

Abstract
The presence of correctly formed disulfide bonds is crucial to the structure and function of proteins that are synthesized in the endoplasmic reticulum (ER). Disulfide bond formation occurs in the ER owing to the presence of several specialized catalysts and a suitable redox potential. Work in yeast has indicated that the ER resident glycoprotein Ero1p provides oxidizing equivalents to newly synthesized proteins via protein disulfide isomerase (PDI). Here we show that Ero1-Lα, the human homolog of Ero1p, exists as a collection of oxidized and reduced forms and covalently binds PDI. We analyzed Ero1-Lα cysteine mutants in the presumed active site C391VGCFKC397. Our results demonstrate that this motif is important for protein folding, structural integrity, protein half-life and the stability of the Ero1-Lα–PDI complex.
Keywords: chaperones/disulfide bonds/endoplasmic reticulum/protein folding/redox
Introduction
Correct introduction of disulfide bonds into proteins in the periplasm or endoplasmic reticulum (ER) is essential for protein function (Raina and Missiakas, 1997). The oxidizing potential of the ER has been assumed to depend on the specific import of oxidized glutathione (GSSG). Within the ER lumen, GSSG was thought to donate disulfide bonds to reduced polypeptides (Hwang et al., 1992). Recent studies in yeast, however, have shown that the presence of the Ero1p protein is required for the folding of disulfide-containing secretory pathway proteins such as CPY and Gas1p (Frand and Kaiser, 1998; Pollard et al., 1998). Reconstitution of an ero1-1 mutant with the functional protein results in increased levels of GSSG in the ER, indicating that reduced glutathione actually competes with reduced protein substrates for oxidation (Cuozzo and Kaiser, 1999).
In yeast, Ero1p was detected in a complex with protein disulfide isomerase (PDI), which itself can be trapped in a complex with the substrate CPY (Frand and Kaiser, 1999). Ero1p is required to keep PDI oxidized and functional (Frand and Kaiser, 1999). These data suggest that electrons flow in a redox reaction chain through the protein substrate, via lumenal PDI to Ero1p, to an as yet unknown final electron acceptor. This system is remarkably similar to that in the bacterial periplasm, the functional analog of the eukaryotic ER (Frand et al., 2000). In bacteria, a number of periplasmic Dsb proteins have been identified which catalyze disulfide bond formation (for a review see Rietsch and Beckwith, 1998). For example, DsbA introduces disulfide bonds into polypeptides and DsbB reoxidizes DsbA by accepting its excess electrons. These are then fed into the electron transport chain via cytochrome bd or bo oxidase (Bardwell et al., 1991; Kamitani et al., 1992; Bader et al., 1999).
The human homolog of Ero1p, Ero1-Lα, has been cloned and shown to be an ER lumenal glycoprotein that can partially complement the yeast ero1-1 mutant (Cabibbo et al., 2000). Ero1-Lα contains the conserved CXXCXXC motif, which has been postulated to have redox activity because it resembles the CXXC thioredoxin-like motif of eukaryotic and prokaryotic disulfide exchange proteins (Martin, 1995; Aslund and Beckwith, 1999). However, the CXXCXXC motif could represent a metal ion-binding site since these residues are characteristic of FeS cluster proteins (Beinert and Kiley, 1999). Oxidation of Fe2+ to Fe3+ in the Ero1-Lα protein could contribute to the electron flow from reduced newly synthesized proteins to the respiratory chain or to another final electron acceptor. Unlike most other redox-active proteins, Ero1-Lα contains 12 additional cysteine residues. Their contribution to the structure and activity of Ero1-Lα is unknown. Neither yeast nor human Ero1-Lα possesses a recognizable ER retention signal, and the mechanism by which these proteins are localized to the ER is not yet understood.
A number of proteins with CXXC motifs are expressed in the ER of higher eukaryotes. These include PDI and Erp57 (Ferrari and Söling, 1999), which contain two thioredoxin-like domains each. Unlike Ero1-Lα, these PDI-like proteins contain few if any cysteine residues outside their active sites. Over a decade ago, PDI was shown to be essential for the co-translational formation of disulfide bonds (Bulleid and Freedman, 1988). The enzyme can be cross-linked to various ER substrates in mammalian cells (e.g. Roth and Pierce, 1987; Reddy et al., 1996). It was only recently, however, that PDI was detected in direct association with newly synthesized proteins in Semliki Forest virus-infected cells (Molinari and Helenius, 1999). These data, combined with genetic evidence in yeast (Holst et al., 1997; Frand and Kaiser, 1999), support the view that PDI is the primary donor of disulfide bonds to newly synthesized ER proteins, whilst Ero1p maintains PDI in an oxidized state. The function of the other ER resident PDI-like proteins is less well understood. Erp57 forms complexes with the ER lectins calreticulin and calnexin (Oliver et al., 1999) and can participate in the folding of major histocompatibility complex (MHC) class I molecules (Hughes and Cresswell, 1998; Lindquist et al., 1998; Morrice and Powis, 1998). Because it is part of a chaperone complex, Erp57 may act specifically on glycosylated proteins (Oliver et al., 1999).
Here, we provide evidence that the intact CXXCXXC motif is required for the folding, stability and structure of Ero1-Lα, which is unique among the redox-active proteins identified so far in the ER. The lifetime of covalent complexes between oxidized Ero1-Lα and PDI also depends on the cysteines in the putative CXXCXXC active site.
