

Abstract
Natural products produced by microorganisms are important starting compounds for drug discovery. Secondary metabolites, including antibiotics, have been isolated from different Streptomyces species. The production of these metabolites depends on the culture conditions. Therefore, the development of a new culture method can facilitate the discovery of new natural products. Here, we show that mycolic acid-containing bacteria can influence the biosynthesis of cryptic natural products in Streptomyces species. The production of red pigment by Streptomyces lividans TK23 was induced by coculture with Tsukamurella pulmonis TP-B0596, which is a mycolic acid-containing bacterium. Only living cells induced this pigment production, which was not mediated by any substances. T. pulmonis could induce natural-product synthesis in other Streptomyces strains too: it altered natural-product biosynthesis in 88.4% of the Streptomyces strains isolated from soil. The other mycolic acid-containing bacteria, Rhodococcus erythropolis and Corynebacterium glutamicum, altered biosynthesis in 87.5 and 90.2% of the Streptomyces strains, respectively. The coculture broth of T. pulmonis and Streptomyces endus S-522 contained a novel antibiotic, which we named alchivemycin A. We concluded that the mycolic acid localized in the outer cell layer of the inducer bacterium influences secondary metabolism in Streptomyces, and this activity is a result of the direct interaction between the mycolic acid-containing bacteria and Streptomyces. We used these results to develop a new coculture method, called the combined-culture method, which facilitates the screening of natural products.
In modern microbiology, single-strain culture is the standard method for cultivating microorganisms. However, owing to the absence of interacting microorganisms that are present in the natural environment, the growth conditions in a flask culture are significantly different from those in the natural environment.
The members of the order Actinomycetales, especially the genus Streptomyces, produce a number of antibiotics and other bioactive natural products. The genomic analysis of some Streptomyces strains revealed the presence of biosynthetic gene clusters for about 30 secondary metabolites, and these data imply that a single Streptomyces strain can produce more than 30 secondary metabolites (1, 9, 12). However, some of these secondary-metabolite genes are not expressed in fermentation culture. To date, various methods (7, 14, 15, 17) have been used to activate genes synthesizing cryptic secondary metabolites. Secondary-metabolite production is affected by environmental factors (5, 17) such as temperature, the presence of hormone-like chemicals (6), and medium composition (3). Therefore, to identify new antibiotics, the isolation of new actinomycetes should be accompanied by the study of the effects of different growth conditions on each isolated strain.
Coculture is an effective method for inducing the production of cryptic metabolites. Some coculture methods have been reported (2, 18); however, these methods often are specific to two bacterial strains, because they are based on the specific mutual interaction between these strains. Therefore, it is difficult to extend these methods to other strains.
To overcome this limitation, we developed a novel fermentation method, the combined-culture method, involving the coculture of two bacterial strains. We found that Streptomyces and mycolic acid-containing bacteria interact with each other for inducing secondary metabolism, and we applied this method to the screening of natural products.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
The bacterial strains used in this study are shown in Table 1. Strains were obtained from the Japan Collection of Microorganisms (JCM; RIKEN BioResource Center, Wako, Saitama, Japan) and NITE Biological Resource Center (NBRC), Department of Biotechnology, National Institute of Technology and Evaluation (Kisarazu, Chiba, Japan). Actinomycetes were isolated from plant tissues according to a previously described method (19). A-3M medium was used for the production of secondary metabolites, and V-22 medium was used for seed culture (14). Bennett's glucose agar, nutrient agar, and mannitol soya flour agar were used for actinomycetes isolation (14). A dialysis-culture flask was purchased from Ouchi Rikakogyo (Tokyo, Japan). A mixed cellulose ester dialysis membrane, ADVANTEC 0.2 μm, was purchased from Toyo Roshi Kaisya (Tokyo, Japan), and regenerated cellulose membrane, Spectra/Por 7 (pore size is about 50 kDa), was purchased from Funakoshi (Tokyo, Japan). For comparing the secondary-metabolite profiles of different combined cultures, we isolated 112 actinomycetes from soil samples collected in Hokuriku district, Japan. The analysis of the 16S rRNA genes of these isolated actinomycetes revealed that all of them were strains of Streptomyces. For antibiotic screening, another 97 actinomycetes were isolated from soil or plant tissue samples collected from Toyama Prefecture, Japan.
TABLE 1.
