

Abstract
We have identified the N-terminus of adenovirus early region 1A (AdE1A) as a region that can regulate the 26S proteasome. Specifically, in vitro and in vivo co-precipitation studies have revealed that the 19S regulatory components of the proteasome, Sug1 (S8) and S4, bind through amino acids (aa) 4–25 of Ad5 E1A. In vivo expression of wild-type (wt) AdE1A, in contrast to the N-terminal AdE1A mutant that does not bind the proteasome, reduces ATPase activity associated with anti-S4 immunoprecipitates relative to mock-infected cells. This reduction in ATPase activity correlates positively with the ability of wt AdE1A, but not the N-terminal deletion mutant, to significantly reduce the ability of HPV16 E6 to target p53 for ubiquitin-mediated proteasomal degradation. AdE1A/proteasomal complexes are present in both the cytoplasm and the nucleus, suggesting that AdE1A interferes with both nuclear and cytoplasmic proteasomal degradation. We have also demonstrated that wt AdE1A and the N-terminal AdE1A deletion mutant are substrates for proteasomal-mediated degradation. AdE1A degradation is not, however, mediated through ubiquitylation, but is regulated through phosphorylation of residues within a C-terminal PEST region (aa 224–238).
Keywords: adenovirus E1A/p53/proteasome/S4/Sug1 (S8)
Introduction
Adenovirus early region 1A (AdE1A) expression is essential for both Ad-induced transformation and a productive Ad infection (Gallimore et al., 1985; Shenk, 1996). AdE1A is expressed as two differentially spliced mRNAs, 12S and 13S. The larger 13S transcript encodes a protein of 289 amino acids in Ad5. The 12S transcript encodes a protein that is identical to the larger 13S molecule, except for a highly conserved region of 46 amino acids which is located towards the C-terminus (conserved region 3; CR3). Other identifiable regions of conservation exist between different virus serotypes. CR1 lies between amino acids 40 and 80, and CR2 lies between amino acids 120 and 139 in Ad5. These regions of the molecule are important in defining interactions with a number of cellular proteins that function primarily in regulating cell growth.
AdE1A binds the retinoblastoma tumour suppressor protein, Rb, and the Rb-related proteins p107 and p130, through interactions with CR1 and CR2 (Whyte et al., 1988; Li et al., 1993). This interaction serves to disrupt Rb binding to E2F, allowing for stimulation of transcription through E2F-responsive promoters. CR3 is essential for transactivation of both viral early, and cellular promoters (Jones, 1995). The transactivating capacity of CR3 resides in its ability to bind transcription factors such as ATF-2 (Liu and Green, 1994), transcriptional mediators such as hSur2 (Boyer et al., 1999) and the basal transcriptional machinery, TATA-box-binding protein (TBP; Geisberg et al., 1994), directly. AdE1A also binds C-terminal-binding protein (CtBP) through a conserved PXDLS motif, located towards the extreme C-terminus of the molecule (Schaeper et al., 1995).
The N-terminus of AdE1A is weakly conserved between virus serotypes. This region of the molecule, however, is important in defining many protein–protein interactions that appear remarkably to be conserved between serotypes. The best characterized N-terminal-binding proteins are p300/CBP (Eckner et al., 1994), although structural elements in CR1 are also required for the interaction. p300/CBP are transcriptional regulators, possessing intrinsic histone-directed acetyltransferase (HAT) activity (Bannister and Kouzarides, 1996; Ogryzko et al., 1996). The arginine residue at position 2 of Ad5E1A is critical in mediating this binding, since mutation at this position to glycine ablates p300/CBP binding (Wang et al., 1993). AdE1A binding to p300/CBP inhibits HAT activity; AdE1A binding to p300/CBP mimics inhibitory phosphorylation of p300/CBP (Ait Si Ali et al., 1998). The N-terminus of AdE1A, in conjunction with elements in CR1 and CR2, is important in determining the ability of AdE1A to induce quiescent cells to enter S phase and to inhibit cell differentiation (Quinlan and Grodzicker, 1987; Howe and Bayley, 1992). Thus, p300/CBP and the Rb family of proteins are implicated in mediating these cellular responses.
The 26S proteasome is the major non-lysosomal proteolytic machinery in eukaryotes, serving to degrade protein substrates targeted specifically by polyubiquitin modification (reviewed by Ciechanover, 1998). The 26S proteasome is assembled from two large multisubunit complexes, the 20S proteasome and the 19S regulatory complex (RC). The 19S RC comprises six structurally related ATPase subunits (S4, S6, S6′, S7, S8 and S10b) which are members of the highly conserved AAA ATPase superfamily of proteins (Zwickl and Baumeister, 1999). It is suspected that these ATPases function to determine the ATP-dependent unfolding and translocation of protein substrates into the 20S proteasomal core. The 19S RC also comprises 12 non-ATPase subunits, with some having limited homology to eukaryotic initiation factor 3 and components of the signalosome (Asano et al., 1997; Glickman et al., 1998; Seeger et al., 1998). The 20S component of the proteasome is composed of a stack of four heptameric rings, comprising 28 subunits, arranged into a cylinder with the proteolytic sites facing the inner chamber. These subunits can be arranged accordingly into two subfamilies, α and β, dependent on their evolutionary relationship to the subunits comprising the Thermoplasma acidophilium proteasome (Baumeister et al., 1998).
The tumour suppressor gene product p53 plays a crucial role in the negative regulation of cell growth in response to DNA damage and other stress-activated signalling pathways (Levine, 1997). In the absence of co-operating oncogenes, AdE1A can up-regulate p53 expression, inducing apoptosis (Debbas and White, 1993; Lowe and Ruley, 1993; Grand et al., 1994; Sabbatini et al., 1995). Stabilization of p53 by AdE1A is achieved post-translationally, as assessed by an increase in the half-life of the p53 protein (Lowe and Ruley, 1993; Grand et al., 1996). Mutational analysis of AdE1A has indicated that the N-terminus and elements in CR1 are required for p53 stabilization, implicating roles for p300/CBP and Rb in this process (Chiou and White, 1997; Querido et al., 1997).
p53 is a target for ubiquitin-mediated degradation through the 26S proteasome (Scheffner et al., 1990; Ciechanover et al., 1994). Mdm2 promotes the degradation of p53 (Haupt et al., 1997; Kubbutat et al., 1997), functioning as a p53-directed E3 ubiquitin ligase (Honda et al., 1997). p14ARF can stabilize p53 by complexing with Mdm2 and specifically inhibiting Mdm2 E3 ubiquitin ligase activity (Honda and Yasuda, 1999). AdE1A can stabilize p53, at least in part, by up-regulating transcription and hence translation of p14ARF (de Stanchina et al., 1998). In contrast, other DNA tumour virus-transforming proteins can de-stabilize p53. Human papillomavirus (HPV) E6 from oncogenic types 16 and 18 binds directly to p53, and targets it for degradation by recruiting E6-AP, a cellular protein which functions as an E3 ubiquitin ligase (Thomas et al., 1999). Adenovirus E1B 55K and E4ORF6 also target p53 for degradation during viral infection (Steegenga et al., 1998).
Sug1 (also called Rtp6p in yeast), originally identified in yeast as a transcriptional activator of GAL4, was shown subsequently to be an integral component of the mammalian 19S RC (S8; Rubin et al., 1996). Determination of the quaternary structure of the 19S RC in vitro suggests that Sug1 forms multimeric complexes with both S4 and S6 ATPases and the non-ATPase component, S1 (Richmond et al., 1997; Gorbea et al., 2000). Mouse Sug1 was identified, however, in a yeast two-hybrid screen as a retinoic acid receptor-binding protein. Indeed, mouse Sug1 binds to a number of other nuclear steroid receptors, including the thyroid receptor, the oestrogen receptor, the vitamin D3 receptor and retinoid-X-receptors (Lee et al., 1995; vom Baur et al., 1996). In these circumstances, Sug1 functions in a ligand-dependent manner as a transcriptional co-activator, presumably by functioning as a 3′–5′ DNA helicase (Fraser et al., 1997). Sug1 interaction with c-Fos (Wang et al., 1996) and TBP (Swaffield et al., 1995) might also impinge on transcriptional regulation.
The work detailed here expands on an earlier report from our laboratory identifying Sug1 as an Ad5- and Ad12 E1A-binding protein (Grand et al., 1999). Using a large panel of Ad mutants, we have now identified the N-terminus of AdE1A, at a site distinct from that required for p300/CBP binding, as the region essential for mediating this interaction. This region of the molecule can also associate independently with another 19S RC, S4. This interaction has important biological consequences, regulating both 19S RC ATPase activity and p53 degradation. Studies carried out to determine the factors governing AdE1A stability have defined phosphorylation of a C-terminal region as crucial in targeting the molecule for proteasomal-mediated degradation. Thus, regulation of proteasomal activity by AdE1A and degradation of AdE1A are separable events: the N-terminus regulates proteasomal activity and the C-terminus targets the molecule for proteasomal-mediated degradation.
Results
Sug1 binds to the N-terminus of AdE1A
In order to define the region on AdE1A required for Sug1 binding, human A549 cells were infected with Ad5 mutants that contain deletions spanning the whole AdE1A gene (Table I). The ability of these AdE1A mutants to interact with Sug1 was assessed by conventional immunoprecipitation and western blotting strategies. Initially, AdE1A deletion mutants spanning the N-terminus, CR1 and CR2 were tested for their ability to bind Sug1, relative to wild-type (wt) Ad5 12S E1A (dl520). Interestingly, the N-terminal mutant dl1101 was unable to bind Sug1 (Figure 1A, ii, compare lane 1 with lane 9, dl520). CR1 deletion mutants dl1102, dl1103 and dl1141 all displayed reduced, but significant binding (Figure 1A, ii, compare lanes 2–4 with lane 9, dl520). Similarly, dl1142 showed reduced ability to bind Sug1 (Figure 1A, ii, compare lane 6 with lane 9, dl520). CR1 mutant dl1105, and CR2 mutants dl1108 and dl1109 all retained wild-type capacity to bind Sug1 (Figure 1A, ii, compare lanes 5, 7 and 8 with lane 9, dl520). Other Ad5 E1A mutants were investigated similarly for their ability to bind Sug1. The C-terminal mutants dl177-9 and dl1135 retained wild-type binding capacity (Figure 1B, ii, compare lanes 1 and 2, respectively, with lane 6, wt Ad5). Similarly, no defect in AdE1A–Sug1 binding was seen when studying specific AdE1A point mutations, e.g. RG2, YH47 and hr3 (Figure 1B, ii, compare lanes 3, 4 and 5 with lane 6, wt Ad5).
Table I. Adenovirus deletion mutantsa used.
| E1A gene | Mutation | Phenotype |
|---|---|---|
| dl520 | 12S E1A | wild-type 12S |
| dl1101 | del 4–25 | does not bind p300/p400 |
| RG2 | R to G at 2 | does not bind p300 |
| dl1504 | del 1–14 | does not bind p300 |
| sub1085 | del 20–24 | N-terminal mutant |
| dl1102 | del 26–35 | does not bind p400 |
| dl1103 | del 30–49 | does not bind p300/p400 |
| dl1104 | del 48–60 | does not bind p300 |
| dl1141 | del 61–69 | does not bind p300 |
| dl1105 | del 70–81 | CR1 mutant |
| dl1142 | del 82–92 | mutant C-terminal to CR1 |
| dl1107 | del 111–123 | does not bind Rb family |
| dl1108 | del 124–127 | does not bind Rb family |
| dl1109 | del 128–138 | binds Rb poorly |
| dl1132 | del 178–192 | exon 2 mutant |
| dl177–9 | del 178–238 | does not bind CtBP |
| dl1135 | del 225–238 | does not bind CtBP |
| YH47 | Y to H at 47 | does not bind Rb |
| hr3 (13S) | M to K at 176 | transactivation impaired |
| wild type | 12S + 13S | wild-type Ad5 |
aAll genes are relative to 12S unless stated otherwise.