Results
Ero1-Lα has three distinct redox states, including a reduced form
Ero1-Lα was shown by immunofluorescence and cell fractionation to be a resident ER glycoprotein in mammalian cells (Cabibbo et al., 2000). We investigated the folding and disulfide bond formation of this ER protein, since it contains 15 cysteine residues, three of which lie in a putative active site, CXXCXXC. An understanding of the folding properties of the protein will provide insight into its structure and function. As the endogenous expression levels of Ero1-Lα are low, we used a semi-permeabilized cell system (Plutner et al., 1992; Pind et al., 1993). This method faithfully reproduces the assembly of collagen and the peptide loading of MHC class I molecules (Wilson et al., 1995), as well as the folding of influenza virus hemagglutinin (HA) and maturation of human immunodeficiency virus (HIV) gp120 (our unpublished observations).
We reconstituted semi-permeabilized HT1080 cells with an Ero1-Lα in vitro translation reaction, allowing the nascent chains access to an intact and functional ER compartment. The advantage of this method is that Ero1-Lα is the only labeled translation product, which guarantees visualization of all conformations of Ero1-Lα without the need for immunoprecipitation. At various time points after the start of translation, the reaction was stopped by cooling the cells on ice. Trapping of the different disulfide-bonded forms of Ero1-Lα was achieved by blocking free sulfhydryl groups with the rapidly alkylating agent N-ethylmaleimide (NEM).
Figure 1A shows the results obtained in a typical experiment. When cell pellet lysates were treated with 50 mM dithiothreitol (DTT), two translation products were visible, the top one representing R, the glycosylated reduced form of Ero1-Lα (Figure 1A, lanes 5–8). The lower band represented non-glycosylated reduced Ero1-Lα, the majority of which was membrane associated but sensitive to protease treatment (not shown). When the same samples were denatured without reduction, disulfide bonds were kept intact. By non-reducing SDS–PAGE, newly synthesized Ero1-Lα was recovered as a translation product resembling R (Figure 1A, lanes 1–4), which indicated that significant disulfide bond formation did not occur during synthesis. The reduced form gradually partitioned into two distinct, more compact oxidized species containing different sets of disulfide bonds (OX1 and OX2, lanes 2–4). Non-glycosylated reduced Ero1-Lα, present after 15 min of translation (Figure 1A, lane 1), also disappeared with time and most probably misfolded into aggregates. Experiments in which the translation inhibitor ATCA (L-azetidine-2-carboxylic acid) or the elongation inhibitor cycloheximide were included after a 15 min translation gave similar results (not shown). These experiments therefore resembled a pulse–chase approach.

Fig. 1. Folding of Ero1-Lα in semi-permeabilized cells. (A) Ero1-Lα mRNA was translated at 30°C in the presence of ∼2 × 106 semi-permeabilized HT1080 cells for the times given, prior to detergent lysis of the cell pellets in sample buffer and analysis of samples by 7.5% SDS–PAGE. On a non-reducing gel (lanes 1–4), newly synthesized Ero1-Lα started as a reduced glycosylated protein R, then formed OX1 and OX2, as well as a 120 kDa complex (*). R formed a doublet on reducing gels (lanes 5–8). The diamond-shaped symbol designates non-glycosylated protein. Ero1-Lα protein was translated for 60 min and the reaction was stopped on ice in the absence of NEM. Reduced material in sample buffer was left unblocked (lane 9) or treated with excess NEM (lane 10). (B) Ero1-Lα mRNA was translated as in (A), but glycosylated proteins were isolated from the lysed ER pellets on concanavalin A–Sepharose beads, with subsequent analysis by non-reducing (lanes 1–2) and reducing (lanes 3–4) SDS–PAGE. (C) Ero1-Lα mRNA was translated for 60 min as in (A) and the pellets were either mock treated (lanes 1 and 3) or digested with Endo H (lanes 2 and 4) for 1 h at 37°C prior to non-reducing (lanes 1 and 2) or reducing (lanes 3 and 4) SDS–PAGE. The deglycosylated forms showed accelerated mobility. (D) Ero1-Lα mRNA (lanes 1 and 3) and Ero1-Lα-myc mRNA (lanes 2 and 4) were translated as in (A) for 60 min and analyzed on non-reducing (lanes 1 and 2) and reducing (lanes 3 and 4) SDS–PAGE. The tagged protein showed decreased mobility. (E) Translation of Ero1-Lα mRNA was performed in the absence of HT1080 cells. In the absence of ER membranes, folding of the non-glycosylated protein occurred (shown by diamond-shaped symbols) but complexes were not formed.
In addition to monomeric Ero1-Lα, a specific band of 120 kDa appeared after 30 min of translation in the upper part of the non-reducing gel (Figure 1A, lanes 3 and 4, asterisk). This complex, which may represent a covalent dimer of Ero1-Lα and PDI, disappeared upon reduction of the samples with DTT (lanes 7 and 8). In some experiments, including this one, an additional complex of ∼80 kDa was detected. These bands must represent relatively mature Ero1-Lα in a disulfide-bonded complex with another, unlabeled, protein.
To verify that the Ero1-Lα bands in the non-reducing gel contained glycosylated protein, we performed a translation experiment as in Figure 1A. For selective recovery of N-glycosylated proteins from the cell pellet, we incubated pellet lysates with concanavalin A–Sepharose beads. R, OX1, OX2 and the 120 kDa complex were captured by the lectin beads, whereas non-glycosylated Ero1-Lα was not (Figure 1B, lanes 1–2). These forms all resolved as a reduced glycosylated doublet upon reduction with DTT (Figure 1B, lanes 3–4). The binding of the 120 kDa complex to concanavalin A suggested that it too contained glycosylated Ero1-Lα. As an independent test for the presence of sugar residues in the four Ero1-Lα bands, cell pellet lysates from a translation experiment were subjected to digestion by endoglycosidase H (Endo H; Figure 1C, lanes 2 and 4). OX1, OX2, R and the 120 kDa complex all shifted down upon Endo H treatment, confirming that they all contained glycosylated protein (Figure 1C, lane 2).
