

Abstract
Hepatitis C virus NS3-4A is a membrane-bound enzyme complex that exhibits serine protease, RNA helicase, and RNA-stimulated ATPase activities. This enzyme complex is essential for viral genome replication and has been recently implicated in virus particle assembly. To help clarify the role of NS4A in these processes, we conducted alanine scanning mutagenesis on the C-terminal acidic domain of NS4A in the context of a chimeric genotype 2a reporter virus. Of 13 mutants tested, two (Y45A and F48A) had severe defects in replication, while seven (K41A, L44A, D49A, E50A, M51A, E52A, and E53A) efficiently replicated but had severe defects in virus particle assembly. Multiple strategies were used to identify second-site mutations that suppressed these NS4A defects. The replication defect of NS4A F48A was partially suppressed by mutation of NS4B I7F, indicating that a genetic interaction between NS4A and NS4B contributes to RNA replication. Furthermore, the virus assembly defect of NS4A K41A was suppressed by NS3 Q221L, a mutation previously implicated in overcoming other virus assembly defects. We therefore examined the known enzymatic activities of wild-type or mutant forms of NS3-4A but did not detect specific defects in the mutants. Taken together, our data reveal interactions between NS4A and NS4B that control genome replication and between NS3 and NS4A that control virus assembly.
Hepatitis C virus (HCV) is an enveloped, positive-strand RNA virus classified within the genus Hepacivirus in the family Flaviviridae (21). The nonsegmented HCV genome is 9.6-kb and encodes a long polyprotein that is cleaved by cellular and viral proteases into at least 10 distinct products (47, 54). The N-terminal region of this polyprotein encodes the viral structural proteins: core and two envelope glycoproteins, E1 and E2. The remainder of the polyprotein encodes nonstructural (NS) proteins: p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B. Together, the NS proteins regulate intracellular aspects of the viral life cycle, including RNA replication, virus assembly, and viral subversion of host antiviral responses.
NS3-4A is an essential enzyme complex of the viral replication machinery and a major target for HCV-specific drug development. The N-terminal domain of NS3 encodes a chymotrypsin-like serine protease that requires NS4A for full activity and is responsible for the cleavage of the NS3/4A, NS4A/4B, NS4B/5A, and NS5A/5B junctions (47). In addition, the NS3-4A serine protease antagonizes host innate antiviral responses by cleaving the cellular messenger IPS-1 (also known as MAVS, Cardif, and VISA) (43, 44, 49, 53). The C-terminal region of NS3 encodes a superfamily 2 helicase/NTPase domain, which is essential for virus genome replication (36, 41). The serine protease and RNA helicase activities are coordinated, such that the helicase domain can influence the protease activity, and the protease domain can influence RNA helicase activity (7, 8, 24, 40). More recently, a number of genetic studies have implicated the NS3 helicase domain in virus particle assembly (28, 52, 58, 69). However, the precise role of NS3-4A helicase activity in RNA replication and virus assembly is not yet clear.
At only 54 amino acids, NS4A is the smallest NS protein. It encodes an N-terminal hydrophobic alpha-helix that anchors NS3-4A to cellular membranes, a middle “cofactor” peptide that forms a beta-strand and contributes to proper folding of the NS3 serine protease domain, and a C-terminal acidic region that forms an alpha-helix at low pH (3, 12, 20, 34, 46, 66, 68). Our prior genetic and biochemical studies have shown that the NS4A C-terminal region contributes to NS3-4A helicase activity and is essential for HCV RNA replication (6, 46).
In addition to its NS3 cofactor activity, NS4A has additional roles that may be important for the viral life cycle. NS4A expression is needed for NS5A hyperphosphorylation (2, 32, 66), and NS4A can interact with cellular factors such as creatine kinase B (29) and elongation factor 1A (39). Overexpression of NS4A in the absence of NS3 inhibits cellular translation (23, 33, 39) and can lead to mitochondrial damage (55, 62), although it is not yet clear whether NS4A exhibits these NS3-independent functions during the viral life cycle. In addition, it has been noted that the sequence of NS4A covaries with multiple viral genes, suggesting that NS4A may play a central role for multiple processes (13).
To further understand the role of NS4A in the HCV life cycle, we conducted alanine-scanning mutagenesis of the NS4A C-terminal acidic region in the chimeric genotype 2a infectious cell culture system. Our results confirm that this region contains important RNA replication determinants and reveal that this region has an essential role in the assembly of viral particles. Further analyses demonstrated genetic interactions between NS3, NS4A, and NS4B in replication and virus assembly. We therefore investigated how specific mutations affect NS3-4A enzymatic activities but did not observe detectable differences relative to the wild type (WT). These data support a role for NS3-4A in virus particle assembly, perhaps through interaction with other proteins.
MATERIALS AND METHODS
Plasmid constructs.
Standard molecular biology methods were used throughout (61). Plasmids were verified by restriction enzyme digestion and/or DNA sequencing at the W. M. Keck Foundation Biotechnology Resource Center at Yale University.
The plasmids pYSGR-JFH/Neo, pJc1, pJc1/GLuc2A, pJc1/GLuc2A(Δcore), and pJc1/GLuc2A(GNN) were recently described (58). To construct the pYSGR-JFH/GLuc replicon, a 280-bp fragment from pYSGR-JFH was amplified with the primers YO-0065 (5′-TCC CGG GAG AGC CAT AGT GGT-3′) and YO-0672 (5′-GTT TAA ACG CGG CCG CGA ATT CTA GAT CTG GGC GAC GGT TGG TGT TTC-3′). The amplification product was cloned into pCR2.1-TOPO (Invitrogen, Carlsbad, CA), sequence verified, and subcloned into pYSGR-JFH by using common AgeI and PmeI sites, yielding pYSGR-JFH-INT. A 572-bp fragment of Gaussia princeps luciferase (GLuc), including its endogenous signal peptide, was then subcloned from pCMV-GLuc (New England Biolabs, Ipswich, MA) into pYSGR-JFH-INT by using common BamHI and NotI restriction sites.
Alanine substitutions were introduced into NS4A as follows. First, pRB96 was constructed by subcloning a 3,043-bp Acc65I/BamHI fragment from pJc1 (58) into pSL1180 (GE Healthcare, Buckinghamshire, United Kingdom). Site-directed mutagenesis was then conducted on pRB96 by using the QuikChange strategy with Pfu DNA polymerase (Stratagene, La Jolla, CA), forward primers (listed in Table 1 ), and complementary reverse primers. Reactions were treated with DpnI (New England Biolabs) and used to transform competent DH5α bacterial cells. After sequence verification, mutations were subcloned into a derivative of pJc1/GLuc2A (58) by using common SpeI and BamHI sites.
TABLE 1.
Primers used for site-directed mutagenesis of NS4A
| NS4A mutation | Primer sequence (5′-3′)a |
|---|---|
| D40A | GTCGTCGTTGCGCCGGCCAAGGAGGTCCTGTAT |
| K41A | AGTCGTCGTTGCGCCGGATGCCGAGGTCCTGTATGAG |
| E42A | GTCGTTGCGCCGGATAAGGCCGTCCTGTATGAGGCT |
| V43A | TGCGCCGGATAAGGAGGCCCTGTATGAGGCTTTTGATG |
| L44A | TGCGCCGGATAAGGAGGTCGCCTATGAGGCTTTTGATG |
| Y45A | GATAAGGAGGTCCTGGCCGAGGCTTTTGATGAGAT |
| E46A | ATAAGGAGGTCCTGTATGCCGCTTTTGATGAGATGGA |
| F48A | AGGTCCTGTATGAGGCTGCCGATGAGATGGAGGAATG |
| D49A | TCCTGTATGAGGCTTTTGCCGAGATGGAGGAATGC |
| E50A | TGTATGAGGCTTTTGATGCCATGGAGGAATGCGCCTC |
| M51A | ATGAGGCTTTTGATGAGGCCGAGGAATGCGCCTCTAG |
| E52A | GAGGCTTTTGATGAGATGGCCGAATGCGCCTCTAGGGCGGCT |
| E53A | AGGCTTTTGATGAGATGGAGGCCTGCGCCTCTAGGGCGGCTC |
The forward primers are shown. Complementary reverse primers were also used in site-directed mutagenesis. Mutation sites are underlined.