Fig. 1. Mapping the binding site on AdE1A for Sug1. (A) Human A549 cells were infected with either dl1101, dl1102, dl1103, dl1141, dl1105, dl1142, dl1108, dl1109 or dl520 (lanes 1–9 and 10–18, respectively) at a multiplicity of 50 p.f.u. per cell. At 24 h post-infection, cell lysates were mock immunoprecipitated with pre-immune rabbit serum (i), or Sug1 was immunoprecipitated from infected cell lysates with an anti-Sug1 pAb (ii). Immunoprecipitates were separated on alkaline urea gels and AdE1A detected using M58 mAb. (B) Human A549 cells were infected with either dl177-9, dl1135, RG2, YH47, hr3 or wild-type Ad5 (lanes 1–6 and 7–12, respectively) at a multiplicity of 50 p.f.u. per cell. At 24 h post-infection, cell lysates were mock immunoprecipitated with pre-immune rabbit serum (i) or Sug1 was immunoprecipitated from infected cell lysates with an anti-Sug1 polyclonal antibody (ii). Immunoprecipitates were separated on alkaline urea gels and AdE1A detected using M58 mAb.
These results were substantiated further by infecting A549 cells with dl1101 and dl520 and performing reciprocal co-immunoprecipitation and western blotting analyses. An anti-AdE1A monoclonal antibody (mAb), which recognizes a C-terminal epitope, was unable to co-precipitate Sug1 from dl1101-infected cells but did co-precipitate Sug1 from dl520-infected cells (Figure 2A). Consistent with this, an anti-Sug1 polyclonal antibody did not co-precipitate AdE1A from dl1101-infected cells, but was capable of co-precipitating AdE1A from dl520-infected cells (Figure 2B). Since AdE1A–Sug1 complexes could not be found in dl1101-infected cells, this identified amino acids 4–25 in AdE1A as the region important in defining the interaction with Sug1.

Fig. 2. The N-terminus of AdE1A binds to Sug1. Human A549 cells were infected with either dl1101 or dl520 at a multiplicity of 50 p.f.u. per cell. At 24 h post-infection, AdE1A was immunoprecipitated from infected cell lysates using M73 mAb. Immunoprecipitated protein complexes were separated by SDS–PAGE; Sug1 was identified by western blotting, using a rabbit anti-Sug1 pAb, (A). Sug1 was immunoprecipitated similarly from infected cell lysates 24 h post-infection, using a rabbit anti-Sug1 polyclonal antibody. Immunoprecipitates were separated on alkaline urea gels; AdE1A was identified by western blotting, using M73 mAb, (B). Human Ad5 293 cells were fractionated as described in Materials and methods. Sug1 was immunoprecipitated from cytoplasmic and nuclear cell equivalents; immunoprecipitates were separated on alkaline urea gels and AdE1A detected using M73 mAb, (C). The partitioning of AdE1A (i), Sug1 (ii), α-actinin (iii) and lamin B (iv) between the cytoplasm and nucleus by western blotting was also performed.
Subcellular fractionation reveals that AdE1A and Sug1 can be found in both cytoplasmic and nuclear fractions (Figure 2C, i and ii). It was therefore considered important to determine the proportion of AdE1A–Sug1 complex found in each compartment. Thus, Sug1 was immunoprecipitated from fractionated Ad5 293 cell equivalents, and co-precipitated AdE1A identified by western blotting. The majority of the AdE1A–Sug1 complex (∼75%) was found in the nuclear fraction (Figure 2C, i) despite a substantial proportion of cellular Sug1 being localized to the cytoplasm (Figure 2C, ii). AdE1A partitions to the nucleus and cytoplasm in approximately equal proportions (Figure 2C, i). There was, however, also clear evidence of AdE1A–Sug1 complex present in the cytoplasm (Figure 2C, i). A qualitative measure of fractionation was determined by western blotting. As expected, soluble α-actinin was found exclusively in the cytoplasm (Figure 2C, ii), whereas lamin B, a component of the nuclear lamina, was found exclusively in the nuclear fraction (Figure 2C, iv).
AdE1A does not affect Sug1-associated DNA helicase activity or ATPase activity in vitro
To determine whether AdE1A affected the known biological properties of Sug1, purified GST–Sug1 preparations were assayed for 3′–5′ DNA helicase activity in the presence of bacterially expressed and purified Ad12 13S E1A. Upon addition of GST, the annealed DNA duplex remained intact when electrophoresed on an acrylamide–TBE gel (Figure 3A, compare lanes 2–5 with control lane 1). However, in the presence of increasing amounts of GST–Sug1, the DNA duplex was unwound, consistent with Sug1-associated DNA helicase activity (Figure 3A, compare lanes 6–9 with control lane 1). Upon the addition of increasing amounts of AdE1A, however, there was no change in intrinsic Sug1 DNA helicase activity (Figure 3B, compare lanes 4–8 with control lane 3). Therefore, perhaps not surprisingly, when intrinsic Sug1 ATPase activity was measured by phosphate release assay, AdE1A had no effect on its activity (data not shown).

Fig. 3. AdE1A does not affect Sug1-associated DNA helicase activity in vitro. (A) DNA helicase activity associated with purified GST–Sug1 preparations. Lane 1, negative control (substrate alone); lanes 2–5, 1, 10, 100 and 200 ng of GST; lanes 6–9, 1, 10, 100 and 200 ng of GST–Sug1; lane 10, positive control (substrate heated to 95°C for 5 min). (B) GST–Sug1 DNA helicase activity assayed in the presence of purified Ad12 13S E1A. Lane 1, negative control; lane 2, 100 ng of GST; lane 3, 100 ng GST–Sug1; lanes 4–8, 100 ng GST–Sug1 with either 1, 10, 100, 200 or 1000 ng of Ad12 13S E1A; lane 9, 1000 ng of Ad12 13S E1A alone; lane 10, positive control.
The N-terminus of AdE1A binds to the 19S proteasome
Although we previously had identified Sug1 as an AdE1A-binding protein, we had not ascertained whether the Sug1 complexed to AdE1A was proteasomal in origin. This is an important consideration since it appears that Sug1 is not only an integral component of the 19S proteasome, but also functions independently of the proteasome. Thus, we chose to investigate whether AdE1A could be found in complexes with any other 19S components. S4 was chosen for this study in the first instance, as it apparently functions exclusively in the 19S RC. Immunoprecipitation of S4 from Ad5 293 cells revealed that AdE1A was found in complexes with S4 (Figure 4A).
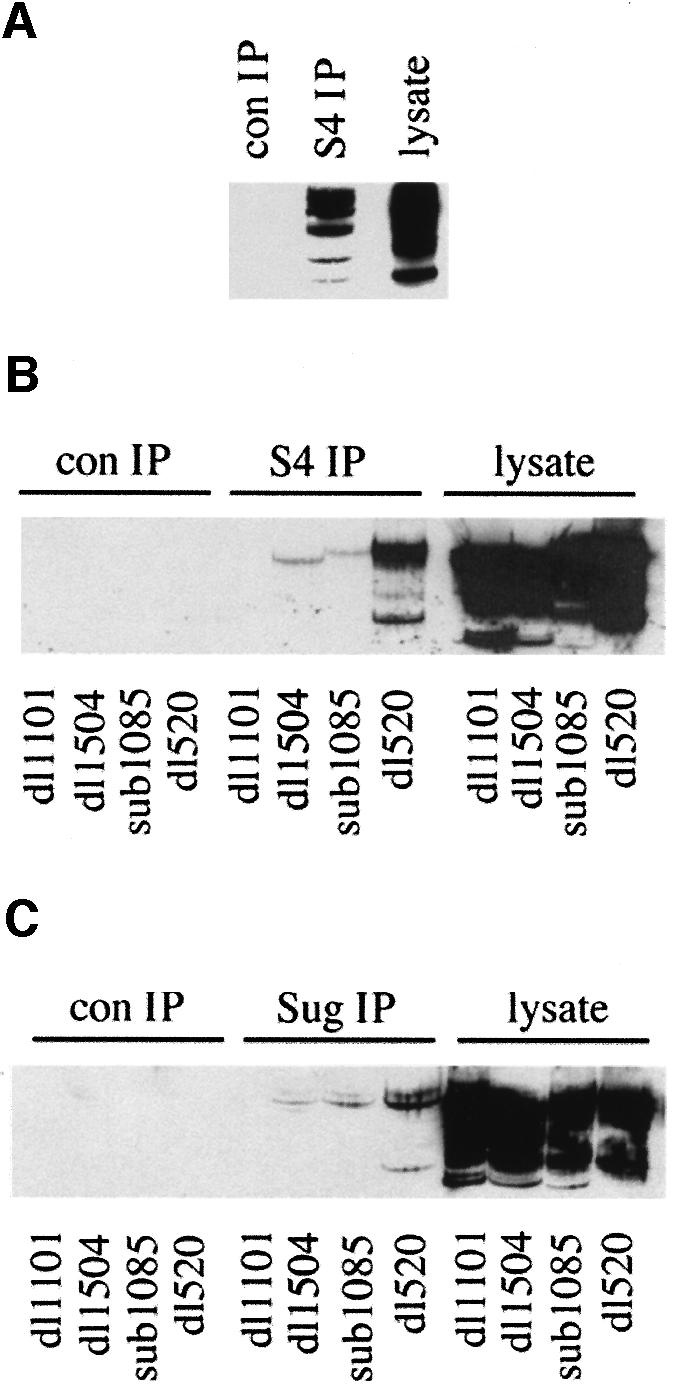

Fig. 4. The N-terminus of AdE1A binds to the 19S proteasome. (A) S4 was immunoprecipitated from human Ad5 293 cells, and protein complexes separated by alkaline urea gels. AdE1A was detected using M73 mAb. (B and C) Human A549 cells were infected with either dl1101, dl1504, sub1085 or dl520 at a multiplicity of 50 p.f.u. per cell. At 24 h post-infection, S4 (B) or Sug1 (C) was immunoprecipitated from infected cell lysates, and protein complexes separated on alkaline urea gels; AdE1A was detected using M73 mAb. (D–F) GST pull-downs separated by SDS–PAGE and subjected to fluorography, demonstrating in vitro binding capabilities between GST–Sug1 and 35S-labelled AdE1A mutants (D), GST–AdE1A mutants and 35S-labelled S4 (E), and 35S-labelled S5b and GST–AdE1A (F).
Fine mapping studies were then employed in an attempt to discriminate S4- and Sug1-binding sites on AdE1A. In an identical scenario to that for Sug1, an anti-S4 polyclonal antibody was unable to immunoprecipitate AdE1A from dl1101-infected cells, but could immunoprecipitate AdE1A from dl520-infected A549 cells (Figure 4B). Two other N-terminal mutants, dl1504 and sub1085, also had severely reduced ability to form complexes with S4 in Ad-infected cells (Figure 4B). Similar experiments indicated that Sug1 had an identical propensity to S4 for these N-terminal AdE1A mutants (Figure 4C). Since AdE1A could be found in complexes with both Sug1 and S4, we employed in vitro GST pull-down experiments to determine which of these 19S components AdE1A targets in vivo. AdE1A was shown to bind both Sug1 and S4 in vitro (Figure 4D and E), identical to the in vivo scenario. Indeed, the N-terminal mutant dl1101 was unable to bind either Sug1 or S4, whereas the RG2 mutant that is unable to bind p300/CBP retained the ability to bind Sug1 and S4 (Figure 4D and E). Significantly, AdE1A was unable to bind to S5b, a non-ATPase component of 19S (Figure 4F), thus indicating the specificity of AdE1A interaction with Sug1 and S4.