We confirmed that Ero1-Lα was present in the 120 kDa complex by using a construct expressing Ero1-Lα tagged with the myc epitope at the C-terminus (Figure 1D, lane 2). As expected, the tagged protein and the 120 kDa complex migrated less rapidly but otherwise behaved the same as the untagged protein (compare lanes 2 and 4 with lanes 1 and 3, respectively). The addition of a C-terminal extension did not perturb the folding and glycosylation of Ero1-Lα, nor the formation of the DTT-sensitive complex.
Upon reduction, with or without Endo H treatment, the reduced, glycosylated Ero1-Lα migrated as a doublet (Figure 1A, lanes 5–8; B, lanes 3–4; C, lanes 3 and 4). The doublet represented two forms of Ero1-Lα with different numbers of cysteine residues accessible to NEM. To demonstrate that the doublet was not a result of in-gel disulfide bond formation, the cell pellet was not alkylated (Figure 1A, lane 9) or alkylated after reduction with DTT (Figure 1A, lane 10). When NEM molecules were not bound to Ero1-Lα, the electrophoretic mobility of the glycosylated and non-glycosylated reduced froms of Ero1-Lα increased (Figure 1A, lane 9). When NEM was added after reduction, the mobility of both bands decreased (Figure 1A, lane 10). Unglycosylated Ero1-Lα (Figure 1A, lanes 5–8) had a mobility similar to that of the reduced, completely alkylated protein (Figure 1A, lane 10), confirming that this form did not become oxidized. The top band of the glycosylated doublet (Figure 1A, lanes 5–8) had a mobility similar to that of reduced alkylated glycosylated Ero1-Lα (Figure 1A, lane 10). The bottom band had a mobility between those of non-alkylated and completely alkylated protein, indicative of an intermediate number of free cysteine residues, as expected for folding intermediates and for proteins in which some cysteine residues remain in the reduced state.
The ER is not essential for proper folding of unglycosylated Ero1-Lα
To examine the contribution of the ER to the folding and complex formation of Ero1-Lα, we translated the protein in the absence of any ER membranes (Figure 1E). The samples obtained after various times of translation were analyzed by non-reducing and reducing SDS–PAGE as before. Without ER membranes, non-glycosylated Ero1-Lα showed a similar folding pattern to that of its glycosylated counterpart inside the ER (Figure 1E, lanes 1–4). Reduction of the samples with DTT collapsed the two oxidized species into a doublet again (Figure 1E, lanes 5–8). The 120 kDa complex was absent (lanes 1–4), confirming that an ER resident component was required for this covalent interaction. Instead, some aggregates appeared on the top of the gel, indicative of less efficient folding. It is interesting that the formation of the compact forms of Ero1-Lα does not have an absolute requirement for the ER or for ER chaperones. It is of course possible that components of the reticulocyte lysate substituted for the ER under the in vitro conditions examined here.
Glycosylated Ero1-Lα forms a covalent disulfide-bonded complex with PDI
To determine whether the Ero1-Lα binding partner in the 120 kDa complex was PDI, Ero1-Lα mRNA was translated in the presence of semi-permeabilized DUKX cells (Figure 2). At each time point, samples were blocked with NEM and split into equal halves. One fraction was analyzed directly by SDS–PAGE (Figure 2A); the other half was immunoprecipitated with an anti-PDI serum (Figure 2B). Samples were analyzed under non-reducing (lanes 1–5) and reducing (lanes 6–10) conditions. After 30 min of translation, the stable Ero1-Lα-containing 120 kDa complex was co-immunoprecipitated with PDI. Cross-linking or acid trapping were not required to isolate this alkylated complex, which must contain ‘cold’, unlabeled PDI and labeled Ero1-Lα. The late appearance of the complex suggested that newly synthesized Ero1-Lα cannot interact with mature PDI. Upon reduction, the Ero1-Lα component of the 120 kDa dimer ran as the glycosylated form of ∼65 kDa. Only a minor fraction of unglycosylated Ero1-Lα interacted with PDI, and free Ero1-Lα was not precipitated by the PDI antiserum. Hence, PDI was bound to Ero1-Lα via a covalent disulfide interchain cross link. In contrast, immunoprecipitations targeting calnexin, calreticulin and Erp72 all failed to recognize any labeled products, demonstrating the specificity of the Ero1-Lα–PDI interaction (data not shown). Although Ero1-Lα may interact transiently with some of these proteins, they did not form a covalent complex with the protein as PDI did.

Fig. 2. PDI interacts specifically with Ero1-Lα. Approximately 2 × 106 CHO DUKX cells were semi-permeabilized and incubated at 30°C in an in vitro translation reaction for the times indicated. Cell pellets were lysed in 1% CHAPS and the clarified lysate was split into two portions for direct analysis by SDS–PAGE (A) or immunoprecipitation with anti-PDI serum (B) prior to analysis by non-reducing (lanes 1–5) and reducing (lanes 6–10) 7.5% SDS–PAGE. Anti-PDI immunoprecipitation selectively recovered glycosylated Ero1-Lα in a complex (*).
We concluded that relatively mature Ero1-Lα associated with mature PDI via an interchain disulfide bond. No special measures, apart from alkylation, were needed to detect a significant amount of this complex. Our results discount the possibility that PDI assists the folding of newly synthesized Ero1-Lα. The amount of complex was the same in a DUKX-derived cell line overexpressing PDI by at least 5-fold, suggesting that Ero1-Lα was the limiting component in the complex (not shown).
Ero1-Lα does not form covalent complexes with the newly synthesized cysteine-rich HIV envelope protein
While its complex formation with PDI suggests that Ero1-Lα accepts electrons from PDI, it is formally possible that Ero1-Lα could interact with other cysteine-rich proteins in the ER. We investigated this further by using the gp120 subunit of HIV-1 envelope as a model substrate in a translation experiment with semi-permeabilized cells. HIV-1 gp120 was chosen as a substrate because it has nine disulfide bonds and it folds extremely slowly (A.Land and I.Braakman, in preparation).