To construct pLenti/core-NS2, the Jc1 core through NS2 region was amplified as a 3,177-bp fragment with KlenTaqLA (DNA Polymerase Technology, Inc., St. Louis, MO) and the primers YO-0353 (5′-TCT AGA GCC ACC ATG AGC ACA AAT CCT AAA CCT C-3′) and YO-0354 (5′-CTC GAG TGT ACA TTA AAG GAG CTT CCA CCC CTT G-3′). The amplified product was cloned into pCR2.1-TOPO (Invitrogen), sequenced, and subcloned into pLenti4/MCS by using compatible XbaI and XhoI sites. pLenti4/MCS was made by annealing oligonucleotides YO-0183 (5′-GAT CCA CTA GTC TGC AGT CCG GAC-3′) and YO-0184 (5′-CAC TAG TCT GCA GTC CGG ACT CGA-3′) and ligating them into pLenti4/EGFP (Invitrogen) cut with BamHI and XhoI.
To prepare purified NS3-4A protein, WT and mutant NS3-4A genes were amplified by PCR and cloned into the pET-SUMO vector (Invitrogen) according to the manufacturer's suggestions.
Cell culture and RNA transfection.
All cell culture work was performed in an enhanced biosafety level 2+ suite licensed by the State of Connecticut Department of Public Health. Huh-7.5 cells (10) were maintained in Dulbecco modified Eagle medium (Invitrogen) containing 10% fetal calf serum (HyClone, Logan, UT) and 1 mM nonessential amino acids (Invitrogen). Cells were transfected with HCV RNA transcripts by electroporation, as previously described (45) or by using the TransIT-mRNA transfection kit (Mirus Bio, Madison, WI).
Huh-7.5[core-NS2] cells, which expressed the Jc1 structural proteins (core, E1, and E2), and p7 and NS2 proteins, were derived by lentivirus transduction. Briefly, the pLenti/core-NS2 lentivirus vector was packaged in 293T cells by using the ViraPower packaging system (Invitrogen) and used to transduce Huh-7.5 cells. Stable core-NS2-expressing cells were selected and maintained by using standard growth medium containing 100 μg of zeocin (InvivoGen, San Diego, CA)/ml.
GLuc2A activity.
Conditioned cell culture medium was collected at various times postelectroporation or at 72 h postinfection, clarified by centrifugation (16,000 × g for 5 min), and mixed with 1/4 volume of Renilla 5× lysis buffer (Promega, Madison, WI) to kill HCV infectivity. GLuc2A activity was measured on a Berthold Centro LB 960 luminescent plate reader with 20-μl sample injected with 50 μl of Gaussia luciferase assay reagent (New England Biolabs), integrated over 10 s.
Replication and infectivity measurements.
To measure the relative replication of GLuc2A reporter viruses, conditioned cell culture media were collected at various times postelectroporation, clarified by centrifugation (16,000 × g for 5 min), and stored at −80°C. To measure relative infectivity, the supernatants were used to infect naive Huh-7.5 cells seeded at 6.4 × 103 cells/well in 96-well plates. After a 6- to 24-h adsorption period, cells were washed three times with Dulbecco phosphate-buffered saline (PBS) and incubated with complete medium for an additional 48 h. Cell culture media were collected, clarified, and assayed as described above.
To measure the relative intracellular infectivity of GLuc2A reporter viruses, cells were harvested by trypsinization at 48 h posttransfection, centrifuged (1,200 × g, 5 min), resuspended in a small volume of complete medium, snap-frozen in liquid nitrogen, and stored at −80°C. Cells were subjected to three rounds of thawing (37°C) and refreezing (−196°C) in liquid nitrogen. Cellular debris was removed by centrifugation (16,000 × g, 5 min), and the supernatants were tested for infectivity as described above.
Colony formation assays were used to measure the replication or infectivity of trans-packaged HCV replicons that express the Neo gene. To measure replication, RNA-transfected Huh-7.5 cells were seeded onto 10-cm dishes. Selection of replicon-containing colonies was performed 3 days postelectroporation by the addition of complete medium supplemented with 1 mg of G418/ml. Selection was carried out for a period of 3 weeks by replacing the G418-containing medium every 3 days. After selection, cells were fixed with 7% formaldehyde and stained with 1% crystal violet in 20% ethanol. G418-resistant colonies were counted and used to calculate transduction efficiencies in units of CFU per microgram of input RNA. To measure the infectivities of trans-packaged replicons, conditioned cell culture media were collected at 48 h postelectroporation from RNA-transfected Huh-7.5[core-NS2] cells, cellular debris was removed by centrifugation (16,000 × g, 5 min), and supernatants were used to infect naive Huh-7.5 cells previously seeded at 5.1 × 105 cells in 10-cm dishes. Selection with G418 was performed as described above.
RNA quantitation.
RNA was extracted from conditioned cell culture media by using the QIAamp viral RNA minikit (Qiagen, Valencia, CA). HCV RNA levels were measured by real-time quantitative reverse transcription (qRT-PCR) as previously described (45). Briefly, reactions were run on a Roche LightCycler 480 with the LightCycler RNA amplification HybProbe kit (Roche Applied Sciences, Mannheim, Germany) containing 2 μl of RNA sample or RNA quantitation standards, 8 mM MgCl2, 375 nM concentrations of each primer, 250 nM probe, and 1 U of RNase inhibitor (United States Biochemicals, Cleveland, OH).
Identification of suppressor mutations.
To select for subgenomic variants that had regained replication or infectivity, G418-resistant colonies were treated with trypsin in glass cloning cylinders and expanded in G418-containing medium. Total RNA was extracted by using TRIzol (Invitrogen) according to the manufacturer's instructions and resuspended in 100 μl of 1 mM sodium citrate (pH 6.4). cDNAs were synthesized by using random hexamers, Superscript III (Invitrogen), and SUPERase-In (Ambion, Austin, TX) at 55°C for 1 h, followed by 99°C for 5 min, and cooled at 4°C. The pYSGR-JFH/GLuc coding regions were amplified by using HCV-specific primers (58) and illustra PuReTaq Ready-To-Go PCR beads (GE Healthcare). PCR products were purified by using QIAquick PCR spin columns and sequenced at the Keck Center. PCRs containing mutations of interest were cloned by using pCR2.1-TOPO (Invitrogen), their sequences were verified, and mutations of interest were subcloned back into pYSGR-JFH, pYSGR-JFH/GLuc, or pJc1/GLuc2A by using common restriction sites.
HCV polyprotein expression.
Huh-7.5 cells were seeded at 5 × 105/well into six-well plates the day before use. Cells were infected for 1 h at a multiplicity of infection of 10 with vTF7-3 (25) and transfected with pJc1/GLuc2A derivatives by using Mirus LT1 reagent according to the manufacturer's recommendations. At 24 h postinfection, the cells were washed twice with Dulbecco PBS, lysed in 0.3 ml of sample buffer (50 mM Tris [pH 6.8], 100 mM dithiothreitol [DTT], 2% [wt/vol] sodium dodecyl sulfate [SDS], 10% [vol/vol] glycerol, 0.1% [wt/vol] bromophenol blue), and homogenized by multiple passes through a 27-gauge needle. Samples (5 μl) were denatured by boiling, resolved by SDS-polyacrylamide gel electrophoresis (PAGE), and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Membranes were blocked in PBS-T (PBS containing 0.1% [vol/vol] Tween 20 [polyoxyethylene sorbitan monolaurate]) containing 5% (wt/vol) dry milk and probed with this blocking buffer containing primary monoclonal antibodies against core (C7-50; Affinity BioReagents, Golden, CO), E2 (3/11; kindly provided by Jane McKeating) (22), NS2 (6H6; kindly provided by Charles M. Rice) (16), NS3 (9G2; Virogen, Watertown, MA), NS5A (9E10) (45), or β-actin (AC-15; Sigma-Aldrich, St. Louis, MO). After several washes with PBS-T, the membranes were probed with horseradish peroxidase-conjugated secondary antibodies and washed repeatedly, and antigens were detected with SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL).
Characterization of NS3-4A enzymology.