The N-terminus of AdE1A mediates the inhibition of ATPase activity associated with anti-S4 immunoprecipitates
To determine the effects of AdE1A upon ATPase activities associated with the 19S proteasome in vivo, A549 cells were infected with dl1101 and dl520. ATPase activities were then assessed by phosphate release assay, performed on anti-S4 and anti-Sug1 immunoprecipitates. ATPase activity associated with anti-S4 immunoprecipitates from dl1101-infected cells did not differ from ATPase activity immunoprecipitated with an anti-S4 polyclonal antibody from mock-infected cells (Figure 5A). Significantly, however, ATPase activity associated with anti-S4 immunoprecipitates from dl520-infected cells was inhibited by ∼20% when compared with ATPase activities from both mock- and dl1101-infected cells.

Fig. 5. AdE1A reduces ATPase activity associated with S4 immunoprecipitates, but does not affect activity associated with Sug1 immunoprecipitates. Human A549 cells were infected with either dl1101 or dl520 at a multiplicity of 50 p.f.u. per cell. At 24 h post-infection, S4 and Sug1 were immunoprecipitated from infected cells, and ATPase activity assayed as described in Materials and methods. (A) Relative ATPase activities associated with Sug1 immunoprecipitates: column 1, mock-infected cells; column 2, dl1101-infected cells; column 3, dl520-infected cells. Relative ATPase activities associated with S4 immunoprecipitates: column 4, mock-infected cells; column 5, dl1101-infected cells; column 6, dl520-infected cells; 100% activity corresponds to cellular ATPase activity associated with Sug1 and S4, respectively, from mock-treated cells. Error bars represent the mean ± SD from three independent experiments (n = 9). (B) AdE1A expression does not alter expression of 26S proteasomal components. Aliquots taken from either mock-infected, dl1101-infected or dl520-infected cells were separated by SDS–PAGE and western blotted for (i) AdE1A, (ii) S4, (iii) Sug1 and (iv) p32.
AdE1A-mediated inhibition of ATPase activity associated with anti-S4 immunoprecipitates was not associated with any change in the level of expression of either S4, Sug1 or the p32 component of the 20S proteasome (Figure 5B, ii, iii and iv). Levels of AdE1A expression were also similar in dl1101- and dl520-infected cells (Figure 5B, i). However, ATPase activity associated with anti-Sug1 immunoprecipitates was not affected following infection with dl520 (Figure 5A). It is important to note, however, that this assay does not distinguish proteasomal Sug1-associated ATPase activity from non-proteasomal Sug1 ATPase activity, such that total Sug1-associated ATPase activity is measured in this instance. Moreover, total Sug1-associated ATPase activity was significantly lower (by approximately two-thirds) than activity associated with S4.
AdE1A reduces the ability of HPV16 E6 to target p53 for degradation
An in vitro degradation assay (see Materials and methods) was utilized to determine the effect of AdE1A on HPV16 E6-mediated degradation of p53. In the absence of HPV16 E6, p53 was highly stable (Figure 6A, i, compare lanes 2–4 with control lane 1) as assessed by SDS–PAGE and fluorography. However, upon the addition of HPV16 E6, p53 was degraded rapidly (Figure 6A, i, compare lanes 5–7 with lanes 2–4). Pre-incubation of reticulocyte lysate with in vitro-translated Ad5 12S E1A reduced the ability of HPV16 E6 to target p53 for degradation (Figure 6A, i, compare lanes 8–10 with lanes 5–7). During this period of incubation, there was no apparent degradation of AdE1A itself, suggesting that AdE1A was not merely competing with p53 for active 26S proteasomes (Figure 6A, ii, compare control lane 11 with lanes 8–10).

Fig. 6. AdE1A reduces the ability of HPV16/18 E6 to target p53 for degradation. (A) (i) In vitro degradation of 35S-labelled p53 was performed as described in Materials and methods. Samples were taken at the appropriate times during the assay, separated by SDS–PAGE and subjected to fluorography to identify remaining p53: lane 1, control; lanes 2–4, no addition; lanes 5–7, addition of HPV16 E6; lanes 8–10, addition of HPV16 E6 + Ad5 E1A. Samples were taken 30, 60 and 120 min after the addition of unprogrammed lysate or HPV16 E6. (ii) As (i), but unlabelled p53 and [35S]Ad5E1A were used instead. SDS–PAGE and fluorography identify AdE1A. Lane 11 corresponds to input levels of Ad5E1A. (B) (i) As (A) (i) except that bacterially expressed Ad12 E1A was used (lanes 8–10) instead. Lanes 11–13, addition of HPV16 E6 + ovalbumin. Samples were taken 15, 30 and 60 min after the addition of unprogrammed lysate or HPV16 E6. (ii) Aliquots were taken and blotted for Ad12 E1A. Lane 14 corresponds to input levels of Ad12 E1A. (C) (i) As (A) (i) except that Ad5 dl1101-13S E1A (lanes 8–10) and Ad5 13S E1A (lanes 11–13) were used instead. (ii) Western blot indicating no degradation of Ad5 dl1101-13S E1A or Ad5 13S E1A during the time course of the experiment. Data shown in (A–C) are representative of several independent experiments. (D) AdE1A expression stabilizes p53 in HeLa cells. HeLa cells were either mock infected or infected with Ad12 dl620 or Ad5 dl1520 at a multiplicity of 50 p.f.u. per cell. At 24 and 48 h post-infection, cells were harvested and proteins were separated by SDS–PAGE and blotted for (i) p53, (ii) Ad12E1A or (iii) Ad5E1A. Lanes 1 and 2, mock-infected cells; lanes 3 and 4, Ad12 dl620-infected cells; lanes 5 and 6, Ad5 dl1520-infected cells.
This experimental procedure was then repeated using purified, unphosphorylated bacterially expressed Ad12 13S E1A, as opposed to in vitro translated (potentially phosphorylated) AdE1A. Bacterially expressed AdE1A was similarly able to inhibit HPV16 E6-mediated degradation of p53 (Figure 6B, i, compare lanes 8–10 with lanes 5–7), in the absence of any degradation of AdE1A (Figure 6B, ii, compare control lane 14 with lanes 8–10). In the same assay, ovalbumin was unable to inhibit HPV16 E6-mediated degradation of p53 (Figure 6B, i, compare lanes 11–13 with lanes 5–7). The N-terminal AdE1A mutant, which is unable to bind the proteasome, was assayed similarly for its ability to affect HPV16 E6-mediated degradation of p53. Significantly, Ad5 dl1101-13S E1A did not affect E6-mediated degradation of p53 (Figure 6C, i, compare lanes 8–10 with lanes 5–7), whereas Ad5 13S E1A did inhibit E6-mediated degradation of p53 (Figure 6C, i, compare lanes 11–13 with lanes 5–7). Ad5 dl1101-13S E1A and Ad5 13S E1A were not substrates for degradation during the course of this experiment (Figure 6C, ii, lanes 1–4, Ad5 dl1101-13S E1A at 15, 30, 60 min and zero time, respectively; lanes 5–8, Ad5 13S E1A at 15, 30, 60 min and zero time, respectively).
HeLa cells, which have multiple integrated copies of HPV18 and hence low levels of p53 due to HPV18 E6-targeted degradation, were used to ascertain whether AdE1A could overcome the ability of HPV18 E6 to degrade p53 in vivo. HeLa cells were infected with Ad12 dl620 and Ad5 dl1520, viruses that express wt AdE1A in the absence of the large E1B proteins. Infection of HeLa cells with either virus stabilized p53 levels (Figure 6D, i–iii), demonstrating that AdE1A can overcome the ability of HPV18 E6 to promote the degradation of p53 in vivo.
Stabilization of AdE1A by proteasome inhibitors
Previous studies have indicated that AdE1A has a very-short half-life. We were therefore interested in determining whether AdE1A was also a substrate for proteasomal-mediated degradation. In order to gauge the contribution of the proteasome in AdE1A degradation, Ad5 293 cells were treated with the proteasomal inhibitor lactacystin. In mock-treated cells, one major AdE1A species was detectable, along with two minor species (denoted by * in Figure 7A) that migrated with reduced mobility on SDS–PAGE. The levels of all AdE1A species were increased substantially after prolonged lactacystin treatment (Figure 7A). To confirm that lactacystin was stabilizing AdE1A at the protein level, the half-life of AdE1A was determined in both the absence and presence of lactacystin. In the absence of lactacystin, AdE1A had a half-life of ∼2 h, whereas in the presence of lactacysin, the half-life was >12 h (Figure 7B).
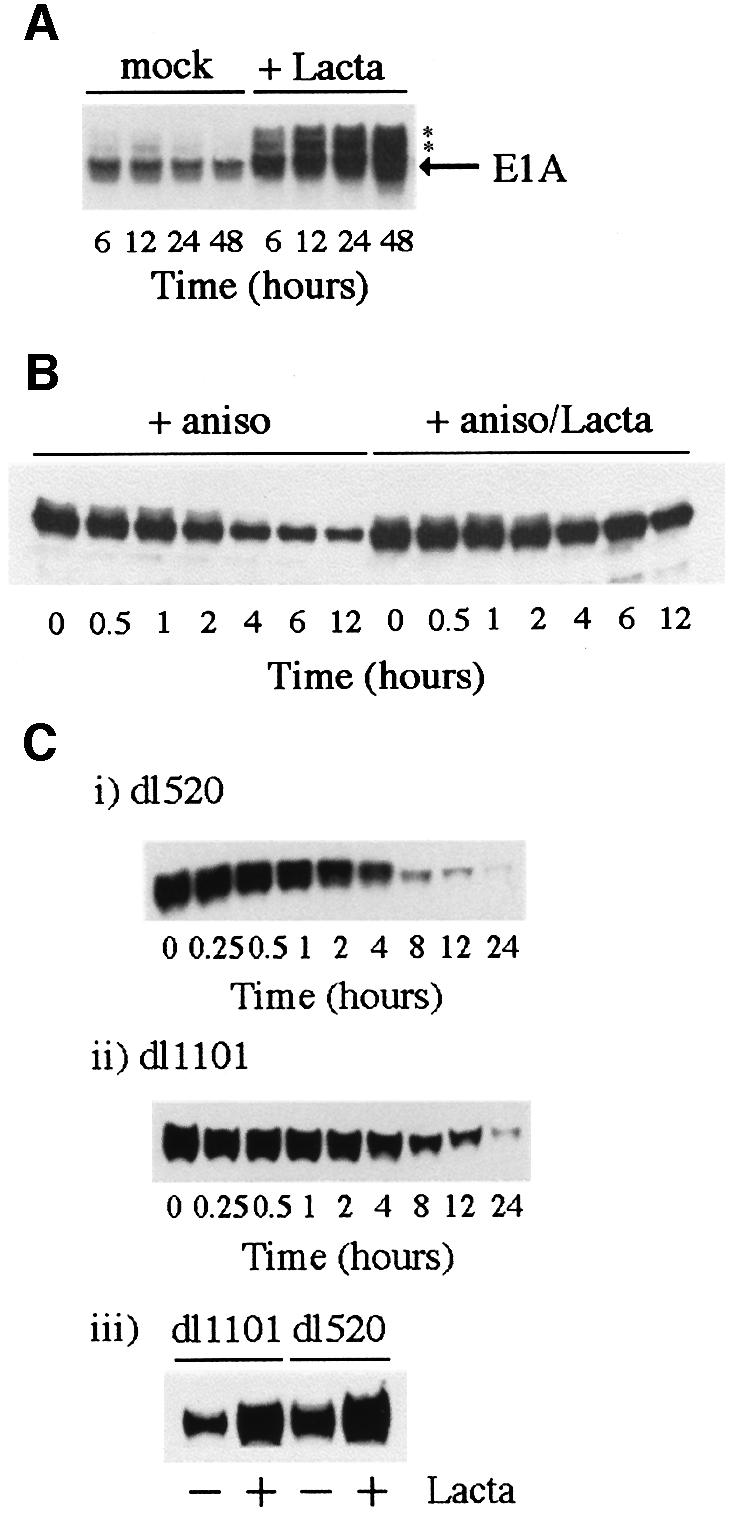
Fig. 7. Factors governing AdE1A stability. (A) Stabilization of AdE1A. Human Ad5 293 cells were either mock treated or treated in the presence of 10 µM lactacystin for the times indicated. Proteins were separated by SDS–PAGE and western blotted for AdE1A using M73 mAb. The asterisk denotes slower migrating, stabilized forms of AdE1A. (B) Half-life of AdE1A in Ad5 293 cells in the absence or presence of lactacystin. Cells were treated with the protein synthesis inhibitor anisomycin (100 µM) in the absence or presence of lactacystin (10 µM). Proteins were separated by SDS–PAGE and western blotted for AdE1A using M73 mAb. (C) The N-terminus of AdE1A affects stability. Human A549 cells were infected with either dl1101 or dl520. At 12 h post-infection, cells were treated with anisomycin to gauge the half-life of different AdE1A species. Proteins were separated by SDS–PAGE and western blotted for AdE1A in Ad5 dl1101-infected cells (i), or Ad5 dl520-infected cells (ii), using M73 mAb. A549 cells were transfected with Ad5 dl1101 and dl520 AdE1A. At 12 h post-infection, cells were treated with 10 µM lactacystin for 12 h. A western blot is presented showing the levels of the respective AdE1A species in mock-treated and lactacystin-treated cells (iii), using M73 mAb.