Bacterial species used in this study and their activities in combined cultures
| Bacterial species | Mycolic acida | Inducing activityb | Red pigment production withc: |
|
|---|---|---|---|---|
| Act | Red | |||
| Streptomyces lividans TK23 | − | 0.2 | ND | |
| Tsukamurella pulmonis TP-B0596 | + | + | 1.9 × 102 | 11.0 |
| Tsukamurella pseudospumae JCM13375T | + | + | 8.0 | 10.9 |
| Tsukamurella spumae JCM12608T | + | + | 38.9 | 14.2 |
| Tsukamurella strandjordii JCM11487T | + | + | 36.3 | 33.3 |
| Corynebacterium glutamicum ATCC13869 | + | + | 11.6 | 38.2 |
| Corynebacterium efficiens NBRC100395T | + | + | 5.7 | 3.0 |
| Rhodococcus erythropolis JCM3201T | + | + | 15.8 | 34.7 |
| Rhodococcus coprophilus JCM3200T | + | + | 31.0 | 14.1 |
| Rhodococcus wratislaviensis JCM9689T | + | + | 3.3 × 102 | 5.0 |
| Rhodococcus zopfii JCM9919T | + | + | 13.6 | 38.2 |
| Gordonia rubripertincta JCM3204T | + | + | 80.4 | 11.9 |
| Gordonia bronchialis NBRC16047T | + | + | 29.5 | 14.8 |
| Nocardia farcinica JCM3088T | + | + | 1.1 × 102 | 7.8 |
| Nocardia asteroides NBRC15531T | + | + | 11.1 | 1.5 |
| Mycobacterium smegmatis NBRC3082 | + | + | 1.1 × 103 | 41.9 |
| Mycobacterium chlorophenolicum NBRC15527T | + | + | 1.3 × 102 | 3.7 |
| Williamsia muralis JCM10676T | + | + | 34.4 | 5.8 |
| Dietzia cinnamea JCM13663T | + | + | 0.4 | 0.8 |
| Dietzia maris JCM6166T | + | − | ND | ND |
| Turicella otitidis JCM12146T | − | − | 0.6 | ND |
| Pseudonocardia autotrophica JCM4348T | − | − | 0.6 | ND |
| Bacillus subtilis ATCC 6633 | − | − | ND | ND |
| Escherichia coli NIHJ JC-2 | − | − | ND | ND |
| Staphylococcus aureus 209P JC-1 | − | − | ND | ND |
| Micrococcus luteus TP-B100 | − | − | ND | ND |
| Saccharomyces cerevisiae TP-F0176 | − | − | ND | ND |
| Candida albicans TP-F0176 | − | − | ND | ND |
Presence of mycolic acid in the outer layer.
Inducing activity of red pigment production in S. lividans.
The concentrations of actinorhodin (Act) and undecylprodigiosin (Red) are indicated as μM culture broth. ND, not detected.
Screening of bacteria that induce red pigment production in Streptomyces lividans.
To assay the induction activity, S. lividans spores were overlaid on Bennett's glucose agar plates. Each inducer bacterium then was inoculated on these plates, and the plates were incubated for 2 to 3 days at 30°C. The production of red pigments around the bacterial colonies was investigated. The spore overlay procedure has been described in our previous paper (14). Among the 400 bacteria used for the red pigment induction assay, 350 were obtained from the actinomycete (TP-A) or bacterial (TP-B) culture library of Toyama Prefectural University, and 50 were newly isolated from soils collected in Toyama Prefecture, Japan.
Assay of red pigment-inducing activities of mycolic acid-containing bacteria.
S. lividans TK23 and mycolic acid-containing bacteria were separately inoculated into test tubes containing 10 ml of V-22 medium. S. lividans was cultured at 30°C for 3 days on a rotary shaker at 200 rpm, and mycolic acid-containing bacteria were cultured at 30°C for 2 days on a rotary shaker at 200 rpm. Three ml of S. lividans culture and 1 ml of the culture of mycolic acid-containing bacteria were transferred into the same 500-ml K-1 flasks containing 100 ml of A-3M medium, and the resulting combined culture was incubated at 30°C for 7 days on a rotary shaker at 200 rpm. The amounts of actinorhodin and undecylprodigiosin were measured separately by the previously described procedures (10). For the measurement of actinorhodin, 1-ml aliquots of culture broths were treated with 200 μl of 6 N KOH (final concentration, 1 N) and vortexed thoroughly. The cell debris was removed by centrifugation (3,000 × g for 5 min), and the A640 values of the supernatants were measured (e640 = 25,350 M−1 cm−1) by using a DU640 spectrophotometer (Beckman Coulter).
For the measurement of undecylprodigiosin, 1-ml aliquots of the culture medium were harvested and centrifuged (15,000 rpm for 2 min), and the cell pellets were resuspended in 1 ml of methanol (pH 1.0). The samples were vortexed thoroughly and centrifuged (15,000 rpm for 2 min). The supernatants were collected and their A530 value was measured (e530 = 100,150 M−1 cm−1).
Preparation of mycolic acid and cell wall fraction and construction of a mycolic acid-deficient mutant strain.
Mycolic acid was extracted according to previously described procedures (21). First, 100 ml of Luria-Bertani culture medium was harvested and centrifuged; the resulting wet cell pellet was resuspended in 20 ml of 10% KOH-MeOH and then hydrolyzed by exposure to 100°C for 2 h. The solution was cooled to room temperature, and the hydrolyzed residues were acidified with 6 N HCl and then extracted using 20 ml of n-hexane. The hexane phase was collected and evaporated in vacuo. An aliquot of the residue was resuspended in 20 ml of benzene-MeOH-H2SO4 (10:20:1) solution and then incubated for 2 h at 100°C. The solution was cooled to room temperature, and the methyl-esterified residue first was extracted using 20 ml of water and n-hexane. Mycolic acid was obtained by the concentration of the hexane phase. To confirm the extraction, an aliquot of the hexane phase was subjected to thin-layer chromatography (TLC) (silica gel 60 F254; Merck); the TLC plates were developed in n-hexane-diethyl ether (4:1) solution and then soaked in 50% H2SO4. The plates were heated at 150°C, and the spots of methylester derivatives of mycolic acid were detected. The cell wall fraction of Tsukamurella pulmonis was extracted according to previously described procedures of the sonication-SDS method (20). The cell wall fraction yields approximately 10% of wet cell weight. In the additional experiments, mycolic acid prepared from the 100-ml culture was put into 10 ml of S. lividans pure culture, and the cell wall fraction prepared from the 500-ml culture was put into 10 and 100 ml of S. lividans pure cultures.
