-IRF-3, Flag-MDA5, and GFP-protease. Cell lysates were subsequently subjected to immunoblotting with anti-Myc, anti-Flag, anti-GFP, and anti-β-actin antibodies. (D) HIV-1 PR inhibits ΔRIG-I-mediated activation of IRF-3. HEK293 cells were transfected with 500 ng of Myc-IRF-3, 1 μg Myc-ΔRIG-I or GFP-TBK1, and 1 μg of GFP-protease expression plasmids as indicated. Whole-cell extracts were analyzed by immunoblotting for IRF-3 pSer-396, RIG-I, GFP-TBK1, GFP-protease, and β-actin. (E) HIV-1 PR downregulates IFN-stimulated gene expression. HEK293 cells were transfected with Myc-ΔRIG-I along with the GFP-protease expression plasmid. cDNA samples were subjected to real-time PCR analysis with primers specific for IFN-β, APOBEC3G, and β-actin. Results are presented as RQs. Values are means ± SEM.
To further characterize the inhibition of the IFN response by the viral protease, the effect of PR on downstream gene activation was evaluated by measuring IRF-3 phosphorylation in the presence of HIV-1 PR, using the phospho-specific Ser-396 (pSer-396) IRF-3 antibody (53). ΔRIG-I coexpression induced IRF-3 Ser-396 phosphorylation (Fig. 6D, lane 3), while the coexpression of PR completely blocked phosphorylation (Fig. 6D, lane 4). On the other hand, TBK1-mediated IRF-3 phosphorylation was not inhibited by PR (Fig. 6D, lanes 5 and 6), again indicating that PR targets RIG-I signaling upstream of TBK1. In addition, mRNA levels of IFN-β as well as the IFN-induced gene APOBEC3G were downregulated in the presence of PR (Fig. 6E). In sum, these results demonstrate that HIV-1 protease targets RIG-I and leads to the ablation of IFN signaling.
HIV-1 PR causes a relocalization of RIG-I to the insoluble membrane fraction followed by lysosome-mediated degradation.
In THP-1 cells infected by HIV-1 (Fig. 7A), RIG-I was not degraded via the proteasomal pathway, nor was RIG-I cleaved by cellular caspases, based on the use of the proteasome inhibitor MG132 or the pancaspase inhibitor Z-VAD. Similar results were obtained with HEK293 cells expressing RIG-I and PR (Fig. 7B). Unexpectedly, the inhibition of the HIV-1 PR activity with the protease inhibitor saquinavir did not restore the expression of RIG-I (Fig. 7C), indicating that the catalytic activity of PR was not required for RIG-I downregulation and, hence, that RIG-I was not cleaved directly by PR.
FIG. 7.

HIV-1 PR downregulates RIG-I protein expression through a proteasome- and caspase-independent mechanism. (A) RIG-I downregulation is proteasome and caspase independent. THP-1 cells were infected de novo with HIV-1 at an MOI of 1 for 6, 24, and 72 h. Cells were incubated with either dimethyl sulfoxide (DMSO), 5 μM MG132 (proteasomal inhibitor), or 50 μM Z-VAD (pancaspase inhibitor), respectively. THP-1 cells infected with Sendai virus (40 hemagglutinating units/ml) for 6 h were used as a control for the induction of RIG-I expression. Whole-cell extracts were subjected to immunoblotting with anti-RIG-I and anti-β-actin antibodies. (B) RIG-I downregulation is proteasome and caspase independent. HEK293 cells were cotransfected with plasmids expressing Myc-RIG-I and GFP-protease and incubated with either 5 μM MG132 (proteasomal inhibitor) or 50 μM Z-VAD (pancaspase inhibitor). Whole-cell extracts were subjected to immunoblotting with anti-Myc, anti-GFP, and anti-β-actin antibodies. (C) HIV-1 PR-mediated downregulation of RIG-I is not dependent on the HIV-1 PR catalytic activity. HEK293 cells were transfected with Myc-RIG-I or GFP-protease expression plasmids and incubated with 5 μM saquinavir. At 24 h posttransfection, whole-cell extracts were subjected to Western blot analysis and immunoblotted for Myc-RIG-I, GFP-protease, and β-actin.