We translated the gp120 mRNA in the presence of HT1080 cells (Figure 3A) or HT1080 cells stably overexpressing Ero1-Lα (C3 cells, Figure 3B). The difference in Ero1-Lα expression was confirmed by immunofluorescence (Figure 3A and B, inset). Gp120 was synthesized as a 120 kDa glycoprotein that shifted down with time as its glycans were trimmed by ER glucosidases and mannosidases (Figure 3, lanes 1–8). The small additional mobility shift in the non-reducing gel was caused by folding and oxidation of the protein (Figure 3, lanes 1–4). Deglycosylation of the reduced samples with Endo H uncovered the slow post-translational cleavage of the signal peptide after 120 min, which correlates with the folding state of the protein (lanes 12) (A.Land and I.Braakman, in preparation). Non-productive, non-glycosylated gp120 did not mature, and eventually formed disulfide-bonded aggregates (lanes 4). Control cells (Figure 3A) and the C3 cells overexpressing Ero1-Lα (Figure 3B) yielded identical results. In these assays, overexpression of Ero1-Lα had no effect on gp120 processing or folding. Additional DTT-sensitive higher molecular weight bands that might correspond to Ero1-Lα–gp120 complexes were absent. Moreover, co-translation of HIV-1 gp120 or influenza virus HA mRNA with Ero1-Lα mRNA in the presence of semi-permeabilized cells also failed to produce any higher molecular weight complexes (not shown). We concluded that overexpressed Ero1-Lα did not interact covalently with these newly synthesized disulfide-containing proteins in the ER.

Fig. 3. Ero1-Lα does not interact with HIV-1 gp120 or alter its maturation rate. Approximately 2 × 106 HT1080 cells (A) and Ero1-Lα-overexpressing HT1080 cells (B) were used as a source of ER in an in vitro translation of gp120 mRNA. Aliquots at given time points were incubated without (lanes 1–8) or with (lanes 9–12) Endo H to remove N-linked glycans. By 120 min, folding and signal peptide cleavage had occurred at the same rate in both cell lines. No additional disulfide-linked complexes had formed. Reduced, non-glycosylated gp120 was also observed (diamond-shaped symbol). The inset shows the difference in ER-specific Ero1-Lα expression between the two cell lines by indirect immunofluorescence using antibody A29.
Ero1-Lα CXXCXXC mutants display aberrant folding
If Ero1-Lα were a functional redox-active protein, like the CXXC-containing thioredoxin family, the CXXCXXC motif might contain active site cysteines (Martin et al., 1993). These cysteines therefore should contribute to Ero1-Lα function but not to protein structure. To determine whether the CXXCXXC motif was involved in PDI binding, the behavior of three cysteine to alanine mutants and their myc-tagged counterparts (C391A, C394A and C397A) was analyzed after translation in the presence of semi-permeabilized HT1080 cells.
Figure 4 reveals that each mutant had very different properties. Glycosylated C391A (Figure 4A, lanes 1–4) ran as the reduced form R. OX1 and OX2 could not be detected. Instead, this mutant existed as a smear of intermediates, which increased in compactness after 60 min of translation. The degree of ‘smeariness’ of OX1 and OX2 had some variation between experiments. In contrast, C394A (Figure 4A, lanes 5–9) had an oxidation pattern almost identical to that of wild-type Ero1-Lα (compare with Figures 1A and 2A). It formed R, OX1 and OX2 (Figure 4A, lanes 5–9). C397A showed a third type of behavior (Figure 4A, lanes 10–14). This mutant did form R and OX1, but it became trapped as OX1 and could hardly progress to OX2. Upon reduction, the intermediates for each mutant were resolved as glycosylated and non-glycosylated reduced Ero1-Lα (Figure 4B). This indicated that these structures were a genuine collection of folding intermediates rather than degradation products and that glycosylation was not altered by the mutations. The same folding behavior was also exhibited by the tagged CXXCXXC mutants (not shown). The three cysteine mutants had a remarkably diverse range of oxidized forms: both C391 and C397 contributed substantially to the structural integrity of oxidized Ero1-Lα whereas C394 did not. We suggest that if the CXXCXXC motif does mediate Ero1-Lα activity, it must function in conjunction with other cysteine residues within the protein. Mutations at a self-contained active site would not be expected to cause such large perturbations in the protein’s architecture.

Fig. 4. Ero1-Lα CXXCXXC mutants have distinct folding patterns. Approximately 2 × 106 HT1080 cells were used per translation to reconstitute the folding of mutants C391A (lanes 1–4), C394A (lanes 5–9) and C397A (lanes 10–14). On non-reducing gels (A), C391A ran as a smear, C394A behaved similar to wild-type protein and C397A was arrested in OX1. Each mutant formed the 120 kDa complex (*). On reducing gels (B), glycosylated (R) and non-glycosylated (diamond-shaped symbol) Ero1-Lα were recovered in each case (lanes 1–14).
To our surprise, each of the mutants still formed the complex with PDI (Figure 4A, lanes 3, 4, 7–9, 13 and 14). Although C391A was incapable of folding into compact oxidized forms, it did form oligomers, albeit with more fuzziness and a lower mobility than that of wild-type Ero1-Lα and C394A. The C397A complex had an intermediate mobility. These positions in the gel corresponded to the electrophoretic mobility of the most prominent redox intermediate of each mutant, suggesting that the most compact form of Ero1-Lα accumulated in a covalent complex with PDI.