Recombinant NS3-4A was purified as previously described with minor modifications (58). Briefly, proteins were expressed and purified from 1 liter of Rosetta 2 (DE3) bacterial cultures. Cell pellets were resuspended in low-salt buffer (25 mM Tris-HCl [pH 8.0], 25 mM NaCl, 20% [vol/vol] glycerol, 5 mM β-mercaptoethanol, 2% [vol/vol] Triton X-100, 250 U of Benzonase [EMD Chemicals, Philadelphia, PA]) and lysed with three passes through a microfluidizer at 15,000 lb/in2. The lysate was adjusted to 500 mM NaCl and 10 mM imidazole and clarified with a 30,000 × g spin for 40 min at 4°C. The supernatant was batch-bound with 2 ml of Qiagen Ni-NTA agarose beads and washed with 20 column volumes of buffer A (25 mM Tris-HCl [pH 8.0], 20% [vol/vol] glycerol, 500 mM NaCl, 5 mM β-mercaptoethanol, 20 mM imidazole, 0.2% [vol/vol] Triton X-100) in a 10-ml Bio-Rad Poly-Prep column. The bound His6-SUMO fusion proteins were eluted with 5 column volumes of buffer B (buffer A containing 160 mM imidazole). The elution was diluted 5-fold with buffer C (10 mM Tris-HCl [pH 8.0], 20% [vol/vol] glycerol, 5 mM β-mercaptoethanol, 0.2% [vol/vol] Triton X-100, 300 mM NaCl) and treated with His6-SUMO protease overnight at 4°C. The digested elution was batch-bound a second time with 5-ml Ni-NTA beads, and the flowthrough was collected and buffer exchanged three times with buffer C on a 50-kDa cutoff Amicon centrifugal ultrafiltration unit (Millipore). Protein preparations were divided into 10-μl aliquots, flash frozen in liquid nitrogen, and stored at −80°C.
Protease assays were performed as previously described (7). Briefly, 60-μl reaction mixtures contained 40 nM NS3-4A, 5 μM RET-S1 substrate, and assay buffer (25 mM morpholinepropanesulfonic acid [MOPS]-NH4+ [pH 6.5], 1% [vol/vol] glycerol, 2 mM DTT, 30 mM NaCl, 0.2% [vol/vol] Triton X-100) containing 3 mM MgCl2. Fluorescence readings (excitation, 350 nm; emission, 500 nm) were collected at 37°C in a temperature-controlled cuvette on a Cary Eclipse spectrophotometer (Varian, Inc., Palo Alto, CA). Steady-state protease rates were calculated during the linear phase of the reactions (10 to 40 s).
RNA unwinding (helicase) assays were performed as previously described (5-7). Briefly, the unwinding substrate used in these assays was RNA2, a 34-bp duplex with a 20-nucleotide (nt) 3′ overhang. NS3-4A (10 pmol) was preincubated with 1 nM duplex substrate for 1 h at 37°C in 100-μl reaction mixtures containing assay buffer and 3 mM MgCl2. Helicase reactions were initiated by adding 6 μl of the preincubation mixtures to 2 μl of ATP-trap mix (25 mM MOPS-NH4+ [pH 6.5], 3 mM MgCl2, 30 mM NaCl, 1 μM top-strand oligonucleotide, and 16 mM ATP).
The ATPase activity of the NS3-4A constructs was monitored by using an ATP/NADH linked assay as previously described (14, 48). Briefly, 90-μl reaction mixtures contained 50 nmol of NS3-4A, 20 μl of 5× enzyme buffer, 0.75 mM MgCl2, and various RNA concentrations in assay buffer. The 5× enzyme buffer contained 1 mM NADH, 100 U of lactic dehydrogenase/ml, 500 U of pyruvate kinase/ml, and 2.5 mM phosphoenolpyruvate in assay buffer. Reactions were allowed to equilibrate to 37°C for 2 min and then initiated by adding 10 μl of 10 mM ATP. Fluorescence readings (excitation, 340 nm; emission, 450 nm) were collected in a temperature-controlled cuvette on a Cary Eclipse spectrophotometer (Varian). Initial velocities were calculated from a linear regression of each time course and corrected for background ATP hydrolysis and NADH oxidation. The initial velocities (v0) at various RNA concentrations were plotted and fit to the following Michaelis-Menten equation: v0 = (Vmax·[S]/(Km + [S])) + B, where B is the basal activity of the helicase.
RESULTS
Mutagenesis of NS4A.
We conducted a reverse genetic analysis of HCV NS4A in the context of a chimeric genotype 2a cDNA that efficiently replicates and produces high titers of infectious virus in cell culture (45, 59). To facilitate rapid measurements of viral replication and infectivity, we used a reporter virus, Jc1/GLuc2A, which expresses a secreted luciferase from Gaussia princeps (Fig. 1A). Previous work established that this reporter virus efficiently replicates and secretes GLuc2A in proportion to virus replication (58). Furthermore, Jc1/GLuc2A can be used to measure infectious virus production by monitoring the output of secreted GLuc2A activity expressed by infected cells. Importantly, the level of GLuc2A expressed by infected cells is proportional to the amount of input virus, indicating that this is a reliable method to measure relative infectivity levels (58).
FIG. 1.

Phenotypes of NS4A mutants. (A) Jc1/GLuc2A reporter construct used in the present study. The sequence of the NS4A C-terminal acidic domain is shown in single-letter amino acid code. Closed circles represent signal peptidase cleavages, the open circle represents a signal peptide peptidase cleavage, the open arrowhead represents NS2-3 cysteine autoprotease cleavage, and closed arrowheads represent NS3-4A serine protease cleavage sites. (B) Replication of NS4A mutants. Cells were transfected with each indicated mutant, and media were collected at 24, 48, 72, and 96 h posttransfection. Values represent the time course of secreted GLuc activity for each transfection, expressed as an average of at least three independent transfections. Error bars represent the standard deviation of the mean. (C) Released infectivity of NS4A mutants. Relative infectivity levels in the media from panel B were quantified by infecting naive cells and monitoring secreted GLuc activity. (D) RNA release of NS4A mutants. The amount of HCV RNA present in the media at 72 h posttransfection was determined by quantitative RT-PCR as described in Materials and Methods. Values represent average RNA quantities from at least three independent transfections. Error bars represent the standard deviation of the mean. (E) Intracellular infectivity of NS4A mutants. Cells were lysed at 48 h posttransfection and used to infect naive cells as described in Materials and Methods. Values represent average relative infectivity measurements from at least three independent transfections. Error bars represent the standard deviation of the mean.
To investigate the role of NS4A in the viral life cycle, we performed alanine scanning mutagenesis on the conserved, acidic C-terminal domain of NS4A. NS4A residues 40 to 46 and residues 48 to 53 were individually mutated to alanine in the context of Jc1/GLuc2A (Fig. 1A). NS4A residue 47 was not mutated because it is already an alanine; the cysteine at NS4A residue 54 was not mutated because it is required for proper NS4A/B cleavage (4, 35, 37, 42, 65). Viral replication levels were determined by measuring the amount of luciferase activity secreted into the cell culture media after electroporating Huh-7.5 cells with WT or mutant Jc1/GLuc2A RNA transcripts. As expected, WT Jc1/GLuc2A showed a time-dependent increase in secreted GLuc2A activity, while Jc1/GLuc2A(GNN), which encodes a catalytically inactive RNA-dependent RNA polymerase, did not express detectable GLuc2A activity (Fig. 1B, compare to “mock” transfected cells). Jc1/GLuc2A(Δcore), which replicates but does not produce infectious particles, expressed lower levels of secreted GLuc2A activity than did the WT, presumably because this genome cannot spread within the culture (58). A majority of NS4A mutants efficiently replicated, expressing secreted GLuc2A levels that were comparable to Jc1/GLuc2A or Jc1/GLuc2A(Δcore) (Fig. 1B). The notable exceptions were the NS4A Y45A and F48A mutants, which had undetectable levels of viral replication. These data are consistent with an essential role for these aromatic residues, as observed in our previous analysis of NS4A in genotype 1b replicons (46).
When assayed for infectious virus production, the NS4A D40A, E42A, V43A, and E46A mutants produced high levels of relative infectivity, similar to WT Jc1/GLuc2A (Fig. 1C). As expected, Jc1/GLuc2A(GNN), pJc1/GLuc2A(Δcore), and mock-transfected cells did not produce infectious virus. Surprisingly, a majority of NS4A mutants showed severe or lethal defects in infectious virus production. The NS4A K41A, L44A, Y45A, F48A, E50A, M51A, E52A, and E53A mutants did not produce detectable levels of infectious virus, whereas the NS4A D49A mutant showed a severe infectivity phenotype (Fig. 1C). These data reveal a novel role for the NS4A acidic region in producing virus particles.