The involvement of HIV-1 viral proteins in cellular trafficking has been well documented; Vpu, for example, was previously shown to sequester the retroviral restriction factor tetherin in a perinuclear compartment (15). The observation that RIG-I turnover was independent of the catalytic activity of PR prompted us to examine the possibility that the cellular localization of RIG-I may be disrupted by PR, thus inhibiting RIG-I antiviral activity. An analysis of RIG-I in the cytosolic versus insoluble cellular fractions revealed that in the presence of PR, >50% of the RIG-I protein was located in the insoluble membranous fraction, whereas RIG-I was located exclusively in the cytosolic fraction in the absence of PR (Fig. 8A). In addition, de novo HIV-1-infected primary monocyte-derived macrophages demonstrated a relocalization of RIG-I in the insoluble fraction at 6 h postinfection, thus indicating that PR-containing virions target RIG-I early during HIV-1 infection (Fig. 8B). Confocal microscopy was next used to visualize RIG-I in the presence or absence of PR in A549 cells, which constitutively express endogenous RIG-I. RIG-I staining was predominantly cytoplasmic in the absence of PR, but in the presence of PR, endogenous RIG-I was localized to a perinuclear compartment. Lysosomal staining using the lysosomal marker LAMP-1 localized RIG-I to the lysosomes only in the presence of PR (Fig. 8C). In addition, following saquinavir treatment, endogenous RIG-I remained perinuclear, further demonstrating that the relocalization of RIG-I is not dependent on protease activity. Finally, the lysosomal inhibitor E64 was used to block RIG-I degradation in the presence of PR. E64 treatment impeded RIG-I degradation by the lysosomes, as the RIG-I protein was restored to levels seen in the absence of PR (Fig. 8D). Altogether, these data demonstrate that the cytoplasmic pool of RIG-I is redirected to the lysosomal compartment by HIV-1 PR, and relocation from the cytosol appears to be sufficient to abolish RIG-I signaling during de novo HIV-1 infection.
FIG. 8.

HIV-1 protease expression causes a relocalization of the cytoplasmic pool of RIG-I to the lysosomes. (A and B) HEK293 cells were cotransfected with plasmids expressing Flag-RIG-I and GFP-protease (A), and MDMs were infected de novo (or not) with HIV-1 at an MOI of 1 for 6, 24, and 72 h (B). Whole-cell extracts isolated from the cytoplasmic fraction versus the insoluble fraction were run on an SDS-PAGE gel and subsequently subjected to immunoblotting with anti-Flag, anti-RIG-I, or anti-β-actin antibodies, respectively. (C) A549 cells were transfected with plasmids expressing GFP or GFP-protease with or without saquinavir (5 μM). At 15 h posttransfection, cells were, fixed, permeabilized and immunostained for endogenous RIG-I (red) or LAMP-1 (gray). Cells were visualized by confocal microscopy. (D) A lysosomal inhibitor restores RIG-I protein expression. HEK293 cells were cotransfected with Myc-RIG-I or GFP-protease expression plasmids and treated with 10 μM E64 (lysosomal-protease inhibitor cocktail). At 24 h posttransfection, expression levels of RIG-I, protease, and β-actin were analyzed by immunoblotting with antibodies against Myc, GFP, and β-actin, respectively.
DISCUSSION
Recent studies highlighted the importance of the RIG-I sensor in the recognition of incoming virus RNA structures (76); an understanding of the evasion strategies used by viruses to circumvent this crucial antiviral response is an important step in the development of novel therapies. To date, there has been no evidence to suggest that RIG-I signaling is triggered by retroviral infection. The present study provides new insights concerning the involvement of RIG-I in HIV-1 infection: (i) the sensing of HIV-1 RNA occurs through a RIG-I-dependent but not MDA5-dependent pathway (in addition to a TLR7-dependent pathway), (ii) HIV-1 gRNA monomers induce a stronger activation of RIG-I than do HIV-1 gRNA dimers, and (iii) despite RIG-I triggering by viral RNA, downstream antiviral gene expression is inhibited by the HIV-1 protease, which causes a depletion of the cytoplasmic fraction of endogenous RIG-I and redistribution to the membranous lysosomal compartment. In support of these conclusions, the intracellular amount of RIG-I was decreased during de novo single-cycle infection by BH10 provirus, but not by the protease-deficient provirus (BH10-PR−) (Fig. 5), and by the ectopic expression of the PR (Fig. 6B). RIG-I downregulation was not mediated by proteasomal degradation or caspase-dependent cleavage (Fig. 7) and was not dependent on protease activity per se, since the protease inhibitor saquinavir did not block RIG-I relocalization (Fig. 7C and 8C). Moreover, RIG-I protein levels were restored by the lysosomal protease inhibitor E64 (Fig. 8D), suggesting that HIV-1 PR relocalized cytoplasmic RIG-I to the lysosomal compartment for degradation. Interestingly, the RIG-I cytoplasmic fraction was relocalized in the insoluble fraction following viral entry in de novo HIV-1-infected MDMs (Fig. 8B). By virtue of removing RIG-I from the cytosol, PR may impede RIG-I interaction with the mitochondrial adaptor MAVS and thus disconnect early innate antiviral signaling.