The mycolic acid-deficient mutant of Corynebacterium glutamicum was gifted by H. Kawasaki. The mycolic acid-deficient mutant was constructed according to the procedure described by Portevin et al. (16). Two independent mutants were gifted, and their red pigment-inducing activities were analyzed.
Analysis of secondary-metabolite profiles and antibiotic screening of the coculture broth.
To assay the secondary metabolites, both combined and pure cultures were incubated at 30°C for 7 days on a rotary shaker at 200 rpm; subsequently, the entire culture broth was extracted with an equal volume of n-butanol, and the secondary metabolites in the extracts were assayed (a flowchart for the combined-culture procedure is shown in Fig. 3A). For secondary-metabolite profile analysis, the extracts were evaporated in vacuo, and the residue was dissolved in dimethyl sulfoxide and subjected to high-performance liquid chromatography (HPLC) analysis. HPLC analysis was performed using an HP 1090 system (Hewlett Packard) and a C18 Rainin Microsorb column (inner diameter, 4.6 mm; length, 100 mm; Rainin Instrument Co., MA). The temperature was set at 30°C and the flow rate at 1.2 ml/min. The solvent was composed of acetonitrile and an aqueous solution of 0.15% KH2PO4 (pH 3.5). The elution profile of secondary metabolites was monitored at 254 nm (see Fig. 4A). The differences in the antibiotic production of the cocultures and pure cultures were determined by studying the HPLC profile. The number of strains with altered secondary-metabolite production was counted, and these data are presented as Venn diagrams (see Fig. 3B to F). The antibacterial activity of each culture broth was measured by performing a paper disc diffusion assay on agar plates. The indicator bacteria were overlaid on nutrient agar plates, and paper discs immersed in the broth extracts then were placed on these plates. Antibacterial activity was estimated by measuring the diameter of the inhibitory zone around the disc.
Purification and structure determination of alchivemycin A.
To purify alchivemycin A, we extracted 3 liters of the combined-culture broth (Streptomyces endus S-522 and T. pulmonis) with 1.5 liters of n-butanol. After the evaporation of n-butanol, the residue was dissolved in methanol and applied to a silica gel column (silica gel 60N; 63 to 210 μm; Kanto Chemical, Japan). The column was eluted with a stepwise gradient of chloroform-methanol (20:1 to ∼0:1). The fractions containing antibiotic activity were eluted using chloroform-methanol (4:1 and 2:1). These fractions were pooled and evaporated to obtain crude alchivemycin A. The crude alchivemycin A was dissolved in dimethyl sulfoxide and applied to a reverse-phase silica gel column (ODS-AM; internal diameter, 200 by 46 mm; YMC, Japan). The column was eluted with a stepwise gradient of acetonitrile and 0.15% K2HPO4 (pH 3.5) (2:8 to 9:1). The fraction containing antibacterial activity (8:2) was evaporated in vacuo. The resulting aqueous layer was extracted with ethyl acetate. The organic layer was dried in vacuo to obtain pure alchivemycin A (25 mg).
1H- and 13C-nuclear magnetic resonance (NMR) spectra were obtained at 750 and 189 MHz in dimethyl sulfoxide-d6. Liquid chromatography-mass spectrometry (LC-MS) spectra were obtained on an AP1165 machine (Applied Biosystems). UV-visible spectra were taken on an HP1090 system. NMR assignment data are shown in Table S1 and Fig. S7 in the supplemental material.
Nucleotide sequence accession numbers.
The 16S rRNA sequences were submitted to the DDBJ database under accession numbers AB564290 (Streptomyces endus S-522) and AB564289 (Tsukamurella pulmonis TP-B0596).
RESULTS
Isolation of bacteria that induce secondary-metabolite production.
In this study, we used the production of two red pigments by Streptomyces lividans TK23 under certain conditions as a indicator of secondary metabolism (10). In Bennett's glucose medium, S. lividans does not produce the two red pigments actinorhodin (23) and undecylprodigiosin (4). We then screened bacterial strains that induce the production of these two pigments on Bennett's glucose plate. Of the 400 inducer strains assayed, 1 bacterial strain, TP-B0596, induced red pigment production (Fig. 1A), even in liquid cultures (Fig. 1B). Microscopic studies revealed that the TP-B0596 and TK23 strains partly intertwined in liquid culture (Fig. 1D). The scanning electron micrograph showed that TP-B0596 was a coryneform bacterium (see Fig. S1a in the supplemental material), and the phylogenetic analysis of the 16S rRNA genes revealed that it was Tsukamurella pulmonis (25).
FIG. 1.