OX2 is the stable native state and the CXXCXXC motif governs ERO1-Lα stability
To confirm our findings, we examined the folding of Ero1-Lα in living cells using pulse–chase experiments (Figure 5). Myc-tagged Ero1-Lα constructs were used because the affinity of the anti-Ero1-Lα serum (A29) used for immunofluorescence was too weak for quantitative immunoprecipitations. The myc tag has no effect on Ero1-Lα folding or PDI complex formation in semi-permeabilized cells, and can be considered relatively conformation insensitive (Figure 1D). HeLa cells were infected with a recombinant vaccinia virus carrying a bacterial T7 polymerase gene (VVT7). Subsequent transfection with the myc-tagged Ero1-Lα construct behind the T7 promoter ensured high level expression of the protein. At 5 h post-infection, the cells were radioactively pulse-labeled for 10 min, chased for the indicated times and lysed in detergent. The clarified lysates were subjected to immunoprecipitation with anti-myc 9E10 monoclonal antibody (mAb) and analyzed on non-reducing (Figure 5A, lanes 1–4) and reducing SDS–PAGE (lanes 5–8).
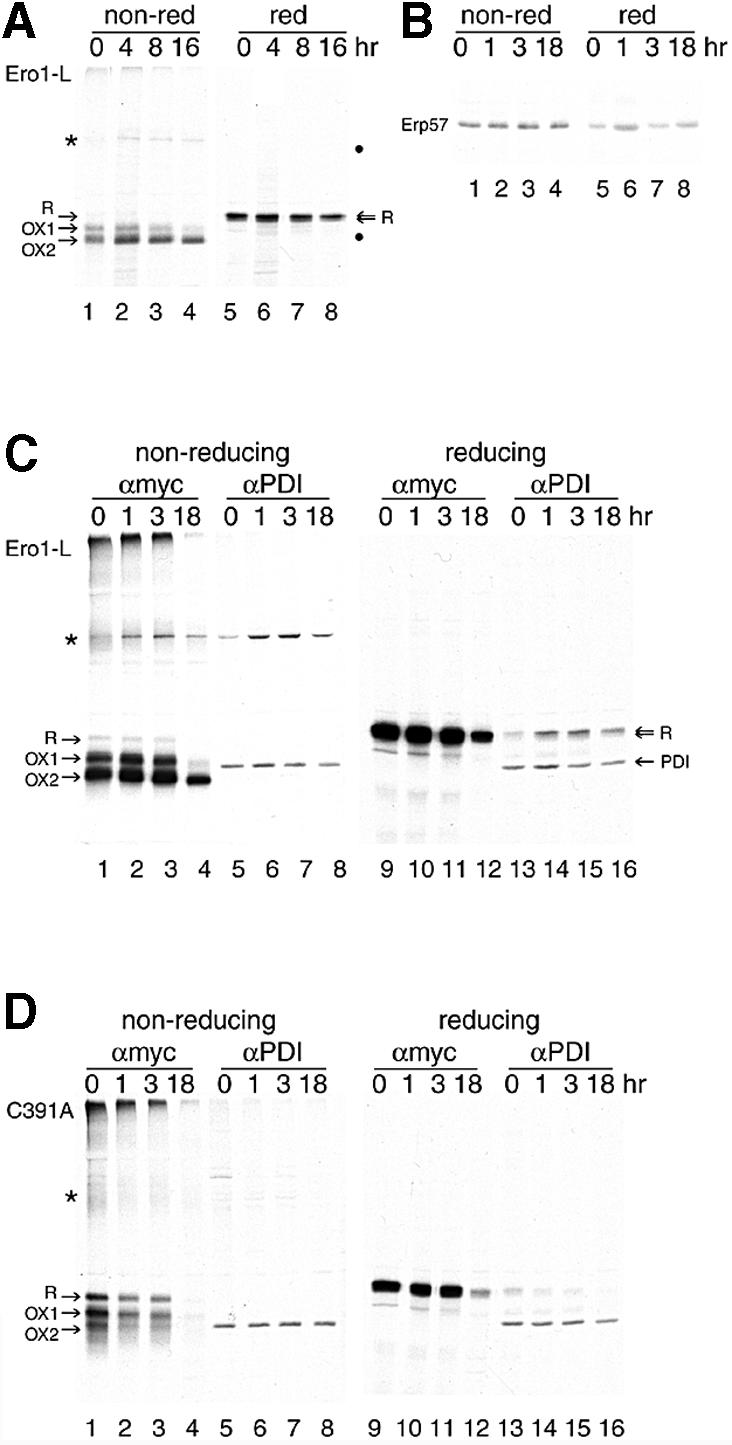
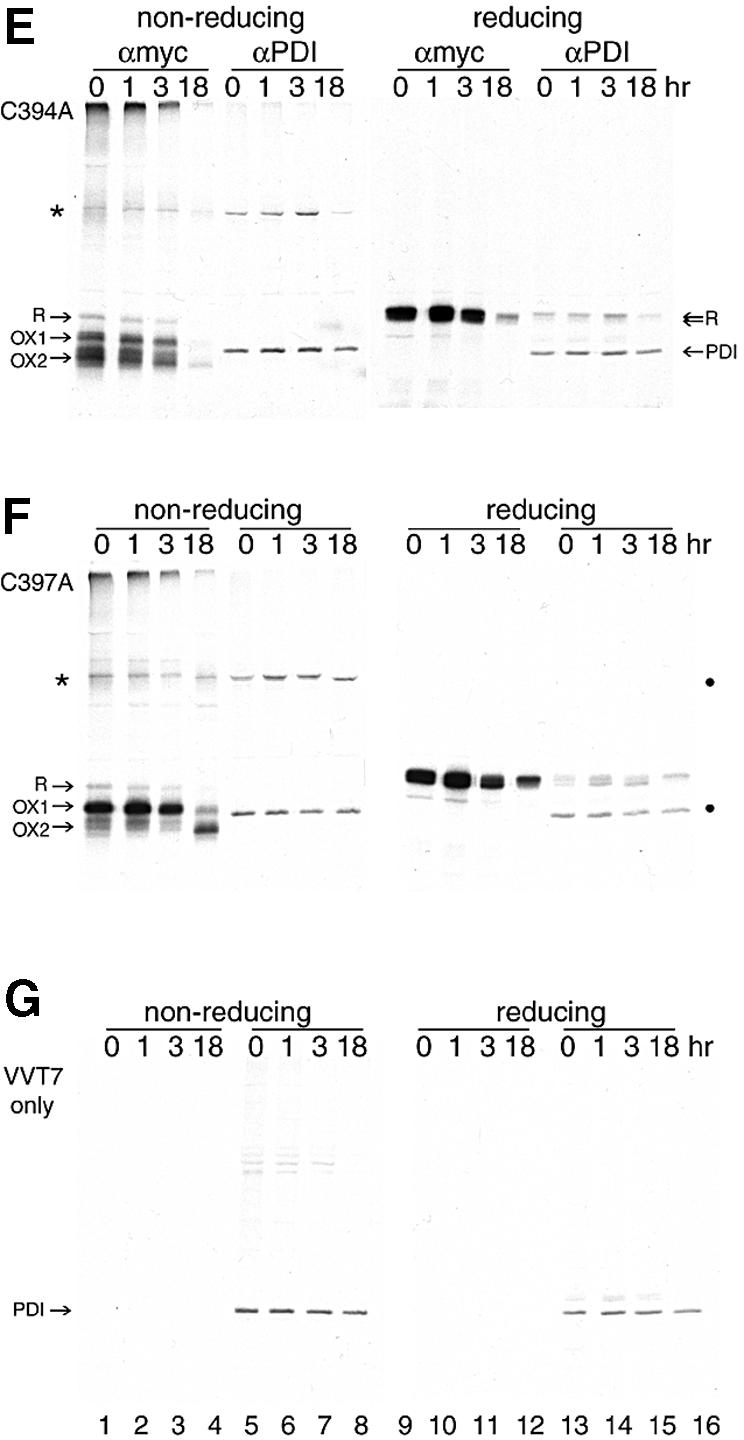
Fig. 5. OX2 is the stable state of Ero1-Lα in the living cell. HeLa cells, grown in 6 cm dishes, were infected with VVT7 (A and C–G) and mock transfected (G) or transfected with pcDNA3.1 encoding (A and C) Ero1-Lα-myc, (D) C391A-myc, (E) C394A-myc or (F) C397A-myc. In (A), immunoprecipitation was with the anti-myc mAb 9E10 (lanes 1–4 non-reducing and lanes 5–8 reducing). In (C–G), immunoprecipitation was with either the anti-myc mAb 9E10 (lanes 1–4 non-reducing and lanes 9–12 reducing) or anti-PDI sera (lanes 5–8 non-reducing and lanes 13–16 reducing). 9E10 specifically isolated Ero1-Lα R, OX1 and OX2, along with the Ero1-Lα–PDI complex (*). Anti-PDI sera immunoprecipitated PDI in all cases, and the Ero1-Lα–PDI complex (*) from Ero1-Lα-myc, C391A-myc, C394A-myc and C397A-myc, but not the mock-transfected cells. In (B), Erp57 was immunoprecipitated from uninfected, untransfected HeLa cells after a pulse–chase and analyzed on non-reducing (lanes 1–4) and reducing (lanes 5–8) SDS–PAGE. ERp57 had no disulfide-bonded intermediates.