Previous studies have identified the first four residues at the extreme N-terminus as important in regulating the stability of Ad5E1A (Simon and Richter, 1990). We were therefore interested in determining whether the N-terminal deletion mutant dl1101 was stabilized relative to Ad5 12S E1A. The half-lives of dl1101 and dl520 were therefore determined in infected A549 cells. Figure 7C (i and ii) illustrates that the N-terminal mutant had a half-life approximately double that of wt Ad5 12S E1A (4 h as opposed to 2 h). The half-life determined for dl1101 is, however, somewhat short of the half-life determined for wt Ad5 12S E1A in the presence of lactacystin (Figure 7B). Indeed, dl1101 can be stabilized further in the presence of lactacystin (Figure 7C, iii), suggesting that other regions within the AdE1A molecule also define stability.
AdE1A is not a substrate for ubiquitylation
To determine whether the stabilized AdE1A species detectable by western blotting were polyubiquitylated forms of AdE1A, Ad5 293 cells were transfected with either a haemagglutinin (HA)-tagged ubiquitin (HA-Ubi) construct or a plasmid expressing HA alone. Transfected cells were either mock-treated or treated with lactacystin for 48 h. HA-Ubi species were then immunoprecipitated from transfected cells. No AdE1A species were co-precipitated with anti-HA antibodies (Figure 8B) despite substantial expression of HA-Ubi (Figure 8A). To confirm that the HA-Ubi construct was functional, Ad5 293 cells were transfected with either HA-Ubi alone, cJun-His6 alone or with both together. At 48 h post-transfection, His6-tagged proteins were purified, separated by SDS–PAGE and western blotted using an anti-HA antibody. In cells transfected with either HA-Ubi or cJun-His6 alone, no HA species could be detected (Figure 8C). However, HA-polyubiquitylated cJun-His6 species were detectable in cells co-transfected with HA-Ubi and cJun-His6 (Figure 8C), indicating the integrity of the constructs and the utility of the experimental system. Experiments performed in Ad5 293 cells using a His6-Ubi construct similarly were unable to demonstrate ubiquitylation of AdE1A, but showed incorporation into known cellular substrates such as Mcm7 (data not shown).
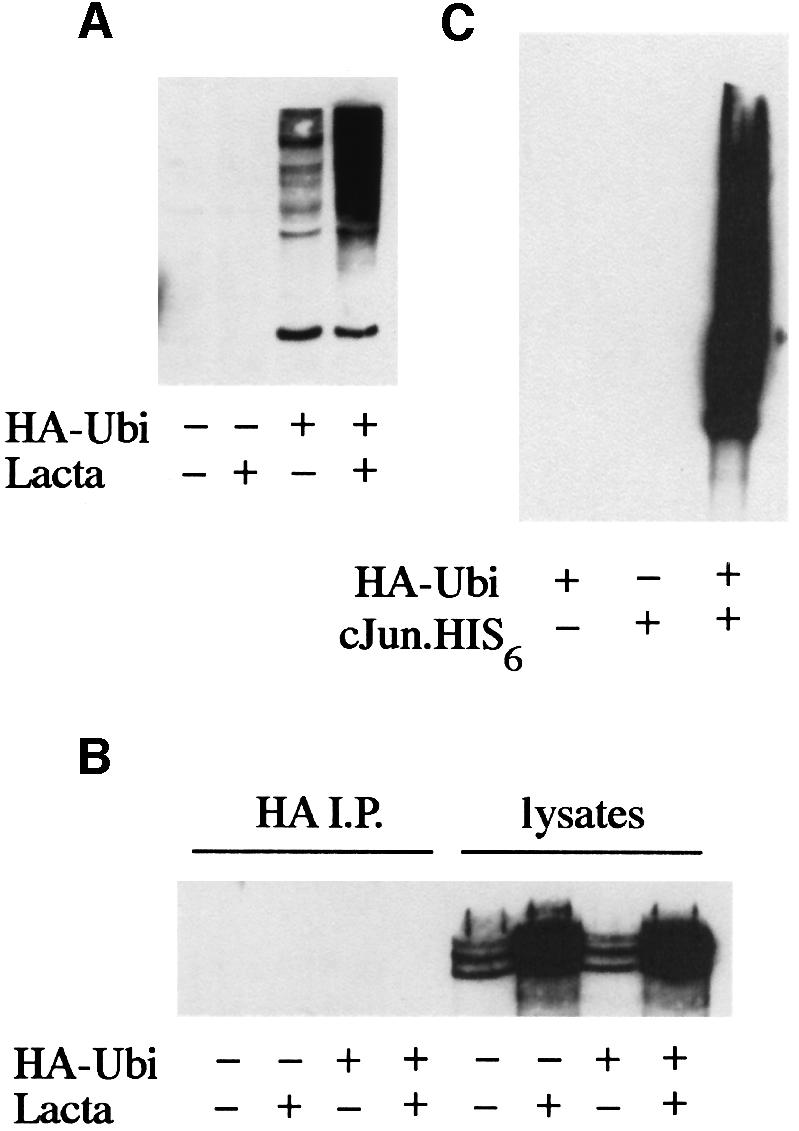
Fig. 8. AdE1A is not a substrate for ubiquitylation. (A) Ad5 293 cells were transfected with 2 µg of an HA-tagged ubiquitin construct (HA-Ubi). At 24 h post-infection, cells were then either mock treated or treated in the presence of lactacystin for an additional 24 h. (A) Immunodetection of HA-Ubi conjugates using an anti-HA mAb (3F10). (B) HA-Ubi conjugates were immunoprecipitated from cell lysates, separated by alkaline urea gels and immunoblotted for AdE1A. No AdE1A was co-precipitated with HA-Ubi. (C) Ad5 293 cells were transfected with 2 µg of HA-Ubi alone (lane 1), 2 µg of cJun-His6 alone (lane 2) or co-transfected with 2 µg each of HA-Ubi and cJun-His6 (lane 3). At 48 h post-transfection, cJun-His6 conjugates were purified, separated by SDS–PAGE and immunoblotted for HA (using 3F10).
Regulation of AdE1A stability through phosphorylation and PEST sequences
To determine whether the slower migrating AdE1A forms on SDS–PAGE (Figure 7A) are phosphorylated species, AdE1A was immunoprecipitated from [32P]orthophosphate-labelled Ad5 293 cells treated in the absence or presence of lactacystin, and separated by SDS–PAGE. Autoradiography revealed that both the major species of AdE1A and the slower migrating stabilized AdE1A form were present as phosphorylated species (Figure 9A, i). Comparison of specific activities of phosphate incorporation into AdE1A relative to absolute AdE1A protein levels (Figure 9A, ii) indicated that the higher molecular weight AdE1A species was hyper-phosphorylated (a/A = 1.67) relative to the low molecular weight form (b/B = 1.19). The high molecular weight form incorporated 40% more phosphate on a molar ratio than the low molecular weight form.

Fig. 9. (A) The minor higher molecular weight AdE1A species is hyperphosphorylated compared with the major lower molecular weight species. Ad5 293 cells were labelled with 100 µCi/ml [32P]orthophosphate for 18 h, in the absence or continued presence of lactacystin (10 µM). AdE1A species were immunoprecipitated from cell lysates, and separated by SDS–PAGE. (A) (i) An autoradiogram of the dried gel, indicating stabilized AdE1A species in the presence of lactacystin; (ii) a corresponding western blot for AdE1A using M73 mAB. (B) (i) Stabilization of AdE1A in Ad5 293 cells by the cdk inhibitor, olomoucine. Ad5 293 cells were treated for 1 h with 100 µM iso-olomoucine or 100 µM olomoucine, prior to treatment with 100 µM anisomycin. Western blot showing the respective levels of AdE1A after the indicated times of treatment, using mAb M73. (ii) Ad5 dl1101-13S and (iii) Ad5 dl1107 are stabilized by olomoucine treatment. A549 cells were transfected with the appropriate AdE1A construct. At 24 h post-infection, cells were pre-treated with 100 µM iso-olomoucine or olomoucine for 1 h prior to treatment with 100 µM anisomycin. Western blots are presented showing the effects of olomoucine on the half-life of different AdE1A species after the indicated times of treatment.
To determine the contribution of phosphorylation to the regulation of AdE1A stability, the half-life of AdE1A and numerous AdE1A deletion mutants was measured in the presence of various selective protein kinase inhibitors (Figure 9; and data not shown). The half-life of AdE1A in Ad5 293 cells was increased substantially (from ∼2 to 6 h) in the presence of the selective cyclin-dependent kinase (cdk) inhibitor olomoucine (Meijer, 1995), relative to iso-olomoucine, an inactive olomoucine analogue (Figure 9B, i). Olomoucine reduced the incorporation of phosphate into AdE1A from Ad5 293 cells by ∼25% (data not shown). This indicates both the selective nature of olomoucine treatment and the contribution of other kinases in regulating AdE1A phosphorylation. Interestingly, olomoucine also stabilized the N-terminal mutant dl1101 (Figure 9B, ii), indicating that phosphorylation of residues that lie outside this N-terminal region is important in governing AdE1A stability. Previous studies have indicated that AdE1A can recruit functional cyclin E–cdk2 and cyclin A–cdk2 complexes through binding the Rb family of proteins. Thus, to ascertain whether cdk recruitment to AdE1A through Rb regulates AdE1A stability, the half-life of an AdE1A deletion mutant dl1107 (Δ111–123), which is unable to bind the Rb family of proteins, was determined in the presence of olomoucine. Significantly, dl1107 was stabilized by olomoucine (Figure 9B, iii), suggesting that recruitment of cdks through Rb does not de-stabilize AdE1A.