We hypothesized that NS4A mutants could release virus particles that are not infectious, exhibit a block in releasing virus particles, or fail to assemble virus particles. To determine whether NS4A mutants release virus particles that are not infectious, we used qRT-PCR to measure the total amount of viral RNA released into the cell culture medium, which serves as a marker for virus particle release. As shown in Fig. 1D, WT Jc1/GLuc2A efficiently released HCV RNA into the cell culture medium, while Jc1/GLuc2A(GNN) and Jc1/GLuc2A(Δcore) did not. Notably, NS4A mutants with defects in infectious virus production did not release HCV RNA into the cell culture medium, while NS4A mutants that produced infectious virus released viral RNA levels that were similar to WT Jc1/GLuc2A (Fig. 1D). Thus, the amount of viral RNA released into the cell culture medium strongly correlated with the amount of relative infectivity released into the cell culture medium, indicating that the noninfectious NS4A mutants do not secrete virus particles.
To determine whether the noninfectious NS4A mutants had defects in virus release or in virus assembly, we examined the relative levels of intracellular infectivity present at 72 h after transfection of Huh-7.5 cells with the panel of NS4A mutants. As shown in Fig. 1E, the levels of intracellular infectivity strongly correlated with the levels of infectivity (Fig. 1C) and RNA (Fig. 1D) released into the cell culture media. These data indicate that NS4A plays an essential role at an early stage of virus assembly, prior to or at the formation of intracellular infectious particles.
Polyprotein processing of NS4A mutants.
Since NS3-4A plays an essential role in HCV polyprotein processing, we considered the possibility that the replication and assembly defects of the NS4A mutants might simply be due to defects in polyprotein processing. We therefore examined the patterns of viral protein expression by the panel of NS4A mutants via Western blot analysis. To overcome differences in protein expression due to the fact that some mutants did not replicate, we used the vaccinia virus T7 RNA polymerase expression system to drive HCV expression in Huh-7.5 cells transfected with the WT or mutant HCV cDNA clones. As shown in Fig. 2, there were no major differences in the accumulation of core, E2, NS2, NS3, or NS5A by any of the NS4A mutants. It was notable that WT and all NS4A mutants expressed two forms of NS5A: an abundant, slow-migrating form and a less-abundant, fast-migrating form. Additional experiments with WT Jc1 virus confirmed that these are different phosphoforms of NS5A (data not shown), which likely represent hyper- and hypophosphorylated forms of NS5A as described by others (reviewed in reference 30). We were unable to examine the expression of E1, NS4A, NS4B, and NS5B proteins because the antibodies available to us were unreactive against the Jc1 proteins (data not shown). Although we cannot rule out differences in NS4A-B processing, the available evidence suggests that these NS4A mutants do not have gross abnormalities in polyprotein processing, which is consistent with our previous analysis of NS4A in the context of the Con1 replicon (46).
FIG. 2.

Polyprotein processing of NS4A mutants. Parallel cultures of Huh-7.5 cells were infected with vaccinia virus vTF7-3 for 1 h, transfected with the indicated Jc1/GLuc2A cDNA clones, and lysed at 24 h postinfection. Proteins were separated by SDS-PAGE and detected by Western blotting as described in Materials and Methods.
Selection of NS4A mutants with restored RNA replication.
Given the error-prone nature of HCV replication, we tried to identify revertants and second-site suppressors for the two NS4A mutants with defects in replication. To do this, we transfected Huh-7.5 cells with the NS4A Y45A and F48A mutant RNAs, serially passed the transfected cells, and monitored HCV replication by measuring luciferase activity, as described above. However, we did not identify viruses capable of replication even after several cell passages. We therefore modified our selection strategy by subcloning the replication-defective NS4A mutations into the subgenomic replicon pYSGR-JFH/Neo (Fig. 3A), which expresses the dominant selectable marker Neo. After RNA transfection and selection with G418, three colonies were obtained for the NS4A Y45A mutant, and four colonies were obtained for the NS4A F48A mutant. Colonies were clonally isolated, expanded, and sequenced. All three NS4A Y45A colonies simply reverted back to the WT and were not analyzed further. Only one of the four NS4A F48A colonies was stable and continued to grow upon passage. Sequence analysis of this variant indicated that it retained the NS4A F48A mutation and had acquired a single additional mutation, NS4B I7F. To determine whether this second-site mutation could suppress the NS4A F48A replication defect, we reconstructed the double mutant in YSGR-JFH/Neo and in the context of YSGR-JFH/GLuc, a subgenomic replicon that expresses the secreted Gaussia luciferase (Fig. 3B). As shown in Fig. 3C, NS4B I7F was able to partially restore stable replication and Neo expression of the NS4A F48A mutant. Similarly, the NS4B I7F partially restored transient replication and GLuc expression of the NS4A F48A mutant (Fig. 3D). It was notable that YSGR-JFH/GLuc expressed high levels of GLuc activity at early times posttransfection and was independent of viral replication, which is consistent with translation from the input RNA. This early expression from input RNA was not seen with Jc1/GLuc2A (Fig. 1), which could reflect the interference of HCV translation by the viral structural genes (11, 56, 67, 70).
FIG. 3.

NS4B I7F partially suppresses the replication defect of subgenomic replicons containing the NS4A F48A mutation. (A) The pYSGR-JFH construct used in the present study and the workflow to identify revertants or suppressor mutants. HCV polyprotein processing sites are annotated as in Fig. 1. (B) pYSGR-JFH/GLuc reporter construct used in this study and workflow of our reporter virus assay. RNA was electroporated into Huh-7.5 cells, and culture medium was harvested at various times posttransfection. Secreted GLuc activity was assayed as detailed in Materials and Methods. (C) Stable replication phenotypes of NS4A F48A and its suppressor NS4B I7F. Colony-forming activity was measured for NS4A F48A and the suppressor NS4B I7F in the context of pYSGR-JFH. Values represent CFU calculated from three independent transfections. Error bars represent the standard deviation from the mean. (D) Transient replication phenotypes of NS4A F48A and its suppressor NS4B I7F. Luciferase activity was measured for NS4A F48A and the suppressor NS4B I7F in the context of pYSGR-JFH/GLuc. Huh 7.5 cells were transfected with the indicated mutants, and media were collected at 6, 12, 24, 48, 72, and 96 h posttransfection. Values represent the time course of secreted GLuc activity for each transfection, expressed as an average of at least three independent transfections. Error bars represent the standard deviation from the mean.
Since the NS4A F48A replication defect was initially identified in Jc1/GLuc2A, we tested whether the NS4B I7F suppressor could restore replication of a full-length HCV genome. Surprisingly, the NS4B mutation only modestly improved the replication of Jc1/GLuc2A NS4A F48A (Fig. 4A) and did not detectably restore virus assembly (Fig. 4B). Thus, the NS4B I7F mutation could only partially restore the replication defect of the NS4A F48A mutant.
FIG. 4.

NS4B I7F partially suppresses the replication defect of infectious genomes containing the NS4A F48A mutation. (A) Replication phenotypes of NS4A F48A and its suppressor NS4B I7F. Huh7.5 cells were transfected with each indicated mutant, and medium was collected at 24, 48, 72, and 96 h posttransfection. Values represent the time course of secreted GLuc activity for each transfection, expressed as an average of at least three independent transfections. Error bars represent the standard deviation of the mean. (B) Infectivity phenotypes of NS4A F48A and its suppressor NS4B I7F. The infectivity levels in the medium from panel A were quantified by infecting naive cells and monitoring secreted GLuc activity.
Selection of NS4A mutants with restored virus assembly.
As above, we tried to select for NS4A assembly-defective variants that could spread in culture by serially passaging transfected cells and assaying the conditioned media for viral infectivity. However, no virus-producing variants arose even after several cell passages. We therefore modified our selection strategy, again by subcloning the NS4A K41A, L44A, D49A, E50A, M51A, E52A, and E53A mutations into the context of YSGR-JFH/Neo. Our strategy was to transfect these constructs into Huh-7.5[core-NS2] cells that stably express the Jc1 core-NS2 genes, serially passage the transfected cells, and identify variant subgenomes that could overcome the block in virus assembly as infectious, trans-packaged Neo-transducing particles (Fig. 5A). The benefits of this approach were that it imposed stringent selective pressure limited to a single round of infection and that it simplified the process of biologically cloning positive hits for further analysis. One caveat to this approach is that it limited the range of potential second-site mutations that could overcome defects in virus assembly to the viral NS3-5B genes and noncoding regions.