The detection of incoming viral RNA by RIG-I is dependent on numerous structural RNA modifications as well as specific secondary or tertiary RNA conformations. Indeed, the base-paired, panhandle structure at the 5′ end of RNA, together with a 5′-PPP moiety, appears to be important for RIG-I activation (61, 62); furthermore, the 5′-PPP-bearing viral RNAs from influenza A and rabies viruses effectively activate RIG-I (2, 26, 54, 57, 58). Other structural features of RNA may also be important for RIG-I recognition, since RNAs lacking 5′-PPP, such as short strands of synthetic poly(I:C), also activate RIG-I (32, 69). In addition, an A-U-rich motif in the 3′-untranslated region of the HCV genome was shown previously to induce RIG-I (60), thus indicating that distinct RNA species can trigger RIG-I in virally infected cells. Interestingly, small self-RNAs generated by the action of the antiviral endoribonuclease RNase L were also shown previously to be recognized by RIG-I (40).
HIV-1 genomic RNA is transcribed via a Tat/RNA polymerase II-dependent mechanism and harbors primarily 5′-capped transcripts. However, the presence of some uncapped (5′-triphosphorylated) RNAs at the 5′ end of HIV-1 RNA cannot be formally excluded; in fact, a mixture of capped and 5′-triphosphorylated RNAs was previously detected in producer cells and virions at early stages after virus entry (44). Several stem-loop structures composed of A-U-rich sequences in the 5′-untranslated region (5′-UTR) of HIV-1 RNA may stimulate RIG-I, due to their propensity to form complex secondary structures. This idea is supported by the observation that RIG-I was activated to a greater extent by HIV-1 monomers than by RNA dimers (Fig. 1B). RNA monomers might allow the stimulatory region to be further exposed, whereas the more condensed form of RNA found in dimers restricts the accessibility of the 5′-UTR for RNA sensors such as RIG-I and TLR7. Another IFN signaling pathway involved in viral RNA degradation, the 2′,5′-oligoadenylate synthetase (OAS)/RNase L pathway, is activated by the 5′ transactivation responsive element (TAR) region present in HIV-1 mRNA (39). Because much of the HIV-1 genomic structure remains uncharacterized (>85%) (75), further structural analysis of the 5′-UTR and the viral genome is required to identify the precise structural determinants recognized by RIG-I.
Despite clear evidence that purified genomic RNA can be recognized by RIG-I, an activation of RIG-I following de novo HIV-1 infection in human macrophages was not observed. The production of IFN was also not detected after infection, suggesting that HIV-1 had developed a strategy to counteract the early innate IFN response. An inhibition of RIG-I signaling occurred through an HIV-1 protease-dependent mechanism, since the coexpression of an active form of RIG-I along with increasing amounts of the PR led to a decrease in IFNB promoter activity in a concentration-dependent manner as well as an inhibition of IRF-3 phosphorylation (Fig. 6).
A major function of HIV-1 protease in the virus life cycle is to process the Gag and Gag-Pol precursors to yield mature virion proteins. The protease is initially synthesized in an inactive form as part of the Gag-Pol precursor and is activated within the virion during the final stages of virus assembly and virus maturation. PR is required for virion maturation, and its inhibition gives rise to immature virions that are unable to reinfect cells (reviewed in reference 11). The active protease present in incoming virions is thus poised to target host antiviral responses. The cleavage of cellular proteins by PR has been well documented; for example, the antiapoptotic protein Bcl-2 is cleaved by HIV-1 protease, leading to apoptosis (4). The death of HIV-1-infected T cells is linked to the cleavage of procaspase 8 to Casp8p41 by protease, which, once cleaved, induces apoptosis (50).