PEST regions previously have been identified as residues critical in defining the stability of a number of eukaryotic proteins (Rogers et al., 1986). Within the primary sequence of AdE1A, four putative PEST regions have been identified. These regions lie between residues 44 and 94, 125 and 149, 177 and 202, and 223 and 244 (in Ad5 13S E1A). To identify whether any of these PEST regions are important in conferring instability on AdE1A, the half-lives of AdE1A deletion mutants lying within these PEST regions were determined. Deletion mutants that lie between residues 44 and 94 did not enhance AdE1A stability. Thus, the half-lives of dll104 (Δ48–60; Figure 10A, i) and dl1105 (Δ70–81; data not shown) were similar to that of wt AdE1A (Figure 10A, iv). The half-lives of deletion mutants dl1108 (Δ124–127; data not shown) and dl1109 (Δ128–138; Figure 10A, ii) were also found not to be significantly different from wt AdE1A (Figure 10A, iv). However, deletion of residues 224–238 [dl1132 (Δ178–192), 12S E1A] substantially enhanced the stability of AdE1A (compare Figure 10A, iii and iv), with a half-life (∼12 h) approaching that seen for AdE1A in Ad5 293 cells treated with lactacystin (Figure 7B). However, dl1132 was not stabilized additionally by the selective cdk inhibitor olomoucine, whereas wt 12S AdE1A (dl520) was (Figure 10B, i and ii, respectively). Taken together, these data suggest that phosphorylation of an as yet unidentified residue(s) within the PEST region 224–238 confers instability on AdE1A in the absence of ubiquitylation.
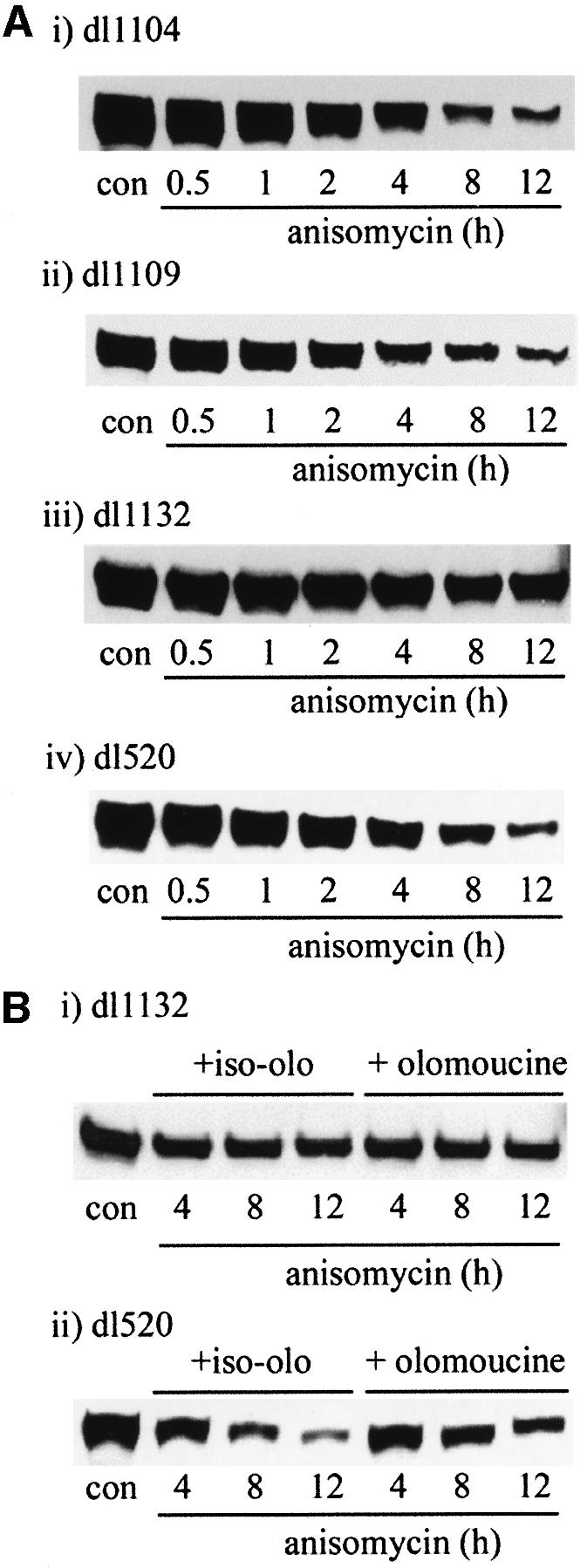
Fig. 10. (A) Role of PEST sequences in AdE1A degradation. A549 cells were infected with 50 p.f.u. per cell with different AdE1A deletion mutants. At 24 h post-infection, cells were treated with 100 µM anisomycin. Western blots are presented showing the effects of anisomycin on the levels of the respective AdE1A species, using mAb M73. AdE1A levels from dl1104 (i), dl1109 (ii), dl1132 (iii) and dl520 (iv) are shown. (B) Olomoucine treatment stabilizes the wild-type AdE1A species, dl520 (ii), but does not stabilize AdE1A from dl1132 (i). A549 cells were infected with 50 p.f.u. per cell with different AdE1A deletion mutants. At 24 h post-infection, cells were treated with 100 µM iso-olomoucine or olomoucine for 1 h, prior to treatment with 100 µM anisomycin. A western blot demonstrating the effects of olomoucine on the stability of the different AdE1A species is presented.
Discussion
The N-terminus of AdE1A is found in complexes with 19S proteasomal components Sug1 and S4
We have identified amino acids 4–25 at the N-terminus of AdE1A as crucial in forming binding sites for both Sug1 and S4 (Figures 1 and 2). Indeed, the fine mapping studies employed thus far have suggested that Sug1 and S4 share a common binding site (Figure 4A and B), residues 15MAASL19 defining at least part of this site. Intriguingly, however, although the binding site for Sug1 and S4 appears to be the same, it does not appear that AdE1A is targeting only one component of the 19S RC and co-precipitating the other 19S components as an inevitable consequence. Indeed, in vitro GST pull-down experiments indicate that the N-terminus of AdE1A can bind Sug1 and S4, apparently independently (Figure 4D and E). In common with the other ATPases that comprise the 19S RC, Sug1 and S4 are members of the AAA ATPase superfamily of proteins and accordingly possess the highest sequence similarity over ∼230 amino acids defining Walker homology sequences and ATPase activities (Patel and Latterich, 1998). Whether AdE1A is targeting a conserved region common to both Sug1 and S4, and will consequently bind all 19S ATPases, awaits further investigation.
Regulation of 26S proteasome function by the N-terminus of AdE1A
To assess the biological significance of AdE1A interaction with the proteasome, we assayed two known functions of the 26S proteasome: first, its ability to function as an ATPase; and secondly its ability to degrade ubiquitylated protein substrates. ATPase activities associated with both anti-Sug1 and anti-S4 immunoprecipitates were unaffected, relative to mock-infected cells, upon infection with the N-terminal AdE1A mutant dl1101 that does not bind the 19S RC (Figure 5). In contrast, however, expression of wt AdE1A inhibited ATPase activity associated with anti-S4 immunoprecipitates by ∼20% compared with mock-infected cells, but, intriguingly, did not affect ATPase activity associated with Sug1 (Figure 5). Although ATPase activities associated with Sug1 represent total cellular Sug1 (i.e. proteasomal and non-proteasomal ATPase activities), the in vivo data (Figure 5) compare favourably with the inability of AdE1A to affect intrinsic Sug1-associated ATPase or DNA helicase activities in vitro (Figure 3).
In light of the observation that AdE1A does not completely ablate ATPase activity associated with anti-S4 immunoprecipitates, two possibilities arise. First, that AdE1A is only targeting a subpopulation of the 19S RC pool to completely inhibit its function; or, alternatively, AdE1A targets all the proteasomal pool and only inhibits a subset of 19S RC ATPase activities. The observation that immunodepletion of AdE1A does not immunodeplete S4 (A.S.Turnell, unpublished observations) suggests that AdE1A only targets a proportion of the total cellular proteasomal pool. This raises the intriguing possibility that AdE1A targeting of the 26S proteasome is highly selective.
To determine whether inhibition of ATPase activity, mediated through the N-terminus of AdE1A, translated to functional inhibition of the 26S proteasome, we assayed the ability of HPV16 E6 to target p53 for 26S proteasomal-mediated degradation in the presence of either wt AdE1A or the N-terminal AdE1A mutant lacking the proteasome-binding site. We have demonstrated here that pre-incubation of 26S proteasomes with AdE1A, in a buffered reticulocyte lysate system, substantially reduces the ability of HPV16 E6 to promote the degradation of p53, in the absence of any significant AdE1A degradation (Figure 6A and B). This considerably extends the half-life of p53 and is in agreement with the ability of AdE1A to stabilize p53 in HeLa cells, which normally express low levels of p53 due to expression of functional HPV 18 genomes (Figure 6D). Significantly, the dl1101 mutant that does not bind the proteasome does not affect the ability of HPV16 E6 to target p53 for ubiquitylated-mediated degradation (Figure 6C).
We postulate that the ability of AdE1A to inhibit HPV E6-mediated degradation of p53 resides in its potential to interact directly with 19S components of the 26S proteasome (Figure 3, summarized in Figure 11A and B) and selectively inhibit 19S RC-associated ATPase activity (Figure 5). This reduction in intrinsic 19S-associated ATPase activity renders the 19S RC unable to target ubiquitylated p53 efficiently to the proteolytic core of 20S. The essential requirement for the N-terminal region of AdE1A in this process (Figure 5) is consistent with the findings of other reports implicating the N-terminus in the stabilization of p53 (Chiou and White, 1997; Querido et al., 1997). Pertinently, a detailed mutagenic analysis of 19S ATPase function in yeast has recently suggested that inhibition of one or more of the ATPase subunits is functionally deleterious and related to specific cell phenotypes (Rubin et al., 1998). This suggests that the inhibition of one or more ATPases by AdE1A would have significant ramifications for proteasomal and, consequently, cellular functions. In this respect, it is interesting to note that the AdE1A mutant dl1101, which does not bind the proteasome, is a transformation-defective mutant (Jelsma et al., 1989). Whether AdE1A targets the proteasome to facilitate cellular transformation is not yet known.

Fig. 11. Figure depicting the relationship between AdE1A and the proteasome. (A) Under normal circumstances, polyubiquitin modification of p53 targets p53 for degradation by the 26S proteasome. (B) The N-terminus of AdE1A inhibits 26S proteasomal-mediated degradation of p53, through binding to and inhibiting the 19S RC. (C and D) Ubiquitin-independent degradation of AdE1A through the 26S proteasome is dependent upon phosphorylation of a C-terminal PEST sequence by an olomoucine-sensitive kinase. Whether AdE1A utilizes a chaperone-like molecule (similar to ODC) as depicted in (D), is currently unknown.
Discrete roles for cytoplasmic and nuclear AdE1A–Sug1 complexes?
Intriguingly, AdE1A–Sug1 complexes can be found in both the cytoplasmic and nuclear compartments (Figure 2C). Whether this relates to discrete cellular functions of these two pools remains to be established. It has been determined previously, however, that Mdm2-mediated degradation of p53 is cytoplasmic (Roth et al., 1998a), whereas Ad5 E1B 55K–E4ORF6 mediates degradation of p53 in the nucleus (Roth et al., 1998b). Conceivably, by interacting with both cytoplasmic and nuclear proteasomes, AdE1A could regulate the degradation of cellular substrates differentially. By stabilizing key cellular proteins, AdE1A potentially could manipulate the intracellular levels of proteins that are required for both transcriptional control and S-phase progression. Relevantly, we have shown previously that human cells infected with viruses expressing only Ad12E1A fail to undergo mitosis, presumably to facilitate viral DNA replication in S phase (Grand et al., 1998b). It is perhaps worth noting that mutation of the mts2+ gene (S4 ATPase homologue in Schizosaccharomyces pombe) blocks cells in mitosis (Gordon et al., 1993). Whether AdE1A prevents mitosis of the host cell during a lytic infection by directly inhibiting proteasome function is an intriguing possibility. In terms of viral infection, the interaction of AdE1A with the proteasome might be controlled temporally, to stabilize p53 (and potentially other proteins) to regulate specific cellular effects, prior to their destruction later in the virus life cycle by E1B 55K and E4ORF6.