FIG. 5.

NS3 Q221L suppresses the assembly defect of subgenomic replicons containing the NS4A K41A mutation. (A) Workflow used to identify revertants or suppressor mutants via trans-packaging. RNAs were electroporated into Huh-7.5[core-NS2] cells, and the cell culture media were collected at each cell passage. The media were used to infect naive cells and productively infected cells were selected with G418. (B) trans-Packaging infectivity phenotypes of NS4A K41A and variants containing NS3 Q221L or NS4B S230P. Colony-forming activity was measured for NS4A K41A and the suppressors NS3 Q221L and NS4B S230P in the context of pYSGR-JFH. Values represent the CFU calculated from three independent transfections. Error bars represent the standard deviation from the mean. (C) Replication phenotypes of NS4A K41A and variants containing NS3 Q221L or NS4B S230P. The luciferase activity was measured for NS4A K41A and the suppressor NS3 Q221L and NS4B S230P in the context of pYSGR-JFH/GLuc. Huh-7.5[core-NS2] cells were electroporated with each indicated mutant genome and media were collected at 6, 12, 24, 48, 72, and 96 h posttransfection. Values represent the time course of secreted GLuc activity for each transfection, expressed as an average of at least three independent transfections. Error bars represent the standard deviation of the mean. (D) trans-Packaging infectivity phenotypes of NS4A K41A and variants containing NS3 Q221L or NS4B S230P. The infectivity levels in the media from panel C were quantified by infecting naive cells and measuring secreted GLuc activity.
Huh-7.5[core-NS2] cells efficiently trans-packaged WT HCV subgenomic replicons (Fig. 5B and D). After serial passage of Huh-7.5[core-NS2] cells transfected with the panel of SGR/JFH-Neo NS4A mutants and selection for infected cells, only a single G418-resistant colony was obtained for the NS4A K41A mutant. In contrast, WT SGR-JFH/Neo continued to efficiently produce trans-packaged Neo-transducing activity at all cell passages (data not shown). The SGR-JFH/Neo NS4A K41A variant was expanded, sequenced, and found to contain two coding changes: NS3 Q221L and NS4B S230P. To determine whether these changes could suppress the assembly defect of the NS4A K41A mutant, we reconstructed these mutations individually and in combination in the SGR-JFH/Neo replicon. As shown in Fig. 5B, the NS3 Q221L mutation fully restored infectious, stable Neo-transducing activity to the NS4A K41A mutant replicon. In contrast, the NS4B mutation partially restored infectious Neo transducing activity, and when both second-site mutations were combined, an intermediate phenotype was observed. These mutations were similarly tested for transient replication and trans-packaging in the SGR-JFH/GLuc replicon. As expected, all of the NS4A K41A mutants replicated efficiently after RNA transfection into Huh-7.5[core-NS2] cells, although the NS4A K41A, NS4A K41A + NS4B S230P, and NS4A K41A + NS3 Q221L + NS4B S230P mutants produced somewhat lower levels of GLuc2A than the WT or NS4A K41A + NS3 Q221L mutant (Fig. 5C). Similar to the results obtained with the Neo-expressing replicon, the NS3 Q221L mutation fully restored trans-packaging of the NS4A K41A mutant, whereas NS4B S230P only slightly restored infectious particle production, and the combination of both second-site suppressors gave an intermediate phenotype (Fig. 5B). These data show that the NS3 Q221L and NS4B S230P mutations could suppress the defect of NS4A K41A in assembling infectious, trans-packaged replicon particles.
To examine whether NS3 Q221L and NS4B S230P could suppress the assembly defect of the NS4A K41A mutant in a cis-packaging full-length genome, we introduced these mutations alone or in combination into the Jc1/GLuc2A reporter virus. As seen above, WT Jc1/GLuc2A and the NS4A K41A + NS3 Q221L double mutant replicated to similar levels, whereas Jc1/GLuc2A Δcore and the NS4A K41A single mutant did not reach the same peak of GLuc2A expression at late time points, which is consistent with their inability to spread. It was notable that both the NS4A K41A + NS4B S230P double mutant and the NS4A K41A + NS3 Q221L + NS4B S230P triple mutant exhibited delayed replication kinetics (Fig. 6A).
FIG. 6.

NS3 Q221L suppresses the assembly defect of full-length genomes containing the NS4A K41A mutation. (A) Replication phenotypes of NS4A K41A and variants containing NS3 Q221L or NS4B S230P. Huh-7.5 cells were transfected with each indicated mutant, and media were collected at 24, 48, 72, and 96 h posttransfection. Values represent the time course of secreted GLuc activity for each transfection, expressed as an average of at least three independent transfections. Error bars represent the standard deviation of the mean. (B) Relative infectivity phenotypes of NS4A K41A and variants containing NS3 Q221L or NS4B S230P. The levels of infectivity levels in the media from panel A were quantified by infecting naive cells and monitoring secreted GLuc activity. Values represent the time course of secreted GLuc activity for each transfection, expressed as an average of at least three independent transfections. Error bars represent the standard deviation of the mean.
When tested for infectious virus production, the NS3 Q221L mutation fully suppressed the virus assembly defect of the NS4A K41A mutant, yielding relative infectivity measurements similar to WT (Fig. 6B). In contrast, NS4B S230P did not significantly suppress the assembly defect in Jc1/GLuc2A NS4A K41A, and the combination of NS4A K41A + NS3 Q221L + NS4B S230P showed only a very modest increase in virus production. Taken together, these data show that the NS3 Q221L mutation suppresses the defect in virus particle assembly caused by the NS4A K41A mutation.
We did not obtain second-site suppressors of other NS4A mutants using the strategy illustrated in Fig. 5A. We therefore examined whether NS3 Q221L could suppress the virus assembly defects of the NS4A L44A, Y45A, F48A, E50A, M51A, E52A, and E53A mutants in the Jc1/GLuc2A genetic background. The NS3 Q221L mutation did not enhance virus production in these mutants (data not shown), indicating that it does not act as a general suppressor for NS4A defects in virus assembly.
Biochemical activities of NS3-4A mutants.
We previously showed that the C-terminal acidic region of NS4A contributes to the RNA-stimulated ATPase activity of NS3-4A (6). Furthermore, we recently found that the NS3 Q221L mutation, which also suppressed specific NS2 defects in virus assembly, reduced the functional affinity of RNA binding (i.e., the amount of RNA needed to stimulate ATPase activity) by NS3-4A (58). We therefore examined the enzymatic properties of purified, recombinant NS3-4A with or without the NS4A K41A mutation and the NS3 Q221L suppressor.
Protease activities were assayed by using a fluorescent resonance energy transfer substrate (RET-S1) peptide (64). As shown in Fig. 7A and Table 2, WT and mutant forms of NS3-4A had similar serine protease activities and cleaved nearly all of the substrate within 3 min. We next examined RNA helicase activities under single-cycle conditions by using a model 34-bp RNA substrate (6, 17). Although the extent of RNA unwinding was slightly reduced for the mutants compared to WT NS3-4A, the rates of unwinding were similar (Fig. 7B and Table 2). Thus, the NS4A K41A, NS3 Q221L, and NS4A K41A + NS3 Q221L mutations had minimal effects on NS3-4A RNA protease and RNA helicase activities.
FIG. 7.