Induction of secondary-metabolite production by mycolic acid-containing bacteria in a coculture with S. lividans TK23. The coculture of T. pulmonis and S. lividans was performed to induce the production of secondary metabolites. (A) Coculture of T. pulmonis and S. lividans in Bennett's medium. S. lividans spores were overlaid, and T. pulmonis was inoculated at the center of the plate (left). S. lividans and T. pulmonis were inoculated in parallel on the medium (right). Sl, S. lividans; Tp, T. pulmonis. Red pigment production was found only at the site of cell-to-cell contact. The organisms were grown at 30°C for 3 days. (B) Liquid medium without bacteria (left), pure culture of S. lividans (center), and coculture (right). The organisms were grown at 30°C for 3 days in A-3M medium. (C) Coculture of T. pulmonis and S. lividans in a dialysis flask. T. pulmonis was grown in the left compartment and S. lividans in the right compartment at 30°C for 7 days. Red pigments were not detected in the broth. (D) Optical micrograph of S. lividans (left), coculture (center), and T. pulmonis (right) at 1,000-fold magnification.
Induction of red pigment production is not mediated by molecular substances but by cell-to-cell interactions.
The liquid culture of T. pulmonis extracted with n-butanol or sterilized using a 0.2-μm filter did not induce pigment production. Moreover, liquid culture broth sterilized in an autoclave at 121°C for 1, 3, 10, or 30 min also did not show this inducing ability (see Fig. S1b in the supplemental material). These results imply that pigment induction was mediated by living cells and not by substances such as microbial hormones (6). We next investigated whether the inducing ability exists when T. pulmonis is cocultivated with S. lividans in a dialysis flask. A dialysis flask consists of two compartments partitioned with a dialysis membrane; this arrangement allows the exchange of small molecules between the compartments during microbial cultivation (13). A 0.2-μm mixed cellulose ester membrane and a regenerated cellulose membrane were used as the dialysis membranes for separation. S. lividans did not produce red pigments in the dialysis culture of the mixed cellulose ester membrane (Fig. 1C) and of the regenerated cellulose membrane (see Fig. S1c and d in the supplemental material), indicating that the pigment-inducing ability of T. pulmonis requires cell-to-cell interactions. On solid culture, cell-to-cell interaction also is needed to induce the pigments. S. lividans and T. pulmonis were inoculated next to one another (Fig. 1A). The red pigment was produced only in part of the cell-to-cell interaction.
Coryneform bacteria are potent inducers of secondary metabolism.
Three closely related strains, T. pseudospumae, T. spumae, and T. strandjordii, also showed pigment-inducing ability (Table 1; also see Fig. S2a in the supplemental material). The genus Tsukamurella belongs to the family Corynebacteriaceae; the members of this family show the presence of mycolic acid in the outer layer of the cells. Therefore, we investigated the relationship between the red pigment induction and mycolic acid. First, we performed experiments using other mycolic acid-containing members of Corynebacteriaceae (the phylogenetic tree is shown in Fig. S3 in the supplemental material). Among the 19 species tested, those belonging to the genera Rhodococcus, Corynebacterium, Nocardia, Mycobacterium, Williamsia, Dietzia, and Gordonia induced pigment formation in S. lividans (Table 1). In contrast, the widely divergent species belonging to the genera Pseudonocardia, Turicella, Escherichia, Staphylococcus, Bacillus, Micrococcus, Saccharomyces, and Candida, which do not possess mycolic acid in the outer layer of the cell, did not induce pigment production (Table 1; also see Fig. S2b in the supplemental material).
Mycolic acid is responsible for inducing red pigment production.
To determine whether mycolic acid is responsible for the pigment-inducing activity, we used a C. glutamicum strain with a disrupted mycolic acid biosynthetic gene, pks13, as a mycolic acid-deficient mutant; the absence of mycolic acid in this mutant was confirmed (see Fig. S4 in the supplemental material), and the mutant had no significant physiological differences from its parental strain. In a coculture with S. lividans, the disruptant did not induce red pigment production (Fig. 2A). We then studied the effect of an inhibitor of mycolic acid biosynthesis on red pigment production. Isoniazid is an inhibitor of mycolic acid synthase (22) and has no effect on the growth of S. lividans at less than 100 mM (11). The addition of isoniazid to the coculture of S. lividans and T. pulmonis inhibited red pigment production (Fig. 2B). Similar results were obtained when isoniazid was added to the cocultures of S. lividans and other mycolic acid-containing species, like Rhodococcus, Corynebacterium, Gordonia, and Nocardia (Rhodococcus erythropolis and Corynebacterium glutamicum; see Fig. S5a in the supplemental material). Actinorhodin and undecylprodigiosin are polyketide compounds, and the possibility of their biosynthesis inhibition by isoniazid was examined by using Streptomyces coelicolor A3(2), which is another actinorhodin and undecylprodigiosin producer. At the concentration of 7.2 mM isoniazid, S. coelicolor A3(2) can produce actinorhodin and undecylprodigiosin (data not shown). This result suggests that isoniazid does not affect the actinorhodin and undecylprodigiosin biosynthetic enzymes, and the decrease in mycolic acid by isoniazid directs the inactivation of the biosynthesis.
FIG. 2.

Effect of mycolic acid on the induction of secondary metabolites in the coculture. (A) Coculture of S. lividans with wild-type C. glutamicum (left) and with Δpks13, the mycolic acid-deficient mutant of C. glutamicum (right). In this experiment, two Δpks13 mutants were used independently for coculture. (B) Effect of isoniazid on metabolite production. Shown are the coculture of S. lividans with T. pulmonis (left) and coculture with added isoniazid (right).