Phosphorylation of a C-terminal PEST region targets AdE1A for proteasomal degradation, in the absence of ubiquitylation
Lactacystin treatment of Ad5 293 cells stabilizes AdE1A at the protein level (Figure 7), suggesting that under normal circumstances AdE1A is targeted for degradation through the proteasome. Proteins normally are targeted for destruction by the 26S proteasome, by specific polyubiquitylation of lysine residues in the substrate protein (Ciechanover, 1998). AdE1A degradation, however, is not dependent on substrate ubiquitylation (Figure 8). Indeed, comparison of the amino acid sequence of AdE1A from different serotypes indicates that not one lysine residue has been conserved during evolution, supporting the notion that ubiquitylation of AdE1A is not a prerequisite for its degradation. Recent evidence supports the notion that ubiquitylation is not essential for 26S proteasomal-mediated degradation. Ornithine decarboxylase (ODC) is targeted for degradation by antizyme, independently of ubiquitylation (Coffino, 1998), whereas c-Jun and p21CIP1, although substrates for ubiquitylation, can also be degraded by the 26S proteasome independently of ubiquitylation (Jariel-Encontre et al., 1995; Sheaff et al., 2000). Whether AdE1A is targeted to the proteasome via a molecular chaperone, akin to ODC, is unknown.
In an extensive series of experiments, we show here that the phosphorylation of a C-terminal PEST region is a major factor in controlling the stability of AdE1A. We have demonstrated that in the presence of the selective cdk inhibitor, olomoucine, AdE1A has a substantially increased half-life (Figure 9B). This suggests that an olomoucine-sensitive kinase, potentially a cdk, destabilizes AdE1A. However, initial inspection revealed that olomoucine also stabilized the N-terminal AdE1A mutant dl1101, suggesting that residues that lie outside of the proteasome-binding site are important in defining stability. AdE1A has been shown previously to recruit active cyclin–cdk complexes through interaction with the Rb family of proteins (Faha et al., 1993). However, deletion of the Rb-binding site affects neither AdE1A stability nor the ability of olomoucine to stabilize AdE1A (Figure 9B). Thus, recruitment of cdks through Rb does not control AdE1A stability. Presumably, the inability of olomoucine to ablate AdE1A degradation completely suggests that olomoucine-insensitive kinases may also contribute to the regulation of AdE1A stability.
PEST motifs were identified originally in highly labile proteins and are characterized by regions rich in P, E, (D), S and T residues that are often flanked by positively charged amino acids (Rogers et al., 1986). Four putative PEST regions exist in AdE1A and lie between residues 44 and 94 (PEST score: 3.5), 125 and 149 (PEST score: 10.8), 177 and 202 (PEST score: 10.9), and 223 and 244 (PEST score: 13.3). Residues 177–202 lie within CR3; 13S has been shown previously to be less stable than 12S (Spindler and Berk, 1984). Our analyses revealed that the PEST sequence between amino acids 223 and 244 (223RECNSSTDSCDSGPSNTPPEIH244) were crucial in regulating the phosphorylation-dependent degradation of AdE1A (Figure 10A and B). Seven potential phosphorylation sites exist within this region. Five of these, residues S227, S228, S231, S234 and S237, have all been identified previously as phosphorylated residues (cited as S.G.Whalen et al., unpublished in Bayley and Mymryk, 1994), although the kinases that phosphorylate them, and their role in AdE1A stability, have not been investigated. This region of AdE1A is not particularly conserved at the level of amino acid identity. However, PEST-find programs revealed that defined PEST sequences were conserved between serotypes in this region of the molecule (data not shown). Extensive mutational analysis will be needed in order to identify the key residues required in mediating this response in Ad5. The identification of these sites and the kinase(s) responsible for their phosphorylation is under investigation. Intriguingly, the intracellular levels of cyclins are controlled in a highly ordered, temporally controlled cell cycle-dependent manner; cyclin levels are also up-regulated by AdE1A expression (through activation of E2F). This suggests that AdE1A could, in theory, control its own destruction in a cell cycle-dependent manner. Relevantly, active cyclin E–cdk2 complexes previously have been shown to promote the phosphorylation-dependent degradation of the cdk inhibitor, p27Kip1 (Vlach et al., 1997).
Interestingly, the N-terminal AdE1A mutant dl1101 that does not interact with 19S components has a half-life approximately double that of wt AdE1A (Figure 7C). However, the half-life of dl1101 is significantly shorter than that of AdE1A from lactacystin-treated Ad5 293 cells (4 and >12 h, respectively). We present data here indicating that phosphorylation of a C-terminal PEST region is more critical in defining the stability of AdE1A (Figure 10, summarized in Figure 11C and D). Deletion of this region considerably extends the half-life of AdE1A to ∼12 h. This compares favourably with the half-life of AdE1A in Ad5 293 cells in the presence of lactacystin. Two possible explanations could account for the increased stability of the N-terminal mutant. By actually contacting the 19S RC, the N-terminus could bring the C-terminal PEST region into the proximity of the proteasome, which upon its phosphorylation is degraded more efficiently than AdE1A not in contact with the proteasome. Alternatively, taken in the context of viral infection or cell cycle regulation, when AdE1A has fulfilled its function in inhibiting proteasome function, AdE1A is degraded subsequently by the proteasome.
The ability of AdE1A to interact with different components of the proteasome raises fundamental questions pertaining not only to viral regulation of the proteasome but also to the normal cellular control mechanisms governing function of the 26S proteasome. Specifically, does the ability of AdE1A to target multiple 19S components define one cellular activity of AdE1A, or does AdE1A target different 19S components to perform different cellular functions, i.e. to stabilize pools of specific proteins in discrete cellular locations? The observations that AdE1A interacts with both cytoplasmic and nuclear pools of proteasomes, and that AdE1A can only stabilize a subset of proteins that are normally targeted for ubiquitin-mediated degradation (A.S.Turnell, unpublished observations), suggests that there is a high degree of selectivity in AdE1A function. Mutagenic analysis of 19S ATPase function in yeast does suggest that differential cellular regulation of ATPases could define discrete proteasomal functions (Rubin et al., 1998) such that targeting different 19S components through a common binding site could define different cellular activities. Moreover, the observation that AdE1A does not fully inhibit ATPase activity associated with anti-S4 immunoprecipitates in vivo intimates that AdE1A is targeting only a proportion of the proteasomal pool. Understanding the complexity and the biological significance of AdE1A–proteasome interactions in the regulation of both viral replication and cellular transformation is a major focus of our laboratory.
Materials and methods
Cells and viruses
Ad5 E1 human embryo kidney cells (HEK) 293 (Ad5 293) cells, human A549 cells, which derive from a small cell lung carcinoma, and human HeLa cells, which derive from a cervical adenocarcinoma, were grown and maintained in HEPES-buffered Dulbecco’s modified Eagle’s medium (DMEM) containing 2 mM glutamine and 8% fetal calf serum (FCS). Wt Ad5 was from the ATCC. The Ad5 E1A deletion mutants used during the course of this study are summarized in Table I. Typically for viral infections, A549 cells were incubated with the appropriate viruses in serum-free medium, at a multiplicity of 50 plaque-forming units (p.f.u.) per cell, for 2 h at 37°C, with intermittent rocking. Cells were then washed once with serum-free medium, and incubated in complete medium for the appropriate times.
Immunoprecipitation
Cells were lysed in 1 ml of buffer containing 50 mM Tris–HCl pH 7.4, 0.825 M NaCl and 1% NP-40. After occasional agitation on ice for 15 min, cell lysates were sonicated, and cleared by centrifugation. Sug1 was immunoprecipitated from 1 mg of protein, using 2 µg/sample of an anti-Sug1 rabbit polyclonal antibody. AdE1A was immunoprecipitated from 1 mg of protein using 2 µg/sample of either purified M73 or M58 mAb. S4 was immunoprecipitated from 1 mg of protein, using an anti-S4 rabbit polyclonal antibody (dilution 1:100). After 2 h mixing at 4°C, 20 µl of packed protein G–Sepharose (Sigma) was added and mixed for a further 2 h. Immunoprecipitates were then washed five times with 1 ml of 50 mM Tris–HCl pH 7.4, 0.5 M NaCl, 5% sucrose (w/v), 1 mM EDTA and 1% NP-40. For co-precipitation western blotting analyses, immunoprecipitates were washed once more with 50 mM Tris–HCl pH 7.4, 0.125 M NaCl and 1% NP-40, prior to resuspending in the appropriate sample buffer, for separation by PAGE.
In vitro GST pull-downs
Ad5 E1A deletion mutants and the 19S components S4 and S5b were in vitro translated, in either the absence or presence of l-[35S]methionine (>1000 Ci/mmol; Amersham), using the TNT®-coupled wheat germ system (Promega), according to the manufacturer’s instructions. GST, GST–Ad5 E1A mutants and GST–Sug1 were all purified as previously described (Grand et al., 1998a). Typically, to assay in vitro binding capacity, 5 µg of GST conjugates were mixed with 10 µl or equivalent proportions of the respective translation mixes for 1 h at 4°C, in 1 ml of buffer containing 50 mM Tris–HCl pH 7.4, 0.825 M NaCl and 1% NP-40. GST complexes were precipitated using 20 µl of packed, pre-swollen, glutathione–Sepharose beads by end-to-end mixing for 1 h at 4°C. Precipitated complexes were washed five times with 1 ml of 50 mM Tris–HCl pH 7.4, 0.5 M NaCl, 5% sucrose (w/v), 1 mM EDTA and 1% NP-40. Beads were washed once more with 50 mM Tris–HCl pH 7.4, 0.125 M NaCl and 1% NP-40, prior to being resuspended in SDS sample buffer, separated by SDS–PAGE and analysed by fluorography.
SDS–PAGE, alkaline urea gels and western blotting
Samples were solubilized in 9 M urea, 50 mM Tris–HCl pH 7.4 and 150 mM β-mercaptoethanol. Samples were subjected to sonication, and cleared by centrifugation. Protein concentrations were determined by Bradford assay (Bio-Rad). For the immunodetection of antigens, 50 µg protein samples were electrophoresed on 12% polyacrylamide gels, run in the presence of 0.1 M Tris, 0.1 M Bicine, 0.1% SDS. For immunodetection of AdE1A from immunoprecipitated samples, proteins were fractionated on 15% polyacrylamide gels run in the presence of 15 mM Tris, 100 mM glycine and 8 M urea (but in the absence of SDS). Separated proteins were transferred to nitrocellulose filters, prior to western blotting and enhanced chemiluminesence (ECL; Amersham) detection of antigens.
Antibodies
Anti-Ad5 E1A mAb M73 was grown as a mouse hybridoma supernatant, whilst purified M58 anti-Ad5 E1A mAb was purchased from Pharmingen. An anti-Sug1 rabbit polyclonal antibody was raised against a synthetic peptide identical to the C-terminus of the molecule (Wang et al., 1996). An anti-S4 rabbit polyclonal antibody was raised similarly against a synthetic peptide identical to the C-terminus of the molecule. The p53 antibodies used in these studies were a rabbit polyclonal antibody (CM-1) and mouse mAbs 1801 and 421. The mAb against the p32 subunit of the 20S proteasome was from Progen. Anti-HA mAbs, 12CA5 (hybridoma supernatant) and 3F10 (Boehringer Mannheim), were utilized during this study. Anti-α-actinin mAb was from Sigma, whereas anti-lamin B mAb was from Oncogene Science.