Protease expression resulted in a reduction in the expression levels of IFN and ISGs; the inability of HIV-1-infected cells to induce IFN-stimulated host restriction factors may indirectly contribute to increased viral replication. For example, the cellular deaminase APOBEC3G incorporates into HIV-1 virions and inhibits viral replication by inducing cytidine-to-uracil deamination in the newly transcribed single-stranded proviral DNA (66). The HIV-1 viral accessory protein Vif counteracts the antiretroviral effects of APOBEC3G by triggering the ubiquitinylation of APOBEC3G, leading to its proteasomal degradation (reviewed in references 12, 16, and 66). Thus, HIV-1 may act on APOBEC3G at two levels: at the transcriptional level, by ablating APOBEC3G induction via the PR-mediated inhibition of RIG-I and the IFN response, and at the protein level, via the Vif-mediated degradation of APOBEC3G. Furthermore, data from studies performed by two different groups have suggested that HIV-1 disrupts IFN signaling at the level of IRF-3 in CD4+ T-cell lines (13, 51). HIV-1 targets IRF-3 for protein depletion through the action of the HIV-1 accessory proteins Vif and Vpr, allowing HIV-1 to evade the host immune response. Similarly, the viral protein Vpu antagonizes the IFN-inducible viral restriction factor tetherin (also called BST-2, HM1.24, or CD317) by reducing its cell surface expression through Vpu-tetherin interactions, resulting in the relocalization and degradation of the restriction factor in perinuclear compartments (15, 71). The trafficking of tetherin from its functional location on the cell surface promotes increased HIV-1 virion release (49). The present study provides an additional example of how HIV-1 may disrupt the host innate response, by relocalizing a crucial RNA-sensing protein away from its functional microenvironment.
The ectopic expression of RIG-I or the helicase domain of RIG-I alone had an inhibitory effect on HIV-1 replication (Fig. 3). Interestingly, RNA helicases are known to participate in different steps of HIV-1 replication, and the DDX30 helicase in particular was previously reported to influence HIV-1 replication by decreasing the infectivity of nascent viral particles (81). As yet, it is not clear whether RIG-I helicase activity inhibits HIV-1 at the level of gRNA recognition or during reverse transcription. In contrast to the reduction in IFN and ISG expression levels, proinflammatory cytokine production was induced by HIV-1 replication early after infection, in part by NF-κB-dependent signaling (reviewed in reference 21). However, a rapid decrease in the expression levels of these proinflammatory cytokines was observed after infection, perhaps related to the expression of viral proteins such as Tat and Vpr (27, 46). This study provides for the first time a mechanism by which HIV-1 evades the activation of RNA sensors. Interestingly, a recent study by Yan et al. demonstrated that the cystolic exonuclease TREX1 suppresses IFN activation following HIV-1 infection. TREX1 binds and digests cytoplasmic HIV-1 DNA that would normally be detected by an unidentified DNA sensor that converges on the adaptor STING and the kinase TBK1 (79). Overall, these recent studies demonstrate the multifaceted strategies used by HIV-1 to suppress viral RNA- and DNA-sensing pathways.
In conclusion, this study demonstrates that HIV-1 genomic RNA is recognized by RIG-I, yet RIG-I signaling is inactivated via a unique PR-dependent mechanism that circumvents the host innate immune response. HIV-1 protease interferes with cytoplasmic RIG-I, leading to a trapped pool of perinuclear RIG-I. The inability to coimmunoprecipitate RIG-I and PR directly suggests that the PR-mediated relocalization of RIG-I may occur via an indirect interaction (data not shown). Since protease inhibitors are used extensively for HIV-1 therapy, and the PR-dependent ablation of RIG-I signaling is independent of protease activity, improved strategies to generate antiprotease drugs will be required to combat this specific mode of host immune response evasion.
Acknowledgments
This research was supported by grants from the Canadian Institute of Health Research (CIHR) and the Canadian Foundation for AIDS Research (CANFAR). M.S. was supported by a CIHR doctoral fellowship. P.N. was supported by an FRSQ doctoral fellowship.