These results indicate that mycolic acid is required for inducing red pigment production in S. lividans. However, the addition of mycolic acid extract obtained from C. glutamicum, T. pulmonis, T. spumae, Rhodococcus zopfii, and Rhodococcus wratislaviensis did not induce red pigment production in S. lividans (see C. glutamicum in Fig. S5b in the supplemental material), and whole-cell-wall fraction extracted from T. pulmonis also did not induce red pigment production in S. lividans (see Fig. S1e in the supplemental material). These results suggest that the mycolic acid present in the outer layer is important for inducing red pigment production, and the outer layer itself influences the inducing activity.
Induction of secondary-metabolite production varies with each strain.
We identified the pigments (actinorhodin or undecylprodigiosin) induced and their concentrations in the coculture broths of each inducer strain and S. lividans (Table 1). Each strain induced a significantly different type and concentration of red pigments. While coculture with T. pulmonis primarily yielded actinorhodin, coculture with C. glutamicum yielded undecylprodigiosin. We then determined whether the pH of each coculture broth was different from that of the pure-culture broth (see Fig. S6 in the supplemental material). The pH values of the cocultures with mycolic acid-containing strains varied from 4.6 to 7.5.
Secondary metabolism in other Streptomyces strains is influenced by mycolic acid-containing bacteria.
We determined whether T. pulmonis influences the secondary metabolism of not only S. lividans but also other Streptomyces strains. For this, we compared the secondary-metabolite profiles, determined using HPLC, between the cocultures and the pure cultures. A total of 112 new Streptomyces strains were isolated for this experiment. The corresponding HPLC peaks were compared between the pure cultures and cocultures; on the basis of this comparison, the HPLC peaks were grouped into the following four basic regulation patterns: increase/decrease in metabolite production, appearance of new metabolites, disappearance of some secondary metabolites, and no difference. These four patterns were then subdivided into eight combinations in each Streptomyces strain, because the strains did not uniformly produce secondary metabolites. For example, in some strains, we found that one metabolite was produced in a greater quantity in coculture than in pure culture, whereas another metabolite was produced in lesser quantity in coculture than in pure culture (Venn diagrams are shown in Fig. 3B to F). In the first regulation pattern, coculture induced an increase or decrease in the height of an existing peak. Coculture with T. pulmonis increased secondary-metabolite production in 61 strains and decreased production in 49 strains (Fig. 3B). In the second regulation pattern, coculture induced the appearance of a new metabolite peak or the disappearance of an existing peak. Of 112 strains, T. pulmonis induced the production of new secondary metabolites in 41 strains and suppressed secondary metabolite production in 12 strains. In the third pattern, which was observed in 13 strains, there were no differences between the profiles of the pure cultures and cocultures with T. pulmonis. A summary of the data described above reveals that T. pulmonis induced the production of new secondary metabolites in 36.6% of strains (41/112) and increased metabolite production in 54.5% of strains (61/112) (Fig. 3B). These results suggest that T. pulmonis can induce secondary-metabolite production in many diverse Streptomyces species. R. erythropolis (Fig. 3C) and C. glutamicum (Fig. 3D) showed the same ability, and they induced the production of new secondary metabolites in 36 and 27 Streptomyces strains, respectively (Fig. 3F). The HPLC profiles showed that some metabolites were induced by all three inducer strains, whereas others were induced by only one or two inducer strains (Fig. 3E and F). These results are consistent with the finding that each mycolic acid-containing strain induces the production of different concentrations of red pigments by S. lividans, and we named our coculture procedure combined culture.