Within the 10 min pulse time, the wild-type Ero1-Lα protein formed the same bands as in the semi-permeabilized cells: R, OX1, OX2 and a band resembling the 120 kDa Ero1-Lα–PDI complex (Figure 5A, lanes 1–4). In addition, some aggregates appeared on the top of the separating gel, indicative of misfolding of a fraction of newly synthesized Ero1-Lα. This is not uncommon in the efficient expression system we used. Whereas OX1 and OX2 were not formed until after synthesis in vitro (Figures 1 and 2A), they were the most prominent bands by the end of the pulse-labeling, indicating that oxidation occurred much more rapidly in vivo. The various Ero1-Lα structures were persistent, but after 8–16 h chase the most compact OX2 form of Ero1-Lα predominated, in addition to the 120 kDa band (Figure 5A, lanes 3 and 4). We therefore regarded OX2 as the most stable state of Ero1-Lα.
A precursor–product relationship between the different Ero1-Lα intermediates was not apparent, not even during the first few hours of chase. The fate of OX1 is not yet known, but it did not change when cells were treated with the proteasome inhibitor I (Traenckner et al., 1994; data not shown). Loss of the myc epitope was unlikely because it is not conformation sensitive. Experiments conducted in the absence of vaccinia virus, using a cytomegalovirus (CMV) promoter, gave similar results, albeit with lower expression (not shown).
Next, we compared the folding pattern of Ero1-Lα with the three CXXCXXC cysteine mutants in living cells. Lysates were subjected to immunoprecipitation with either anti-myc 9E10 mAb (Figure 5C–F, lanes 1–4 and 9–12) or anti-PDI serum (Figure 5C–F, lanes 5–8 and 13–16) and examined by non-reducing (lanes 1–8) and reducing (lanes 9–16) SDS–PAGE. Cell lysates were also analyzed to verify equal labeling. The 120 kDa band was immunoprecipitated with both antibodies. The molecular weight of the complex, its resistance to SDS and its dissociation into PDI and Ero1-Lα upon reduction with DTT (Figure 5C, lanes 13–16) indicated that it was indeed an Ero1-Lα–PDI disulfide-linked dimer. PDI itself was labeled in this experiment and ran between Ero1-Lα OX1 and OX2 (lanes 5–8 and 13–16). Upon reduction of the immunoprecipitated samples, Ero1-Lα was released from the 120 kDa complex but the amount of PDI retrieved remained the same (Figure 5C, compare lanes 5–8 with lanes 13–16). This indicates that in intact cells, newly synthesized Ero1-Lα was bound to pre-existing unlabeled PDI and not to newly synthesized PDI. Clearly, PDI itself had no detectable disulfide-mediated folding pathway when compared with Ero1-Lα under non-reducing conditions, despite its two putative structural cysteines in addition to the CXXC motif (Figure 5C, lanes 5–8). Similarly, the PDI-like protein Erp57 had no detectable disulfide-mediated folding pathway either (Figure 5B, lanes 1–4), indicating that the oxidation pattern of Ero1-Lα was unique amongst these proteins.