We thank the members of the Molecular Oncology Group and the McGill AIDS Center, especially Jorge Martinez-Cajas and Maureen Oliveira, for helpful discussions and reagents. We also thank Robert Silverman and Shizuo Akira for reagents used in this study.
Footnotes
Published ahead of print on 17 November 2010.
REFERENCES
- 1.Baenziger, S., et al. 2009. Triggering TLR7 in mice induces immune activation and lymphoid system disruption, resembling HIV-mediated pathology. Blood 113:377-388. [DOI] [PubMed] [Google Scholar]
- 2.Baum, A., R. Sachidanandam, and A. Garcia-Sastre. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:16303-16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beignon, A. S., et al. 2005. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 115:3265-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanco, R., L. Carrasco, and I. Ventoso. 2003. Cell killing by HIV-1 protease. J. Biol. Chem. 278:1086-1093. [DOI] [PubMed] [Google Scholar]
- 5.Boasso, A., and G. M. Shearer. 2008. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin. Immunol. 126:235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowie, A. G., and L. Unterholzner. 2008. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8:911-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenchley, J. M., et al. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12:1365-1371. [DOI] [PubMed] [Google Scholar]
- 8.Chin, M. P., J. Chen, O. A. Nikolaitchik, and W. S. Hu. 2007. Molecular determinants of HIV-1 intersubtype recombination potential. Virology 363:437-446. [DOI] [PubMed] [Google Scholar]
- 9.Chin, M. P., T. D. Rhodes, J. Chen, W. Fu, and W. S. Hu. 2005. Identification of a major restriction in HIV-1 intersubtype recombination. Proc. Natl. Acad. Sci. U. S. A. 102:9002-9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clerzius, G., et al. 2009. ADAR1 interacts with PKR during human immunodeficiency virus infection of lymphocytes and contributes to viral replication. J. Virol. 83:10119-10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coffin, J. M. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed]
- 12.Cullen, B. R. 2006. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J. Virol. 80:1067-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doehle, B. P., F. Hladik, J. P. McNevin, M. J. McElrath, and M. Gale, Jr. 2009. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 83:10395-10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douville, R. N., and J. Hiscott. 2010. The interface between the innate interferon response and expression of host retroviral restriction factors. Cytokine 52:108-115. [DOI] [PubMed] [Google Scholar]
- 15.Dube, M., et al. 2010. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 6:e1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehrlich, E. S., and X. F. Yu. 2006. Lentiviral Vif: viral hijacker of the ubiquitin-proteasome system. Int. J. Hematol. 83:208-212. [DOI] [PubMed] [Google Scholar]
- 17.Frigault, M. M., J. Lacoste, J. L. Swift, and C. M. Brown. 2009. Live-cell microscopy—tips and tools. J. Cell Sci. 122:753-767. [DOI] [PubMed] [Google Scholar]
- 18.Fu, W., R. J. Gorelick, and A. Rein. 1994. Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J. Virol. 68:5013-5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gringhuis, S. I., et al. 2010. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 11:419-426. [DOI] [PubMed] [Google Scholar]
- 20.Heil, F., et al. 2004. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 303:1526-1529. [DOI] [PubMed] [Google Scholar]
- 21.Herbein, G., and A. Varin. 2010. The macrophage in HIV-1 infection: from activation to deactivation? Retrovirology 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herbeuval, J. P., and G. M. Shearer. 2007. HIV-1 immunopathogenesis: how good interferon turns bad. Clin. Immunol. 123:121-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiscott, J., R. Lin, P. Nakhaei, and S. Paz. 2006. MasterCARD: a priceless link to innate immunity. Trends Mol. Med. 12:53-56. [DOI] [PubMed] [Google Scholar]
- 24.Honda, K., and T. Taniguchi. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6:644-658. [DOI] [PubMed] [Google Scholar]
- 25.Hong, S., J. Cao, and Y. T. Tu. 2009. Evolution of HIV-1 in a patient population failing multiple-drug therapy. Microbiol. Immunol. 53:535-539. [DOI] [PubMed] [Google Scholar]
- 26.Hornung, V., et al. 2006. 5′-triphosphate RNA is the ligand for RIG-I. Science 314:994-997. [DOI] [PubMed] [Google Scholar]