FIG. 3.
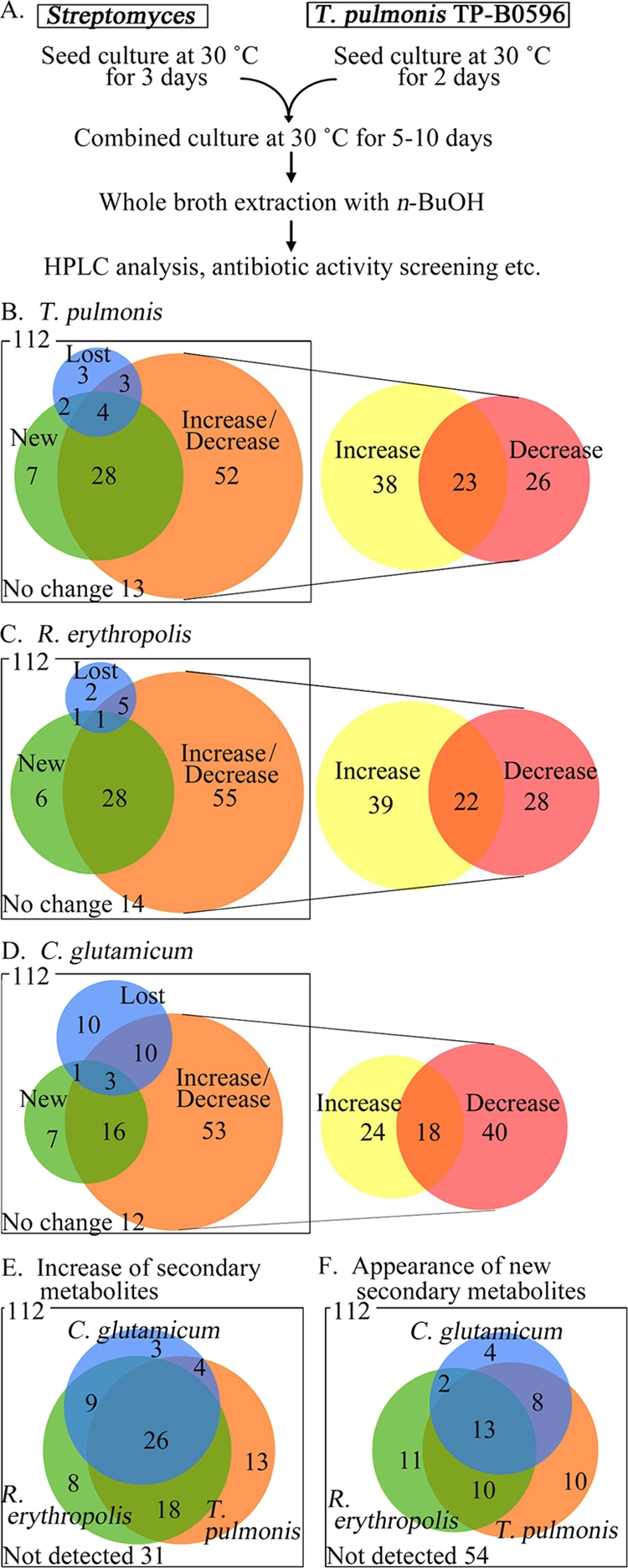
Effects of the coculture of Streptomyces isolated from soil. Metabolite profiles of pure and combined cultures of Streptomyces. (A) Flowchart of the coculture procedure. (B to F) The results of the comparison between the secondary-metabolite profiles of the pure and cocultures are shown in a Venn diagram. (B to D) Comparison between the HPLC profiles of a pure culture of Streptomyces and cocultures. Each profile was classified according to the increase/decrease in metabolite production, appearance of new metabolites, and disappearance of some secondary metabolites. The increase/decrease part is subclassified into increase, both increase and decrease, and decrease in the right panel. (E and F) Comparison of the HPLC profiles of cocultures of Streptomyces with T. pulmonis, R. erythropolis, or C. glutamicum. The numerals represent the number of strains. New, new secondary metabolites appeared in the coculture; increase/decrease, the secondary-metabolite levels in the coculture were higher/lower than the levels in the pure culture; lost, secondary-metabolite production was absent in the coculture; no change, there was no difference between the secondary-metabolite levels in the pure culture and cocultures.
Discovery of a new antibiotic, alchivemycin A, by combined-culture screening.
We then used the combined culture for antibiotic screening. We isolated 97 new actinomycetes and analyzed the extracts of the combined cultures by using antibiotic assays against six microorganisms. The results of these assays were compared to the corresponding results for pure cultures. T. pulmonis induced or activated antibiotic production in 11 of the 97 strains studied (Table 2). In 10 of these 11 strains, the antibiotic production was initiated only in combined culture, and antibiotic activity was increased in the remaining strain. We then identified the antibiotics induced by combined culture. In a combined culture with T. pulmonis, S-522 showed specific antibiotic activity against Micrococcus luteus, whereas the S-522 or T. pulmonis pure cultures did not show this activity (Table 2). We studied the HPLC profiles of the n-butanol extracts of S-522 cultures that were incubated with T. pulmonis (Fig. 4A) or C. glutamicum (Fig. 4B) or without T. pulmonis (Fig. 4C).
TABLE 2.
Paper disc diffusion assays for antibacterial activities of different actinomycete culture brothsa
| Bacterial strain | T. pulmonis | E. coli | B. subtilis | S. aureus | M. luteus | C. albicans | S. cerevisiae |
|---|---|---|---|---|---|---|---|
| S. lividans | + | 10 | 10 | ||||
| TK23 | − | ||||||
| S. coelicolor | |||||||
| A3(2) | + | 11 | 12 | ||||
| − | |||||||
| S501 | − | 19 | 22 | ||||
| − | |||||||
| S510 | + | 23 | 22 | ||||
| − | |||||||
| S522 | + | 40 | |||||
| − | |||||||
| S536 | + | 16 | 12 | 14 | |||
| − | |||||||
| S558 | + | 11 | 17 | 15 | 19 | 17 | 18 |
| − | |||||||
| S566 | + | 19 | 16 | 22 | 16 | 20 | |
| − | |||||||
| S573 | + | 25 | 23 | 20 | |||
| − | 15 | 13 | 12 | ||||
| S576 | + | 16 | 15 | 13 | |||
| − | 20 | ||||||
| S589 | + | 15 | |||||
| − |
Actinomycetes isolated from the soil and plant tissues were cultured as pure cultures (−) or combined cultures with T. pulmonis(+). The culture medium was extracted using n-butanol and assayed by a paper disc method (diameter of the disc, 10 mm). The numbers represent the diameter of the inhibitory zones (mm).
FIG. 4.

Production of a novel antibiotic, alchivemycin A, in a combined culture of Streptomyces endus S-522 and T. pulmonis or C. glutamicum. Shown is the production of alchivemycin A in a combined culture of S. endus S-522 and T. pulmonis or C. glutamicum. (A to D) HPLC profiles of the secondary metabolites produced by S-522 cultured with or without T. pulmonis. Shown are the culture broths of the combined culture with T. pulmonis (A), combined culture with C. glutamicum (B), S-522 pure culture (C), and T. pulmonis pure culture (D). The arrow shows the peak for alchivemycin A. Elution was performed with a linear gradient, as indicated on the scale at the right. (E) Chemical structure of alchivemycin A.