ATPase assay
ATPase activities were assayed on anti-S4 and anti-Sug1 immunoprecipitates that had been collected on 20 µl of packed protein G–Sepharose beads. Immunoprecipitates were washed four times with 1 ml of 50 mM Tris–HCl pH 7.4, 0.5 M NaCl, 5% sucrose (w/v), 1 mM EDTA and 1% NP-40. Immunoprecipitates subsequently were washed twice more with 1 ml of 20 mM Tris–HCl pH 7.4. Washed beads were then resuspended in 30 µl of 5 µM ATP (+ 5 µCi/incubation [γ-32P]ATP; 3000 Ci/mmol, Amersham) in a solution containing 20 mM Tris–HCl pH 7.4, 100 µM MgCl2, 2 mM dithiothreitol (DTT). Reactions proceeded for 15 min at 30°C prior to quantification of released phosphate.
Phosphate release assay
For quantifying ATPase activities, released phosphate was measured as described (Shacter, 1984). For each time point, a 20 µl aliquot was taken from the reaction and added to 200 µl of 5 mM silicotungstate, 1 mM H2SO4 in a 1.5 ml microcentrifuge tube. To this, 300 µl of isobutanol:toluene (1:1 v/v) was added, followed by 40 µl of 5% ammonium molybdate solution containing 2 M H2SO4. After vortexing and centrifugation, 150 µl of the upper organic phase was added to 4 ml of scintillant and released phosphate quantified by liquid scintillation counting (Packard).
Transfections
Forty-five minutes prior to transfection, plasmid DNA and LipofectAMINE were mixed together in serum-free DMEM, according to the manufacturer’s instructions (Boehringer Mannheim). Prior to transfection, cells were washed twice in serum-free medium. Transfection was achieved by incubating the cells with the complex of plasmid DNA and LipofectAMINE for 6 h. Post-transfection, cells were washed and then maintained in DMEM, containing 8% FCS and 2 mM glutamine.
Protein purification of His6-tagged ubiquitylated proteins
This was essentially as described (Treier et al., 1994), but with slight modifications. At varying times post-transfection, Ad5 293 cells were lysed in 2 ml of 6 M guanidinium-HCl, 0.1 M sodium phosphate pH 8.0 and 5 mM imidazole per 10 cm dish. The viscosity of the lysates was reduced by sonication. A 6 ml aliquot of cleared lysates was incubated with 200 µl (packed beads) of Ni2+-NTA–agarose (Qiagen) and mixed for 4 h at room temperature by rotation. The slurry was applied to a glass pasteur pippette, sealed with glass wool. The column was then washed successively and bound proteins eluted (Treier et al., 1994). Eluate was precipitated by trichloroacetic acid on ice with 10 µg of bovine serum albumin (BSA) added as carrier. Precipitated proteins were resuspended in SDS sample buffer for further analysis by SDS–PAGE.
DNA helicase assay
A 2 µg aliquot of a 47mer oligonucleotide was end labelled with [γ-32P]ATP (3000 Ci/mmol; Amersham) using polynucleotide kinase (PNK) in PNK buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM spermidine and 1 mM EDTA) for 1 h at 37°C. The 32P-labelled oligonucleotide was then added to 1 µg of M13mp19 single-stranded DNA in annealing buffer (40 mM Tris–HCl pH 7.5, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT), incubated at 95°C for 2 min, then incubated for 15 min at 65°C, and subsequently cooled to 32°C. The annealed duplex substrate was purified from non-annealed 32P-labelled oligonucleotide by loading the sample onto a Sepharose 4B column and eluting with 200 µl aliquots of ice-cold STE (10 mM Tris–HCl pH 7.4, 1 mM EDTA, 0.1 M NaCl). DNA helicase assays were performed using ∼3000 c.p.m. of labelled substrate incubated in 20 mM Tris–HCl pH 8, 2 mM MgCl2, 50 mM KCl, 5 mM ATP, 8 mM DTT, 4% (w/v) sucrose and 20 µg/ml BSA. DNA helicase activity was assessed by separating annealed from unwound oligonucleotide on acrylamide–TBE gels, drying the gel and visualizing by autoradiography. GST–Sug1 and AdE1A were purified as previously described (Grand et al., 1998a).
In vitro degradation assay
Proteins were in vitro translated using the TNT®-coupled rabbit reticulocyte system (Promega) according to the manufacturer’s instructions, in either the absence or presence of l-[35S]methionine (Amersham). Human wt p53 and HPV16 E6 were under the control of the SP6 promoter, whereas Ad5 12S E1A, dl1101-13S and Ad5 13S E1A were all under the control of a T7 promoter. Bacterial Ad12 13S E1A was purified as previously described (Grand et al., 1998a). HPV16 E6-mediated degradation of p53 was performed basically as described (Crook et al., 1991), with minor modifications. In vitro translated p53 and HPV16 E6 (at a ratio of 2:1) were incubated in degradation buffer (25 mM Tris–HCl pH 7.5, 100 mM NaCl and 3 mM DTT) containing additional unprogrammed rabbit reticulocyte lysate to give a final concentration of 40% (v/v) reticulocyte lysate in each incubation. In incubations containing either in vitro translated AdE1A, bacterially expressed AdE1A or ovalbumin, rabbit reticulocye lysates were pre-incubated with AdE1A or ovalbumin for 20 min prior to the addition of p53 and HPV16 E6 (which similarly had been pre-incubated with AdE1A or ovalbumin, at the same ratio according to the amount of reticulocyte lysate present). After the desired time, aliquots were either mixed with SDS sample buffer and subjected to SDS–PAGE, or immunoprecipitated and then subjected to SDS–PAGE. After Coomassie staining and fluorography, gels were dried and subjected to autoradiography.
Protein half-life studies
To inhibit proteasome function in vivo, cells were treated with 10 µM lactacystin (Affiniti). To assess the effect of proteasome inhibition on AdE1A half-life in Ad5 293 cells, cells were treated in the absence or presence of 10 µM lactacystin for 1 h, and then subsequently (in the continued absence or presence of 10 µM lactacystin) treated with the protein synthesis inhibitor anisomycin (100 µM; Sigma) for various times. To measure the half-life of AdE1A in infected A549 cells, cells were infected with the appropriate viruses for 2 h, 18 h prior to incubation with anisomycin (100 µM). To investigate the role of phosphorylation in the control of AdE1A half-life, 100 µM olomoucine or iso-olomoucine (Alexis® Biochemicals) was added to experimental cells 1 h prior to the addition of anisomycin, and maintained in this medium throughout the time course of the experiment.
Cell fractionation
Ad5 293 cells were harvested in ice-cold saline and pelleted by centrifugation. One volume of pelleted cells was resuspended in 5 vols of homogenization buffer (25 mM Tris pH 7.4, 0.32 M sucrose, 10 mM EDTA), incubated on ice for 15 min, prior to lysis of the plasma membrane by 10 strokes of a loose-fitting dounce homogenizer. Intact nuclei were pelleted by centrifugation at 3000 r.p.m. for 10 min. Nuclei were washed once in homogenization buffer and re-pelleted (nuclear fraction). Intact nuclei were resuspended in 2 vols of 20 mM Tris pH 7.4, 1 mM β-mercaptoethanol, and sonicated. Pooled supernatants were centrifuged at 30 000 r.p.m. for 1 h, and the soluble, cytoplasmic fraction was collected. Prior to immunoprecipitation, samples were diluted 1:1 with high salt buffer (20 mM Tris pH 7.4, 1% NP-40, 1.25 M NaCl).
Acknowledgments
Acknowledgements
We are grateful to Professor Betty Moran (Temple University, PA, USA) for generously providing some of the AdE1A mutant viruses used in this study. We would also like to thank Professor Phil Branton (McGill University, Montreal, Canada) for supplying some of the AdE1A constructs used in this study, Professor Dirk Bohmann (EMBL, Heidelberg, Germany) for supplying the ubiquitin constructs, Dr Sally Roberts (CRC Institute for Cancer Studies, the University of Birmingham, UK) for supplying HPV16 E6 and p53 constructs for in vitro translation, and Professor David Lane (University of Dundee, UK) for supplying antibodies against p53. A special thank you to Dr Poonam Taneja (Vanderbilt University, Nashville, TN, USA) for supplying reagents and useful advice on the DNA helicase assay. This work was funded by the CRC (London, UK); P.H.G. is a CRC Gibb Fellow. C.G. was supported by a National Institutes of Health grant GM37009 to Martin Rechsteiner.