- 27.Ito, M., et al. 1998. HIV type 1 Tat protein inhibits interleukin 12 production by human peripheral blood mononuclear cells. AIDS Res. Hum. Retroviruses 14:845-849. [DOI] [PubMed] [Google Scholar]
- 28.Jalalirad, M., and M. Laughrea. 2010. Formation of immature and mature genomic RNA dimers in wild-type and protease-inactive HIV-1: differential roles of the Gag polyprotein, nucleocapsid proteins NCp15, NCp9, NCp7, and the dimerization initiation site. Virology 407:225-236. [DOI] [PubMed] [Google Scholar]
- 29.Kafaie, J., R. Song, L. Abrahamyan, A. J. Mouland, and M. Laughrea. 2008. Mapping of nucleocapsid residues important for HIV-1 genomic RNA dimerization and packaging. Virology 375:592-610. [DOI] [PubMed] [Google Scholar]
- 30.Kang, D. C., et al. 2002. mda-5: an interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. U. S. A. 99:637-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato, H., et al. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19-28. [DOI] [PubMed] [Google Scholar]
- 32.Kato, H., et al. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato, H., et al. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101-105. [DOI] [PubMed] [Google Scholar]
- 34.Kawai, T., and S. Akira. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131-137. [DOI] [PubMed] [Google Scholar]
- 35.Kawai, T., et al. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981-988. [DOI] [PubMed] [Google Scholar]
- 36.Kirchhoff, F. 2010. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8:55-67. [DOI] [PubMed] [Google Scholar]
- 37.Li, K., Z. Chen, N. Kato, M. Gale, Jr., and S. M. Lemon. 2005. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 280:16739-16747. [DOI] [PubMed] [Google Scholar]
- 38.Lin, R., et al. 2006. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 80:6072-6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maitra, R. K., et al. 1994. HIV-1 TAR RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology 204:823-827. [DOI] [PubMed] [Google Scholar]
- 40.Malathi, K., B. Dong, M. Gale, Jr., and R. H. Silverman. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maniatis, T., et al. 1998. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 63:609-620. [DOI] [PubMed] [Google Scholar]
- 42.Matikainen, S., et al. 2006. Tumor necrosis factor alpha enhances influenza A virus-induced expression of antiviral cytokines by activating RIG-I gene expression. J. Virol. 80:3515-3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meier, A., et al. 2007. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 81:8180-8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menees, T. M., B. Muller, and H. G. Krausslich. 2007. The major 5′ end of HIV type 1 RNA corresponds to G456. AIDS Res. Hum. Retroviruses 23:1042-1048. [DOI] [PubMed] [Google Scholar]
- 45.Meylan, E., et al. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167-1172. [DOI] [PubMed] [Google Scholar]
- 46.Mirani, M., et al. 2002. HIV-1 protein Vpr suppresses IL-12 production from human monocytes by enhancing glucocorticoid action: potential implications of Vpr coactivator activity for the innate and cellular immunity deficits observed in HIV-1 infection. J. Immunol. 169:6361-6368. [DOI] [PubMed] [Google Scholar]
- 47.Moore, M. D., and W. S. Hu. 2009. HIV-1 RNA dimerization: it takes two to tango. AIDS Rev. 11:91-102. [PMC free article] [PubMed] [Google Scholar]
- 48.Nakhaei, P., P. Genin, A. Civas, and J. Hiscott. 2009. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin. Immunol. 21:215-222. [DOI] [PubMed] [Google Scholar]
- 49.Neil, S. J., T. Zang, and P. D. Bieniasz. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425-430. [DOI] [PubMed] [Google Scholar]
- 50.Nie, Z., et al. 2002. HIV-1 protease processes procaspase 8 to cause mitochondrial release of cytochrome c, caspase cleavage and nuclear fragmentation. Cell Death Differ. 9:1172-1184. [DOI] [PubMed] [Google Scholar]
- 51.Okumura, A., et al. 2008. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology 373:85-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papon, L., et al. 2009. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 393:311-318. [DOI] [PubMed] [Google Scholar]
- 53.Paz, S., et al. 2006. Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell. Mol. Biol. (Noisy-le-Grand) 52:17-28. [PubMed] [Google Scholar]
- 54.Pichlmair, A., et al. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997-1001. [DOI] [PubMed] [Google Scholar]