The myc-tagged Ero1-Lα CXXCXXC mutants behaved initially as seen in Figure 4, with C391A being diffuse, C394A being similar to wild-type and C397A being trapped in OX1 (Figure 5D–F, lanes 1–3). After an 18 h chase, however, both C391A and C394A were unstable and were almost completely degraded or secreted, whereas the majority of C397A eventually reached OX2 and maintained its interaction in a complex with PDI (Figure 5F, lane 4). These data clearly demonstrate that the CXXCXXC mutations severely compromised the stability of Ero1-Lα. In the case of C391A, the complex with PDI was considerably weaker and smeared out (Figure 5D, lanes 1 and 5), corresponding to the diffuse nature of its oxidized forms. The fact that the ‘smeared’ form of C391A was found in the 120 kDa complex and the relative absence of R in wild-type and the other two mutants, strongly suggested that it was oxidized rather than reduced Ero1-Lα that interacted with PDI. Importantly, the reduced R form of Ero1-Lα was more prominent in the C391A mutant (Figure 5D, lanes 1–3) than in wild-type Ero1-Lα (Figure 5C, lanes 1–3) and the C394A and C397A mutants (Figure 5E and F, respectively, lanes 1–3). This pointed to a role for C391 in the oxidation of Ero1-Lα.
The complex between PDI and the Ero1-Lα mutant C391A was lost (Figure 5D, lanes 5–8 and 13–16), whereas mutant C394A, despite being unstable, still maintained the complex with PDI (Figure 5E, lanes 5–8 and 13–16). A close examination of mutant C397A revealed important details about the nature of the PDI-bound form of Ero1-Lα (Figure 5F). C397A was present as OX1 during the first 3 h of chase (lanes 1–3) and this form could bind to PDI (lanes 5–7 and 13–15). After the overnight chase, only OX2 remained. OX2 could also bind PDI (Figure 5F, lanes 4, 8 and 16). OX2 binding resulted in a shift down in the migration of the Ero1-Lα–PDI complex (compare lane 7 with 8) and enrichment in the upper reduced band of the Ero1-Lα doublet released from the complex (Figure 5F, lanes 15 and 16). In both wild-type Ero1-Lα and mutant C394A, the top band in the reduced Ero1-Lα doublet was always enriched in the PDI complex (Figure 5C and E, lanes 13–15, and Figure 2B, lanes 8–10). Surprisingly, the most compact state of Ero1-Lα had the largest number of cysteines available to bind NEM (Figure 1A, lanes 9 and 10). This feature may prove to be important in Ero1-Lα-mediated electron transfer processes.
Control HeLa cells infected with only VVT7 showed similar PDI labeling to the infected, transfected cells (Figure 5G, lanes 13–16, and lysates not shown). In the untransfected cells, no proteins were immunoprecipitated with 9E10 (lanes 1–4), whereas PDI (but not the Ero1-Lα–PDI complex) was immunoprecipitated with the anti-PDI serum (lanes 5–8 and 13–16). Some endogenous DTT-sensitive PDI complexes of unknown composition were also immunoprecipitated. The expression level of PDI was unaffected by the infection/transfection procedure.
Taken together, the results of Figures 4 and 5 imply that the Ero1-Lα–PDI interaction did not require any single cysteine in the putative active site. C391 had the most striking effect upon the maintenance of the complex, and all three CXXCXXC cysteines were important for the folding of Ero1-Lα. The folding process may be affected because these cysteines form essential transient disulfide bonds in Ero1-Lα folding intermediates, or because they are part of a native disulfide bond. The possible involvement of other cysteines in the structure and function of Ero1-Lα is intriguing. A full analysis of all the Ero1-Lα cysteine residues by mutagenesis coupled to in vitro and in vivo oxidation assays is required to resolve this issue fully. Nevertheless, it is clear that C391, C394 and C397 make an unexpectedly substantial contribution to the structure, stability and function of oxidized Ero1-Lα.
Discussion
To address the question of Ero1-Lα folding in the ER, we have used in vitro translation with semi-permeabilized cells and pulse–chase approaches. The combination of these two techniques has enabled us to dissect early events that occur too rapidly to be seen in living cells, and late events that cannot be followed with semi-permeabilized cells. The behavior of Ero1-Lα was quite unlike that of PDI and Erp57, whose active site CXXC motifs do not contribute to the gross structure of either protein (Figure 5; Ferrari and Söling, 1999). Glycosylated monomeric Ero1-Lα appeared as three bands on a non-reducing gel: the reduced form R, the oxidized form OX1 and the more compact form OX2 (Figures 1A and B, and 5). The oxidized forms in particular did not show a clear precursor–product relationship in vivo, suggesting that they were in equilibrium with each other. If so, the accumulation of exogenous Ero1-Lα in the OX2 native state after a long chase implies that older Ero1-Lα existed mostly as a more reduced but nonetheless more compact structure. The long half-life of Ero1-Lα, and in particular the OX2 form, is compatible with a role for the protein in ER homeostasis. Our experiments indicated that the CXXCXXC motif is crucial for determining the longevity of Ero1-Lα.