References
- Ait Si Ali S. et al. (1998) Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature, 396, 184–186. [DOI] [PubMed] [Google Scholar]
- Asano K., Vornlocher, H.P., Richter-Cook,N.J., Merrick,W.C., Hinnebusch,A.G. and Hershey,J.W. (1997) Structure of cDNAs encoding human eukaryotic initiation factor 3 subunits. Possible roles in RNA binding and macromolecular assembly. J. Biol. Chem., 272, 27042–27052. [DOI] [PubMed] [Google Scholar]
- Bannister A.J. and Kouzarides,T. (1996) The CBP co-activator is a histone acetyltransferase. Nature, 384, 641–643. [DOI] [PubMed] [Google Scholar]
- Baumeister W., Walz,J., Zuhl,F. and Seemuller,E. (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell, 92, 367–380. [DOI] [PubMed] [Google Scholar]
- Bayley S.T. and Mymryk,J.S. (1994) Adenovirus E1A proteins and transformation. Int. J. Oncol., 5, 425–444. [DOI] [PubMed] [Google Scholar]
- Boyer T.G., Martin,M.E.D., Lees,E., Ricciardi,R.P. and Berk,A.J. (1999) Mammalian Srb mediator complex is targeted by adenovirus E1A. Nature, 399, 276–279. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. (1998) The ubiquitin–proteasome pathway: on protein death and cell life. EMBO J., 17, 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A., Shkedy,D., Oren,M. and Bercovich,D. (1994) Degradation of the tumor suppressor protein p53 by the ubiquitin-mediated proteolytic system requires a novel species of ubiquitin carrier protein—E2. J. Biol. Chem., 269, 9582–9589. [PubMed] [Google Scholar]
- Chiou S.K. and White,E. (1997) p300 binding by E1A cosegregates with p53 induction but is dispensable for apoptosis. J. Virol., 71, 3515–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffino P. (1998) Degradation of ornithine decarboxylase. In Peters,J., Harris,J. and Finley,D. (eds), Ubiquitin and the Biology of the Cell. Plenum Press, New York, NY, pp. 411–428. [Google Scholar]
- Crook T., Tidy,J. and Vousden,K.H. (1991) Degradation of p53 can be targeted by HPV E6 sequences distinct to those required for p53 binding and transactivation. Cell, 67, 547–556. [DOI] [PubMed] [Google Scholar]
- Debbas M. and White,E. (1993) Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev., 7, 546–554. [DOI] [PubMed] [Google Scholar]
- de Stanchina E.M. et al. (1998) E1A signalling to p53 involves the p19ARF tumor suppressor. Genes Dev., 12, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kDa protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- Faha B., Harlow,E. and Lees,E. (1993) The adenovirus E1A-associated kinase consists of cyclin E–p33cdk2 and cyclin A–p33cdk2. J. Virol., 67, 2456–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser R.A., Rossignol,M., Heard,D.J., Egly,J.-M. and Chambon,P. (1997) Sug1, a putative transcriptional mediator and subunit of the PA700 proteasome regulatory complex, is a DNA helicase. J. Biol. Chem., 272, 7122–7126. [DOI] [PubMed] [Google Scholar]
- Gallimore P.H., Byrd,P.J. and Grand,R.J.A. (1985) Adenovirus genes involved in transformation. What determines the oncogenic phenotype? Symp. Soc. Gen. Microbiol., 37, 126–172. [Google Scholar]
- Geisberg J.V., Lee,W.S., Berk,A.J. and Ricciardi,R.P. (1994) The zinc finger region of the adenovirus E1A transactivating domain complexes with the TaTa box binding protein. Proc. Natl Acad. Sci. USA, 91, 2488–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M.H., Rubin,D.M., Coux,O., Wefes,I., Pfeifer,G., Cjeka,Z., Baumeister,W., Fried,V.A. and Finley,D. (1998) A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9–signalosome and eIF3. Cell, 94, 615–623. [DOI] [PubMed] [Google Scholar]
- Gorbea C., Taillandier,D., and Rechsteiner,M. (2000) Mapping subunit contacts in the regulatory complex of the 26S proteasome. J Biol. Chem., 275, 875–882. [DOI] [PubMed] [Google Scholar]
- Gordon C., McGurk,G., Dillon,P., Rosen,C. and Hastie,N.D. (1993) Defective mitosis due to a mutation in the gene for a fission yeast 26S protease subunit. Nature, 366, 355–357. [DOI] [PubMed] [Google Scholar]
- Grand R.J.A., Grant,M.L. and Gallimore,P.H. (1994) Enhanced expression of p53 in human cells infected with mutant adenoviruses. Virology, 203, 229–240. [DOI] [PubMed] [Google Scholar]
- Grand R.J.A., Owen,D., Rookes,S.M. and Gallimore,P.H. (1996) Control of p53 expression by adenovirus 12 early region 1A and early region 1B 54K proteins. Virology, 218, 23–34. [DOI] [PubMed] [Google Scholar]
- Grand R.J.A., Gash,L., Milner,A.E., Molloy,D.P., Szestak,T., Turnell,A.S. and Gallimore,P.H. (1998a) Regeneration of the binding properties of adenovirus 12 early region 1A proteins after preparation under denaturing conditions. Virology, 244, 230–242. [DOI] [PubMed] [Google Scholar]
- Grand R.J.A., Ibrahim,A.P., Taylor,A.M.R., Milner,A.E., Gregory,C.D., Gallimore,P.H. and Turnell,A.S. (1998b) Human cells arrest in S phase in response to adenovirus 12 E1A. Virology, 244, 330–342. [DOI] [PubMed] [Google Scholar]
- Grand R.J.A., Turnell,A.S., Mason,G.G.F., Wang,W., Milner,A.E., Mymryk,J.S., Rookes,S.M., Rivett,A.J. and Gallimore,P.H. (1999) Adenovirus early region 1A protein binds to mammalian Sug1—a regulatory component of the proteasome. Oncogene, 18, 449–458. [DOI] [PubMed] [Google Scholar]
- Haupt Y., Maya,R., Kazaz,A. and Oren,M. (1997) Mdm2 promotes the rapid degradation of p53. Nature, 387, 296–299. [DOI] [PubMed] [Google Scholar]
- Honda R. and Yasuda,H. (1999) Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J., 18, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R., Tanaka,H. and Yasuda,H. (1997) Oncoprotein Mdm2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett., 420, 25–27. [DOI] [PubMed] [Google Scholar]
- Howe J.A. and Bayley,S.T. (1992) Effects of Ad5 E1A mutant viruses on the cell cycle in relation to the binding of cellular proteins including the retinoblastoma protein and cyclin A. Virology, 186, 15–24. [DOI] [PubMed] [Google Scholar]
- Jariel-Encontre I., Pariat,M., Martin,F., Carillo,S., Salvat,C. and Piechaczyk,M. (1995) Ubiquitinylation is not an absolute requirement for degradation of c-Jun protein by the 26S proteasome. J. Biol. Chem., 270, 11623–11627. [DOI] [PubMed] [Google Scholar]
- Jelsma T.N., Howe,J.A., Mymryk,J.S., Evelegh,C.M., Cunniff,N.F.A. and Bayley,S.T. (1989) Sequences in E1A proteins of human adenovirus 5 required for cell transformation, repression of a transcriptional enhancer, and induction of proliferating cell nuclear antigen. Virology, 171, 120–130. [DOI] [PubMed] [Google Scholar]
- Jones N. (1995) Transcriptional modulation by the adenovirus E1A gene. Curr. Top. Microbiol. Immunol., 199, 59–80. [DOI] [PubMed] [Google Scholar]
- Kubbutat M.H.G., Jones,S.N. and Vousden,K.H. (1997) Regulation of p53 stability by Mdm2. Nature, 387, 299–303. [DOI] [PubMed] [Google Scholar]
- Lee J.W., Ryan,F., Swaffield,J.C., Johnston,S.A. Moore,D.D. (1995) Interaction of thyroid hormone receptor with a conserved transcriptional mediator. Nature, 374, 91–94. [DOI] [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Li Y., Graham,C., Lacy,S., Duncan,A.M.V. and Whyte,P. (1993) The adenovirus E1A-associated 130kDa-protein is encoded by a member of the retinoblastoma gene family and physically interacts with cyclins A and E. Genes Dev., 7, 2366–2377. [DOI] [PubMed] [Google Scholar]
- Liu F. and Green,M.R. (1994) Promoter targeting by adenovirus E1A through interaction with different cellular DNA-binding domains. Nature, 368, 520–525. [DOI] [PubMed] [Google Scholar]
- Lowe S.W. and Ruley,H.E. (1993) Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev., 7, 535–545. [DOI] [PubMed] [Google Scholar]
- Meijer L. (1995) Chemical inhibitors of cyclin-dependent kinases. Prog. Cell Cycle Res., 1, 351–363. [DOI] [PubMed] [Google Scholar]
- Ogryzko V.V., Schiltz,R.L., Russanova,V., Howard,B.H. and Nakatani,Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- Patel S. and Latterich,M. (1998) The AAA team: related ATPases with diverse functions. Trends Cell Biol., 8, 65–71. [PubMed] [Google Scholar]
- Querido E., Teodoro,J.G. and Branton,P.E. (1997) Accumulation of p53 induced by the adenovirus E1A protein requires regions involved in the stimulation of DNA synthesis. J. Virol., 71, 3526–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan M.P. and Grodzicker,T. (1987) Adenovirus E1A 12S protein induces DNA synthesis and proliferation in primary epithelial cells in both the absence and presence of serum. J. Virol., 61, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond C., Gorbea,C. and Rechsteiner,M. (1997) Specific interactions between ATPase subunits of the 26S protease. J. Biol. Chem., 272, 13403–13411. [DOI] [PubMed] [Google Scholar]
- Rogers S., Wells,R. and Rechsteiner,M. (1986) Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science, 234, 364–368. [DOI] [PubMed] [Google Scholar]
- Roth J., Dobbelstein,M., Freedman,D.A., Shenk,T. and Levine,A.J. (1998a) Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J., 17, 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J., Konig,C., Wienzeck,S., Weigel,S., Ristea,S., and Dobbelstein,M. (1998b) Inactivation of p53 but not p73 by adenovirus type 5 E1B 55-kilodalton and E4 34-kilodalton oncoproteins. J. Virol., 72, 8510–8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin D.M., Coux,O., Wefes,I., Hengartner,C., Young,R.A. Goldberg,A.L. and Finley,D. (1996) Identification of the gal4 suppressor Sug1 as a subunit of the yeast 26S proteasome. Nature, 379, 655–657. [DOI] [PubMed] [Google Scholar]
- Rubin D.M., Glickman,M.H. and Larsen,C.N., Dhruvakumar,S. and Finley,D. (1998) Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J., 17, 4909–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbatini P., Lin,J.-Y., Levine,A.J. and White,E. (1995) Essential role for p53-mediated transcription in E1A-induced apoptosis. Genes Dev., 9, 2184–2192. [DOI] [PubMed] [Google Scholar]
- Schaeper U., Boyd,J.M., Verma,S., Uhlmann,E., Subramanian,T. and Chinnadurai,G. (1995) Molecular cloning and characterization of a cellular phosphoprotein that interacts with a conserved C-terminal domain of adenovirus E1A involved in the negative modulation of oncogenic transformation. Proc. Natl Acad. Sci. USA, 92, 10467–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M., Werness,B.A., Huibregtse,J.M., Levine,A.J. and Howley,P.M. (1990) The E6 oncoprotein encoded by HPV type-16 and type-18 promotes the degradation of p53. Cell, 63, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Seeger M., Kraft,R., Ferrell,K., Bech-Otschir,D., Dumdey,R., Schade,R., Gordon,C., Naumann,M. and Dubiel,W. (1998) A novel protein complex involved in signal transduction possessing: similarities to 26S proteasome subunits. FASEB J., 12, 469–478. [PubMed] [Google Scholar]
- Shacter E. (1984) Organic extraction of Pi with isobutanol/toluene. Anal. Biochem., 138, 416–420. [DOI] [PubMed] [Google Scholar]
- Sheaff R.J. Singer,J.D., Swanger,J., Smitherman,M., Roberts,J.M. and Clurman,B.E. (2000) Proteasomal turnover of p21CIP1 does not require p21CIP1 ubiquitination. Mol. Cell, 5, 403–410. [DOI] [PubMed] [Google Scholar]
- Shenk T. (1996) Adenovirodae: the viruses and their replication. In Fields,B.N. et al. (eds), Fields Virology. 3rd edn. Lippincott-Raven Publishers, Philadelphia, PA, pp. 2111–2148. [Google Scholar]
- Simon R. and Richter,J.D. (1990) The degradation sequence of adenovirus E1A consists of the amino-terminal tetrapeptide Met-Arg-His-Ile. Mol. Cell. Biol., 10, 5609–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler K.R. and Berk,A.J. (1984) Rapid intracellular turnover of adenovirus 5 early region 1A proteins. J. Virol., 52, 706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegenga W.T., Riteco,N., Jochemsen,A.G., Fallaux,F.J. and Bos,J.L. (1998) The large E1B protein together with the E4ORF6 protein target p53 for active degradation in adenovirus infected cells. Oncogene, 16, 349–357. [DOI] [PubMed] [Google Scholar]
- Swaffield J.C., Melcher,K. and Johnston,S.A. (1995) A highly conserved ATPase protein as a mediator between acidic activation domains and the TATA-binding protein Nature, 374, 88–91. [DOI] [PubMed] [Google Scholar]
- Thomas M., Pim,D. and Banks,L. (1999) The role of the E6–p53 interaction in the molecular pathogenesis of HPV Oncogene, 18, 7690–7700. [DOI] [PubMed] [Google Scholar]
- Treier M., Staszewski,L.M. and Bohmann,D. (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell, 78, 787–798. [DOI] [PubMed] [Google Scholar]
- Vlach J., Hennecke,S. and Amati,B. (1997) Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. EMBO J., 16, 5334–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vom Baur E.V. et al. (1996) Differential ligand-dependent interactions between AF-2 activating domain of nuclear receptors and the putative transcriptional intermediary factors mSUG1 and TIF1. EMBO J., 15, 110–124. [PMC free article] [PubMed] [Google Scholar]
- Wang H.G.H., Rikitake,Y., Carter,M.C., Yaciuk,P., Abraham,S.E., Zerler,B. and Moran,E. (1993) Identification of specific adenovirus-E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J. Virol., 67, 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Chevray,P.M. and Nathans,D. (1996) Mammalian Sug1 and c-Fos in the nuclear 26S proteasome. Proc. Natl Acad. Sci. USA, 93, 8236–8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte P., Buchkovich,K.J., Horowitz,J.M., Friend,S.H., Raybuck,M., Weinberg,R.A. and Harlow, E. (1988) Association between oncogene and anti-oncogene: the adenovirus E1A protein binds to the retinoblastoma gene product. Nature, 334, 124–129. [DOI] [PubMed] [Google Scholar]
- Zwickl P. and Baumeister,W. (1999) AAA-ATPase at the crossroads of protein life and cell death. Nature Cell Biol., 1, E97–E98. [DOI] [PubMed] [Google Scholar]