- 55.Plumet, S., et al. 2007. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS One 2:e279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Randall, R. E., and S. Goodbourn. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1-47. [DOI] [PubMed] [Google Scholar]
- 57.Rehwinkel, J. 2010. Exposing viruses: RNA patterns sensed by RIG-I-like receptors. J. Clin. Immunol. 30:491-495. [DOI] [PubMed] [Google Scholar]
- 58.Rehwinkel, J., et al. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397-408. [DOI] [PubMed] [Google Scholar]
- 59.Romieu-Mourez, R., et al. 2006. Distinct roles for IFN regulatory factor (IRF)-3 and IRF-7 in the activation of antitumor properties of human macrophages. Cancer Res. 66:10576-10585. [DOI] [PubMed] [Google Scholar]
- 60.Saito, T., D. M. Owen, F. Jiang, J. Marcotrigiano, and M. Gale, Jr. 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schlee, M., et al. 2009. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31:25-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt, A., et al. 2009. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. U. S. A. 106:12067-12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Servant, M. J., N. Grandvaux, B. R. tenOever, D. Duguay, R. Lin, and J. Hiscott. 2003. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 278:9441-9447. [DOI] [PubMed] [Google Scholar]
- 64.Seth, R. B., L. Sun, C. K. Ea, and Z. J. Chen. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669-682. [DOI] [PubMed] [Google Scholar]
- 65.Sharma, S., B. R. tenOever, N. Grandvaux, G. P. Zhou, R. Lin, and J. Hiscott. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148-1151. [DOI] [PubMed] [Google Scholar]
- 66.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646-650. [DOI] [PubMed] [Google Scholar]
- 67.Solis, M., et al. 2006. Gene expression profiling of the host response to HIV-1 B, C, or A/E infection in monocyte-derived dendritic cells. Virology 352:86-99. [DOI] [PubMed] [Google Scholar]
- 68.Song, R., J. Kafaie, L. Yang, and M. Laughrea. 2007. HIV-1 viral RNA is selected in the form of monomers that dimerize in a three-step protease-dependent process; the DIS of stem-loop 1 initiates viral RNA dimerization. J. Mol. Biol. 371:1084-1098. [DOI] [PubMed] [Google Scholar]
- 69.Takahasi, K., et al. 2008. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 29:428-440. [DOI] [PubMed] [Google Scholar]
- 70.Vahey, M. T., et al. 2002. Impact of viral infection on the gene expression profiles of proliferating normal human peripheral blood mononuclear cells infected with HIV type 1 RF. AIDS Res. Hum. Retroviruses 18:179-192. [DOI] [PubMed] [Google Scholar]
- 71.Van Damme, N., et al. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vazquez, N., et al. 2005. Human immunodeficiency virus type 1-induced macrophage gene expression includes the p21 gene, a target for viral regulation. J. Virol. 79:4479-4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vendrame, D., M. Sourisseau, V. Perrin, O. Schwartz, and F. Mammano. 2009. Partial inhibition of human immunodeficiency virus replication by type I interferons: impact of cell-to-cell viral transfer. J. Virol. 83:10527-10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang, F., et al. 2008. RIG-I mediates the co-induction of tumor necrosis factor and type I interferon elicited by myxoma virus in primary human macrophages. PLoS Pathog. 4:e1000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watts, J. M., et al. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460:711-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilkins, C., and M. Gale, Jr. 2010. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 22:41-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Woelk, C. H., et al. 2004. Interferon gene expression following HIV type 1 infection of monocyte-derived macrophages. AIDS Res. Hum. Retroviruses 20:1210-1222. [DOI] [PubMed] [Google Scholar]
- 78.Xu, L. G., et al. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727-740. [DOI] [PubMed] [Google Scholar]
- 79.Yan, N., A. D. Regalado-Magdos, B. Stiggelbout, M. A. Lee-Kirsch, and J. Lieberman. 2010. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11:1005-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yoneyama, M., et al. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730-737. [DOI] [PubMed] [Google Scholar]
- 81.Zhou, Y., et al. 2008. The packaging of human immunodeficiency virus type 1 RNA is restricted by overexpression of an RNA helicase DHX30. Virology 372:97-106. [DOI] [PubMed] [Google Scholar]