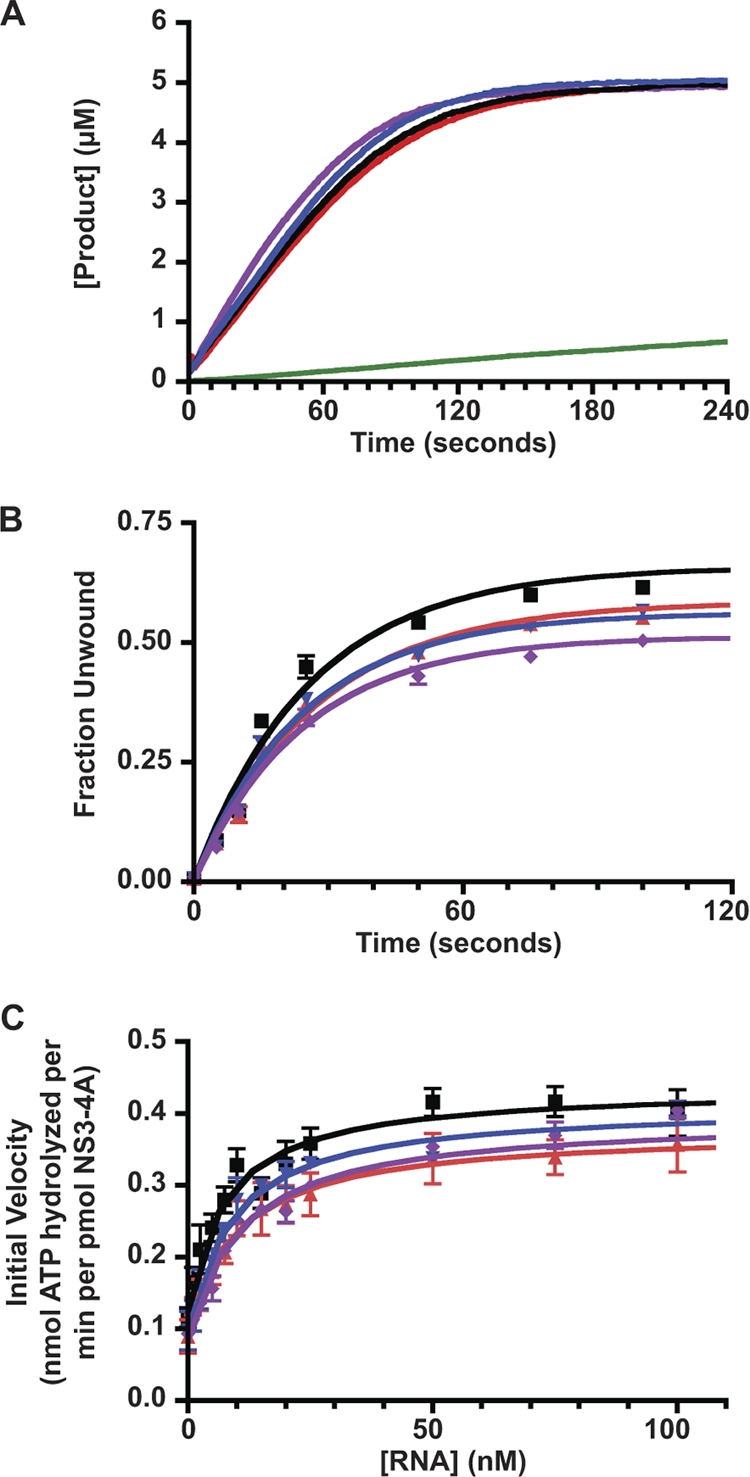
Enzymatic activities of WT and mutant forms of NS3-4A. (A) Serine protease activities of WT (black), NS4A K41A (red), NS3 Q221L (blue), and NS4A K41A + NS3 Q221L (purple). The data were collected at 1-s intervals as described in Materials and Methods. Values represent averages from three independent experiments, and a buffer control (no NS3-4A) is shown in green. (B) RNA unwinding activities of WT (black), NS4A K41A (red), NS3 Q221L (blue), and NS4A K41A + NS3 Q221L (purple) forms of NS3-4A. Values represent the average of three independent experiments. Error bars represent the standard deviation from the mean. (C) RNA-stimulated ATPase activities of WT (black), NS4A K41A (red), NS3 Q221L (blue), and NS4A K41A + NS3 Q221L (purple) forms of NS3-4A. Values represent the average of three independent experiments. Error bars represent standard deviation from the mean.
TABLE 2.
Enzymatic properties of WT and mutant forms of NS3-4A
| NS3-4A protein | Avg ± SDa |
||||
|---|---|---|---|---|---|
| Protease activity (nM/s) | RNA unwinding |
RNA-stimulated ATPase |
|||
| Fraction unwound | kobs (s−1) | Km (nM) | kcatb | ||
| WT | 48.4 ± 2.1 | 0.67 ± 0.02 | 0.029 ± 0.003 | 8.0 ± 1.7 | 0.31 ± 0.02 |
| NS4A K41A | 48.0 ± 3.4 | 0.58 ± 0.01 | 0.038 ± 0.002 | 10.1 ± 2.4 | 0.28 ± 0.02 |
| NS3 Q221L | 51.0 ± 2.3 | 0.57 ± 0.01 | 0.036 ± 0.004 | 8.8 ± 1.4 | 0.32 ± 0.01 |
| NS4A K41A + NS3 Q221L | 58.6 ± 2.1 | 0.51 ± 0.01 | 0.035 ± 0.003 | 10.8 ± 1.5 | 0.31 ± 0.01 |
The data shown are averages from three experiments.
The kcat values are expressed as nanomoles of ATP hydrolyzed per minute per picomole of NS3-4A.
We examined the RNA-stimulated ATPase activity of WT and mutant forms of NS3-4A by using a real-time, coupled ATPase assay (15, 38, 48). This assay utilizes the highly efficient enzymes pyruvate kinase and lactate dehydrogenase to couple ATP hydrolysis to NADH oxidation, which can be monitored fluorometrically. Note that the coupling enzymes have much faster kinetics than NS3-4A ATPase and were not limiting under our assay conditions. The advantages of this assay included the ability to accurately measure ATPase activity in real-time over a large number of samples without the use of radioactivity. Notably, WT NS3-4A yielded readings similar to those obtained with our previous assay (8, 58), validating the use of the coupled assay. As shown in Fig. 7C and Table 2, WT and mutant forms of NS3-4A containing the NS3 Q221L and the NS4A K41A + NS3 Q221L mutations had similar ATPase activities and responses to RNA stimulation.
DISCUSSION
Our major finding here is that HCV NS4A has dual roles in RNA replication and virus assembly. This conclusion was supported by data demonstrating that genotype 2a full-length genomes and subgenomic replicons containing NS4A Y45A or F48A mutations had severe defects in genome replication, while genomes and replicons containing the NS4A K41A, L44A, F48A, D49A, E50A, M51A, E52A, or E53A mutations replicated but did not assemble virus particles. These data complement and extend our prior genetic analysis of the NS4A C-terminal region with a genotype 1b subgenomic replicon (46). Specifically, mutation of either conserved aromatic residue Y45 or F48 was lethal for genome replication in both genetic backgrounds. In contrast, other NS4A mutations gave severe replication defects in genotype 1b replicons but had little effect on genotype 2a genomes or replicons. These included the NS4A D40A (previously, D1697A and D1697R), K41A (R1697A, R1697AD), E46A (R1703A, R1703D), D49A (D1706A, D1706R), E50A (E1707A, E1707R), and M51A (M1708A) mutations. Taken together, our parallel genetic studies indicate that NS4A Y45 and F48 are essential for replication in at least two viral isolates, while other NS4A acidic domain residues may be differentially required for genome replication or virus assembly in a virus genotype- or strain-dependent manner.
One clue as to the differential effect of NS4A mutations on genome replication may come from NS4A's effects on NS5A phosphorylation. In our prior study with genotype 1b NS4A mutants, defects in genome replication strongly correlated with defects in NS5A hyperphosphorylation (46). Furthermore, RNA replication and NS5A hyperphosphorylation could be coordinately restored by second-site suppressor mutations in NS3 (46). In the present study, genotype 2a NS4A mutants had fewer defects in RNA replication (Fig. 1) and did not exhibit detectable differences in NS5A hyperphosphorylation (Fig. 2). NS5A phosphorylation plays an important but unknown role in HCV genome replication (reviewed in reference 30). Thus, genotype 2a replication may be less sensitive to changes in NS4A because hyperphosphorylation of NS5A may be less dependent on NS4A in this genetic background.
By using a forward genetic approach, we identified a second-site mutation, NS4B I7F, as a partial suppressor of the NS4A F48A replication defect. This was interesting, since this suppressor mutation introduced a phenylalanine 13 residues downstream of the original NS4A F48A mutation in the viral polyprotein. This could reflect a requirement to maintain a phenylalanine residue in this region of the polyprotein, perhaps stabilizing the protein fold or a protein-protein interaction. It could serve a role prior to NS4A-B cleavage, or it could function following cleavage, residing in the N-terminal, putative amphipathic helix 1 of NS4B (19). NS4B plays an essential role in HCV genome replication by inducing membrane rearrangements and may form a scaffold for replicase assembly (18, 27). Although the topology of NS4B is not fully understood, the N-terminal region of NS4B can apparently translocate from the cytosol to the ER lumen in a posttranslational manner (51). This topological shift is inhibited by coexpression of NS5A (50), suggesting that the N terminus of NS4B interacts with other viral NS proteins. This premise is further supported by genetic studies performed with genotype 1a and 1b replicons, which showed that the N-terminal region of NS4B contains important determinants of RNA replication efficiency and that replication defects caused by changes in this region could be suppressed by second-site mutations in NS3 and NS4A (9, 57). Intriguingly, we previously identified two of these NS3 mutations—NS3 Q86R (Q1112R in the Con1 polyprotein) and NS3 S343R (S1369R)—as suppressors of replication defects caused by mutations in the NS4A C-terminal acidic domain of genotype 1b replicons (46). Thus, multiple studies in different genetic backgrounds indicate that genetic interactions between NS3, NS4A, and NS4B contribute to RNA replication.