Strain S-522 was isolated from a leaf of Allium tuberosum, and the phylogenetic analysis of the 16S rRNA genes identified it as Streptomyces endus S-522. The HPLC profiles showed that the secondary-metabolite pattern of the combined culture was different from that of the pure S-522 culture. The antibiotic secreted by this strain was identified as the peak that eluted at 23 min (Fig. 4A and B). After purification, 25 mg of pure antibiotic was obtained from 3 liters of combined-culture broth. The chemical structure of the antibiotic was studied using NMR and MS (see Table S1 and Fig. S7 in the supplemental material; the absolute configuration will be described in another paper). On the basis of these analyses, we concluded that this antibiotic has a novel chemical structure and named it alchivemycin A (Fig. 4E). The molecular formula and mass-to-charge ratio (m/z) of alchivemycin A are C35H53NO10 and 648.3754 [M+H]+, respectively. This antibiotic contains a heterocyclic chromophore that has not been described previously and shows specific antibiotic activity against Micrococcus luteus at a concentration of 0.06 μg/ml (Table 3).
TABLE 3.
Antibacterial activities of alchivemycin Aa
| Organism | Relevant characteristic | Alchivemycin A MIC (μg/ml) |
|---|---|---|
| Micrococcus luteus | Gram positive | 0.06 |
| Bacillus subtilis | Gram positive | 40 |
| Staphylococcus aureus | Gram positive | >50 |
| Escherichia coli | Gram negative | >50 |
| Saccharomyces cerevisiae | Yeast cell | >50 |
| Candida albicans | Yeast cell | >50 |
The MICs were determined by the 2-fold sequential dilution method in heart infusion broth (Difco). The inhibition concentration was determined by measuring the absorbance at 600 nm.
DISCUSSION
In this study, we found that T. pulmonis TP-B0596 induces the secondary metabolism of different Streptomyces species. Further analysis revealed that other species belonging to the Tsukamurella genus and related genera have similar activity against Streptomyces. Some substances, such as the γ-butyrolactone autoregulators (6), desferrioxamine E (24), and goadsporin (14), have been reported to induce secondary metabolism in Streptomyces. Dialysis cultures and the addition of some components prepared from the mycolic acid-containing bacteria to Streptomyces pure culture revealed that molecular substances do not mediate this inducing ability. Therefore, the induction mechanism in a combined culture is different from substance-mediated induction. In a liquid culture, Streptomyces grows in filaments and T. pulmonis aggregates to pellet form. In a solid culture, red pigment production was found only at the site of cell-to-cell contact. These results indicate that the inducer bacteria and Streptomyces intertwine in the combined culture, and the inducer bacteria form cell-to-cell interactions with Streptomyces via mycolic acid and therefore influence the secondary metabolism.
The mycolic acid-containing members of Corynebacteriaceae induced pigment formation in S. lividans. In contrast, a wide variety of mycolic acid-deficient bacteria, including the C. glutamicum mycolic acid-deficient mutants, cannot induce pigment production. Mycolic acids are located in the outer layer of the bacterium belonging to Corynebacteriaceae. They are bound to arabinogalactan, trehalose, and proteins, and they are important components of the highly impermeable outer barrier (8). However, the addition of purified mycolic acids into the pure culture of S. lividans had no effect on red pigment production. These results revealed that mycolic acid alone is not sufficient and the intact cells are required for the induction. We therefore conclude that the mycolic acid that exists in the outer layer is important for inducing red pigment production, and the outer layer itself influences the inducing activity.
Each mycolic acid-containing strain induces different changes in secondary metabolism. In a combined culture, we find that the combined culture broth of each inducer strain had a different pH. However, there is no relationship between pH values and red pigment productivity.
On the basis of our observations, the main factors affecting the red pigment induction mechanism in the combined culture are (i) the direct cell-to-cell interactions between Streptomyces and mycolic acid-containing bacteria, and (ii) the changes in medium composition due to the primary metabolism of each inducer bacterium.
The changes in the combined culture reflect the changes in secondary metabolism induced by environmental conditions. Ten of the 18 mycolic acid-containing bacteria having the ability to induce secondary metabolism in Streptomyces were isolated from the soil samples (see Table S2 in the supplemental material). In the natural environment, these mycolic acid-containing bacteria may influence secondary metabolism in Streptomyces, which is one of the major inhabitant strains of soil. The determination of the molecular changes occurring in combined cultures will elucidate not only the process of the induction of secondary-metabolite production but also the environmental bacterial interactions.
We used our combined-culture method for screening bioactive compounds. The combined culture of S. endus S-522 and T. pulmonis yielded a novel antibiotic, alchivemycin A. We concluded that alchivemycin is produced by S. endus, with the following two reasons: (i) the production of these metabolites also was detected in the coculture of S. endus and C. glutamicum, and (ii) we did not detect alchivemycin in the coculture containing the other Streptomyces strains and T. pulmonis.
Combined culture is an easy method for inducing the production of cryptic antibiotics, because it only involves the addition of a mycolic acid-containing bacterium to a pure culture of an actinomycete. We believe that our combined-culture method will prove very useful for the screening of natural products.