Surprisingly, the majority of NS4A mutations had defects in virus assembly (Fig. 1), indicating that the NS4A acidic region contributes to this process. Consistent with this idea, we previously identified an NS4A mutation (E42G) that suppressed a defect in virus assembly caused by a mutation in NS2 (58). To better clarify the role of NS4A in virus assembly, we tried to identify second-site mutations that suppressed these defects. We took advantage of the fact that HCV subgenomes can be packaged in trans (1, 56, 63) to select for trans-packaged replicons containing mutations of interest. This strategy yielded a mutation in NS3 (Q221L) that fully restored assembly of the NS4A K41 mutant in cis and in trans, but did not suppress the assembly defects of other NS4A mutants. Interestingly, we previously identified NS3 Q221L as a suppressor of specific NS2 defects in virus assembly (58), and Ma et al. identified this as a cell-culture-adaptive mutation that enhanced the production of genotype 1a/2a chimeric virus particles (52). Thus, NS3 Q221L can suppress multiple virus assembly defects but does not act as a general suppressor of all virus assembly defects, suggesting that this mutation may compensate for a common set of defects within a specific pathway.
We also identified a second-site mutation, NS4B S230P, that partially restored the trans-packaging of this NS4A mutant but did not allow assembly of full-length genomes in cis. Interestingly, this mutation decreased the level of virus assembly when combined with the NS3 Q221L suppressor. This mutation likely perturbs the C-terminal alpha helix of NS4B, a region previously implicated in NS4B membrane association, RNA replication, and virus assembly (9, 26, 31, 57). Thus, perhaps this mutation shifts the balance between RNA replication versus virus assembly. Consistent with this, Pietschmann et al. recently demonstrated that cell-culture-adaptive mutations that enhance HCV genome replication can antagonize virus assembly (60).
To understand the molecular basis of how NS3-4A participates in virus assembly, we characterized the enzymatic activities of NS3-4A containing the NS4A K41A mutation with or without the NS3 Q221L suppressor. Only subtle differences were found between the WT and mutant forms of NS3-4A. This was surprising, since we previously found that the NS3 Q221L mutation increased the amount of RNA needed to stimulate NS3-4A ATPase activity (58). This discrepancy may be attributable to methodological differences between our studies. First, the coupled ATPase assay used here yielded results with lower variance, and the increased throughput of this assay allowed us to measure a wider range of RNA concentrations. Second, NS3-4A was preincubated with RNA for a shorter period of time prior to adding ATP, which may enhance the stability of this protein complex in solution. To summarize, NS4A K41A and NS3 Q221L have only modest effects on the enzymatic properties of NS3-4A.
So how does NS3-4A contribute to virus assembly and why do mutations in these genes cause virus assembly phenotypes? It is notable that NS3 mutations affecting virus assembly map to residues on the surface the RNA helicase domain, distal from residues important for RNA helicase activity (28, 52, 58, 69). Thus, it seems likely that these mutations affect the interaction of NS3-4A with itself or with other molecules rather than directly modulating helicase activity. In this regard, NS3 is an RNA-binding protein that has coordinated serine protease and RNA helicase activities. Because NS4A stabilizes the serine protease fold and contributes to RNA helicase activity, it likely helps to coordinate intramolecular interactions within NS3 and may contribute to NS3-4A binding to RNA or other proteins. Clearly, an important next step will be to define NS3-4A interaction partners that are relevant for virus assembly.
Acknowledgments
We thank J. Bloom, N. Counihan, and K. Stapleford for helpful discussions and comments on the manuscript. We are grateful to C. M. Rice and J. McKeating for providing antibodies; R. Beran for providing pRB96; C. Peters for technical support; and P. Flynn, Y. Jiang, and T. Drozdz for administrative support.
This study was funded through NIH grants AI087925 (to B.D.L.) and AI089826 (to A.M.P. and B.D.L.).
Footnotes
Published ahead of print on 3 November 2010.
REFERENCES
- 1.Adair, R., et al. 2009. Expression of hepatitis C virus (HCV) structural proteins in trans facilitates encapsidation and transmission of HCV subgenomic RNA. J. Gen. Virol. 90:833-842. [DOI] [PubMed] [Google Scholar]
- 2.Asabe, S. I., et al. 1997. The N-terminal region of hepatitis C virus-encoded NS5A is important for NS4A-dependent phosphorylation. J. Virol. 71:790-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartenschlager, R., L. Ahlborn-Laake, J. Mous, and H. Jacobsen. 1994. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J. Virol. 68:5045-5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartenschlager, R., L. Ahlborn-Laake, K. Yasargil, J. Mous, and H. Jacobsen. 1995. Substrate determinants for cleavage in cis and in trans by the hepatitis C virus NS3 proteinase. J. Virol. 69:198-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beran, R. K., M. M. Bruno, H. A. Bowers, E. Jankowsky, and A. M. Pyle. 2006. Robust translocation along a molecular monorail: the NS3 helicase from hepatitis C virus traverses unusually large disruptions in its track. J. Mol. Biol. 358:974-982. [DOI] [PubMed] [Google Scholar]
- 6.Beran, R. K., B. D. Lindenbach, and A. M. Pyle. 2009. The NS4A protein of hepatitis C virus promotes RNA-coupled ATP hydrolysis by the NS3 helicase. J. Virol. 83:3268-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beran, R. K., and A. M. Pyle. 2008. Hepatitis C viral NS3-4A protease activity is enhanced by the NS3 helicase. J. Biol. Chem. 283:29929-29937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beran, R. K., V. Serebrov, and A. M. Pyle. 2007. The serine protease domain of hepatitis C viral NS3 activates RNA helicase activity by promoting the binding of RNA substrate. J. Biol. Chem. 282:34913-34920. [DOI] [PubMed] [Google Scholar]
- 9.Blight, K. J. 2007. Allelic variation in the hepatitis C virus NS4B protein dramatically influences RNA replication. J. Virol. 81:5724-5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blight, K. J., J. A. McKeating, and C. M. Rice. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001-13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boni, S., J. P. Lavergne, S. Boulant, and A. Cahour. 2005. Hepatitis C virus core protein acts as a trans-modulating factor on internal translation initiation of the viral RNA. J. Biol. Chem. 280:17737-17748. [DOI] [PubMed] [Google Scholar]
- 12.Brass, V., et al. 2008. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc. Natl. Acad. Sci. U. S. A. 105:14545-14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campo, D. S., Z. Dimitrova, R. J. Mitchell, J. Lara, and Y. Khudyakov. 2008. Coordinated evolution of the hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 105:9685-9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De La Cruz, E. M., H. L. Sweeney, and E. M. Ostap. 2000. ADP inhibition of myosin V ATPase activity. Biophys. J. 79:1524-1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De La Cruz, E. M., A. L. Wells, S. S. Rosenfeld, E. M. Ostap, and H. L. Sweeney. 1999. The kinetic mechanism of myosin V. Proc. Natl. Acad. Sci. U. S. A. 96:13726-13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dentzer, T. G., I. C. Lorenz, M. J. Evans, and C. M. Rice. 2009. Determinants of the hepatitis C virus nonstructural protein 2 protease domain required for production of infectious virus. J. Virol. 83:12702-12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dumont, S., et al. 2006. RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature 439:105-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egger, et al. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974-5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elazar, M., P. Liu, C. M. Rice, and J. S. Glenn. 2004. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J. Virol. 78:11393-11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Failla, C., L. Tomei, and R. De Francesco. 1994. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 68:3753-3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fauquet, C., M. A. Mayo, J. Maniloff, U. Desselberger, and L. A. Ball (ed.). 2005. Virus taxonomy. Eighth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, London, England.