Supplementary Material
Acknowledgments
We thank Yoko Kanamori and Kanako Kami for their technical assistance. We thank Hisashi Kawasaki for providing the C. glutamicum Δpks13 mutant. We thank Noriko Saito for the SEM micrographs.
This work was supported in part by grants in aid from the New Energy and Industrial Technology Development Organization (NEDO) of Japan (to H.O.).
Footnotes
Published ahead of print on 19 November 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Bentley, S. D., et al. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141-147. [DOI] [PubMed] [Google Scholar]
- 2.Cueto, M., et al. 2001. Pestalone, a new antibiotic produced by a marine fungus in response to bacterial challenge. J. Nat. Prod. 64:1444-1446. [DOI] [PubMed] [Google Scholar]
- 3.Doull, J. L., and L. C. Vining. 1990. Nutritional control of actinorhodin production by Streptomyces coelicolor A3(2): suppressive effects of nitrogen and phosphate. Appl. Microbiol. Biotechnol. 32:449-454. [DOI] [PubMed] [Google Scholar]
- 4.Feitelson, J. S., F. Malpartida, and D. A. Hopwood. 1985. Genetic and biochemical characterization of the red gene cluster of Streptomyces coelicolor A3(2). J. Gen. Microbiol. 131:2431-2441. [DOI] [PubMed] [Google Scholar]
- 5.Flardh, K., and M. J. Buttner. 2009. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat. Rev. Microbiol. 7:36-49. [DOI] [PubMed] [Google Scholar]
- 6.Horinouchi, S. 2007. Mining and polishing of the treasure trove in the bacterial genus streptomyces. Biosci. Biotechnol. Biochem. 71:283-299. [DOI] [PubMed] [Google Scholar]
- 7.Hosaka, T., et al. 2009. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat. Biotechnol. 27:462-464. [DOI] [PubMed] [Google Scholar]
- 8.Huc, E., et al. 2010. O-mycoloylated proteins from Corynebacterium: an unprecedented post-translational modification in bacteria. J. Biol. Chem. 285:21908-21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikeda, H., et al. 2003. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 21:526-531. [DOI] [PubMed] [Google Scholar]
- 10.Kieser, T., M. J. Bibb, M. J. Buttner, K. F. Chater, and D. A. Hopwood. 2000. Practical streptomyces genetics. The John Innes Foundation, Norwich.
- 11.McLean, K. J., et al. 2002. Azole antifungals are potent inhibitors of cytochrome P450 mono-oxygenases and bacterial growth in mycobacteria and streptomycetes. Microbiology 148:2937-2949. [DOI] [PubMed] [Google Scholar]
- 12.Ohnishi, Y., et al. 2008. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 190:4050-4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno, M., et al. 1999. Establishing the independent culture of a strictly symbiotic bacterium Symbiobacterium thermophilum from its supporting Bacillus strain. Biosci. Biotechnol. Biochem. 63:1083-1090. [DOI] [PubMed] [Google Scholar]
- 14.Onaka, H., H. Tabata, Y. Igarashi, Y. Sato, and T. Furumai. 2001. Goadsporin, a chemical substance which promotes secondary metabolism and morphogenesis in streptomycetes. I. Purification and characterization. J. Antibiot. (Tokyo). 54:1036-1044. [DOI] [PubMed] [Google Scholar]
- 15.Onaka, H., S. Taniguchi, H. Ikeda, Y. Igarashi, and T. Furumai. 2003. pTOYAMAcos, pTYM18, and pTYM19, actinomycete-Escherichia coli integrating vectors for heterologous gene expression. J. Antibiot. (Tokyo). 56:950-956. [DOI] [PubMed] [Google Scholar]
- 16.Portevin, D., et al. 2004. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. U. S. A. 101:314-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scherlach, K., and C. Hertweck. 2009. Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 7:1753-1760. [DOI] [PubMed] [Google Scholar]
- 18.Schroeckh, V., et al. 2009. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. U. S. A. 106:14558-14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu, M., et al. 2000. Studies on endophytic actinomycetes (I) Streptomyces sp. isolated from Rhododendron and its antifungal activity. J. Gen. Plant Pathol. 66:360-366. [Google Scholar]
- 20.Takeuchi, M., and A. Yokota. 1989. Cell-wall polysaccharides in coryneform bacteria. J. Gen. Appl. Microbiol. 35:233-252. [Google Scholar]
- 21.Tomiyasu, I. 1982. Mycolic acid composition and thermally adaptative changes in Nocardia asteroides. J. Bacteriol. 151:828-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vilcheze, C., et al. 2000. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 182:4059-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright, L. F., and D. A. Hopwood. 1976. Actinorhodin is a chromosomally-determined antibiotic in Streptomyces coelicolar A3(2). J. Gen. Microbiol. 96:289-297. [DOI] [PubMed] [Google Scholar]
- 24.Yamanaka, K., et al. 2005. Desferrioxamine E produced by Streptomyces griseus stimulates growth and development of Streptomyces tanashiensis. Microbiology 151:2899-2905. [DOI] [PubMed] [Google Scholar]
- 25.Yassin, A. F., et al. 1996. Tsukamurella pulmonis sp. nov. Int. J. Syst. Bacteriol. 46:429-436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.