- 22.Flint, M., et al. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235-6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Florese, R. H., M. Nagano-Fujii, Y. Iwanaga, R. Hidajat, and H. Hotta. 2002. Inhibition of protein synthesis by the nonstructural proteins NS4A and NS4B of hepatitis C virus. Virus Res. 90:119-131. [DOI] [PubMed] [Google Scholar]
- 24.Frick, D. N., R. S. Rypma, A. M. Lam, and B. Gu. 2004. The nonstructural protein 3 protease/helicase requires an intact protease domain to unwind duplex RNA efficiently. J. Biol. Chem. 279:1269-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuerst, T. R., E. G. Niles, F. W. Studier, and B. Moss. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 83:8122-8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gouttenoire, J., R. Montserret, A. Kennel, F. Penin, and D. Moradpour. 2009. An amphipathic alpha-helix at the C terminus of hepatitis C virus nonstructural protein 4B mediates membrane association. J. Virol. 83:11378-11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gouttenoire, J., F. Penin, and D. Moradpour. 2010. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev. Med. Virol. 20:117-129. [DOI] [PubMed] [Google Scholar]
- 28.Han, Q., et al. 2009. Compensatory mutations in NS3 and NS5A proteins enhance the virus production capability of hepatitis C reporter virus. Virus Res. 145:63-73. [DOI] [PubMed] [Google Scholar]
- 29.Hara, H., et al. 2009. Involvement of creatine kinase B in hepatitis C virus genome replication through interaction with the viral NS4A protein. J. Virol. 83:5137-5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang, Y., K. Staschke, R. De Francesco, and S. L. Tan. 2007. Phosphorylation of hepatitis C virus NS5A nonstructural protein: a new paradigm for phosphorylation-dependent viral RNA replication? Virology 364:1-9. [DOI] [PubMed] [Google Scholar]
- 31.Jones, D. M., A. H. Patel, P. Targett-Adams, and J. McLauchlan. 2009. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J. Virol. 83:2163-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaneko, T., et al. 1994. Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem. Biophys. Res. Commun. 205:320-326. [DOI] [PubMed] [Google Scholar]
- 33.Kato, J., et al. 2002. Hepatitis C virus NS4A and NS4B proteins suppress translation in vivo. J. Med. Virol. 66:187-199. [DOI] [PubMed] [Google Scholar]
- 34.Kim, J. L., et al. 1996. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 87:343-355. [DOI] [PubMed] [Google Scholar]
- 35.Kolykhalov, A. A., E. V. Agapov, and C. M. Rice. 1994. Specificity of the hepatitis C virus NS3 serine protease: effects of substitutions at the 3/4A, 4A/4B, 4B/5A, and 5A/5B cleavage sites on polyprotein processing. J. Virol. 68:7525-7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolykhalov, A. A., K. Mihalik, S. M. Feinstone, and C. M. Rice. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74:2046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komoda, Y., et al. 1994. Substrate requirements of hepatitis C virus serine proteinase for intermolecular polypeptide cleavage in Escherichia coli. J. Virol. 68:7351-7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kornberg, A., and W. E. Pricer, Jr. 1951. Enzymatic phosphorylation of adenosine and 2,6-diaminopurine riboside. J. Biol. Chem. 193:481-495. [PubMed] [Google Scholar]
- 39.Kou, Y. H., et al. 2006. Hepatitis C virus NS4A inhibits cap-dependent and the viral IRES-mediated translation through interacting with eukaryotic elongation factor 1A. J. Biomed Sci. 13:861-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuang, W. F., et al. 2004. Hepatitis C virus NS3 RNA helicase activity is modulated by the two domains of NS3 and NS4A. Biochem. Biophys. Res. Commun. 317:211-217. [DOI] [PubMed] [Google Scholar]
- 41.Lam, A. M., and D. N. Frick. 2006. Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J. Virol. 80:404-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leinbach, S. S., et al. 1994. Substrate specificity of the NS3 serine proteinase of hepatitis C virus as determined by mutagenesis at the NS3/NS4A junction. Virology 204:163-169. [DOI] [PubMed] [Google Scholar]
- 43.Li, X. D., L. Sun, R. B. Seth, G. Pineda, and Z. J. Chen. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U. S. A. 102:17717-17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin, R., et al. 2006. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 80:6072-6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindenbach, B. D., et al. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 46.Lindenbach, B. D., et al. 2007. The C terminus of hepatitis C virus NS4A encodes an electrostatic switch that regulates NS5A hyperphosphorylation and viral replication. J. Virol. 81:8905-8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindenbach, B. D., H. J. Thiel, and C. M. Rice. 2007. Flaviviridae: the viruses and their replication, p. 1101-1152. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 1. Lippincott-Raven Publishers, Philadelphia, PA. [Google Scholar]
- 48.Lindsley, J. E. 2001. Use of a real-time, coupled assay to measure the ATPase activity of DNA topoisomerase II. Methods Mol. Biol. 95:57-64. [DOI] [PubMed] [Google Scholar]
- 49.Loo, Y. M., et al. 2006. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 103:6001-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lundin, M., H. Lindstrom, C. Gronwall, and M. A. Persson. 2006. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J. Gen. Virol. 87:3263-3272. [DOI] [PubMed] [Google Scholar]
- 51.Lundin, M., M. Monne, A. Widell, G. Von Heijne, and M. A. Persson. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J. Virol. 77:5428-5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma, Y., J. Yates, Y. Liang, S. M. Lemon, and M. Yi. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 82:7624-7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meylan, E., et al. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167-1172. [DOI] [PubMed] [Google Scholar]
- 54.Moradpour, D., F. Penin, and C. M. Rice. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453-463. [DOI] [PubMed] [Google Scholar]
- 55.Nomura-Takigawa, Y., et al. 2006. Non-structural protein 4A of hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 87:1935-1945. [DOI] [PubMed] [Google Scholar]
- 56.Pacini, L., R. Graziani, L. Bartholomew, R. De Francesco, and G. Paonessa. 2009. Naturally occurring hepatitis C virus subgenomic deletion mutants replicate efficiently in Huh-7 cells and are trans-packaged in vitro to generate infectious defective particles. J. Virol. 83:9079-9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paredes, A. M., and K. J. Blight. 2008. A genetic interaction between hepatitis C virus NS4B and NS3 is important for RNA replication. J. Virol. 82:10671-10683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phan, T., R. K. Beran, C. Peters, I. C. Lorenz, and B. D. Lindenbach. 2009. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 83:8379-8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pietschmann, T., et al. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408-7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pietschmann, T., M. et al. 2009. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 5:e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed., vol. 1. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- 62.Selimovic, D., and M. Hassan. 2008. Inhibition of hepatitis C virus (HCV) core protein-induced cell growth by nonstructural protein 4A (NS4A) is mediated by mitochondrial dysregulation. Bosn. J. Basic Med. Sci. 8:4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steinmann, E., C. Brohm, S. Kallis, R. Bartenschlager, and T. Pietschmann. 2008. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J. Virol. 82:7034-7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taliani, M., et al. 1996. A continuous assay of hepatitis C virus protease based on resonance energy transfer depsipeptide substrates. Anal. Biochem. 240:60-67. [DOI] [PubMed] [Google Scholar]
- 65.Tanji, Y., M. Hijikata, Y. Hirowatari, and K. Shimotohno. 1994. Hepatitis C virus polyprotein processing: kinetics and mutagenic analysis of serine proteinase-dependent cleavage. J. Virol. 68:8418-8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanji, Y., M. Hijikata, S. Satoh, T. Kaneko, and K. Shimotohno. 1995. Hepatitis C virus-encoded nonstructural protein NS4A has versatile functions in viral protein processing. J. Virol. 69:1575-1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang, T. H., R. C. Rijnbrand, and S. M. Lemon. 2000. Core protein-coding sequence, but not core protein, modulates the efficiency of cap-independent translation directed by the internal ribosome entry site of hepatitis C virus. J. Virol. 74:11347-11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan, Y., et al. 1998. Complex of NS3 protease and NS4A peptide of BK strain hepatitis C virus: a 2.2 Å resolution structure in a hexagonal crystal form. Protein Sci. 7:837-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yi, M., Y. Ma, J. Yates, and S. M. Lemon. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81:629-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang, J., O. Yamada, H. Yoshida, T. Iwai, and H. Araki. 2002. Autogenous translational inhibition of core protein: implication for switch from translation to RNA replication in hepatitis C virus. Virology 293:141-150. [DOI] [PubMed] [Google Scholar]