Ero1-Lα specifically associated with PDI through an intermolecular disulfide bond. Whereas acid precipitation is required to trap Ero1-Lα–PDI complexes in yeast (Frand and Kaiser, 1999), we found that only alkylation with NEM was necessary in mammalian cells. Straightforward immunoprecipitation was sufficient to visualize the covalent Ero1-Lα–PDI complex in a variety of transfected cells and in an in vitro translation system (Figures 1, 2 and 5). The difference between yeast and mammalian cells suggests a stronger or longer-lived complex between human Ero1-Lα and PDI. We are certain that Ero1-Lα was in the complex, because the mobility of the complex decreased when the myc-tagged form of Ero1-Lα was used, because its mobility increased when glycans were removed and because Ero1-Lα was the only labeled protein present in vitro. The presence of PDI in the complex was clearly demonstrated with the clean and specific immunoprecipitation of PDI and the complex.
The PDI molecules with which Ero1-Lα interacted were already present in the ER in vitro, suggesting a mature PDI protein in the complex. The interaction began after the formation of Ero1-Lα OX2, indicating that PDI was not acting as a folding enzyme for newly synthesized Ero1-Lα. Although it is possible that PDI interacts as a chaperone with misfolded Ero1-Lα molecules (Gillece et al., 1999), our data do not support this interpretation: the Ero1-Lα–PDI interaction started late and was stronger with wild-type Ero1-Lα than with the Ero1-Lα mutants (Figure 5).
At least two other DTT-sensitive bands were also detected in immunoprecipitates of Ero1-Lα (Figure 5) as well as in semi-permeabilized cells (Figure 1), but we have not yet elucidated the identity of these Ero1-Lα-binding partners. Antibodies against calnexin, calreticulin, Erp72 and Erp57 could not immunoprecipitate these complexes (not shown). In a functionality test, overexpression of Ero1-Lα had no effect on the maturation of the model substrate protein HIV gp120, lending support to the idea that Ero1-Lα itself was not a chaperone (Figure 3). In a variety of experiments, Ero1-Lα, when translated with semi-permeabilized cells in vitro, did not interact covalently with newly synthesized co-translated PDI, gp120, influenza virus HA or Erp72 (data not shown).
It is interesting that unglycosylated Ero1-Lα attained OX1-like and OX2-like states in the absence of an ER environment (Figure 1E). Although this situation is unlikely to be encountered in vivo, it suggests that Ero1-Lα structure can be independent of chaperone activity and glycosylation status, although in the presence of ER membranes the unglycosylated portion of Ero1-Lα did not fold properly. Chaperone-independent folding of Ero1-Lα, however, would be a useful feature for a protein whose expression may be required for homeostasis of the ER lumen.
The discrete steps in the oxidation of Ero1-Lα are highly suggestive of two alternative structures that may be gated by domain or loop closure. Such a proposal is not without precedent: the disulfide-dependent folding pathway of influenza HA in the ER, for example, proceeds via two discrete intermediates, IT1 and IT2 (Braakman et al., 1991, 1992). These intermediates correspond to the formation of two loops in the protein which have been observed in a number of crystal structures of the HA molecule (Wilson et al., 1981; Bullough et al., 1994; Chen et al., 1998; I.Braakman and A.Helenius, in preparation). To our surprise, some structural features of Ero1-Lα were affected directly by the removal of cysteines in the CXXCXXC motif of Ero1-Lα. The three cysteine mutants displayed notably different folding patterns, and diminished stability compared with wild-type Ero1-Lα. The CXXCXXC motif was crucial for the compact structure of Ero1-Lα, and is possibly engaged in native disulfide bond formation. The CXXCXXC cysteine residues may be part of a structural disulfide bond, or they may be important during the folding process of Ero1-Lα. Not one of these cysteines is absolutely required for formation of the PDI complex, suggesting that either another cysteine in the molecule has this task, or that more than one active site cysteine can form the complex. All the possible cysteine mutants of both Ero1-Lα and PDI will be analyzed in order to discern the precise nature of these interactions.
The human Ero1-Lα gene can partially complement the phenotype of the yeast temperature-sensitive ero1-1 strain. In this assay, the C391A mutant has an effect similar to that of wild-type Ero1-Lα, whereas neither growth at 37°C nor DTT resistance can be restored by the C394A and C397A mutants (Cabibbo et al., 2000). The data we obtained from Figure 5 add considerably to the interpretation of these results. The finding that C394A and C397A can bind to PDI but cannot complement the yeast mutant implies that the function of human Ero1-Lα involves more than just binding to PDI. Cysteines 394 and 397 may be important for the release of Ero1-Lα from PDI or for its recruitment to an as yet unknown electron acceptor. The complementation of the ero1-1 strain by the C391A mutant indicates that either a very low amount or rapid cycling of the Ero1-Lα–PDI complex may be sufficient to support partial protein function, or that the human gene complements through an indirect effect on the yeast mutant Ero1p. It will therefore be interesting to analyze the behavior of the human Ero1-Lα gene and the CXXCXXC mutants in a yeast ero1 knock-out strain.
The Ero1-Lα CXXCXXC motif evidently controls the half-life of the protein, the stability of its interaction with PDI and its structure. Considering the remarkable and possibly diverse properties of this motif, an additional role in metal binding cannot be excluded.
We made the unexpected observation that the most compact form of Ero1-Lα also contained the highest number of free cysteines, implying that more than one cysteine residue participated in the redox process of Ero1-Lα. It is possible that the three forms of Ero1-Lα may contribute to a redox cycle. Oxidized glycosylated Ero1-Lα may be recruited to PDI, where it can donate disulfides (accepting electrons), before being released from PDI in a more reduced form, which does not necessarily correspond to R or OX1. The more reduced form could then be re-oxidized by an as yet unknown ER electron acceptor, possibly in the ER membrane. This model would be consistent with the biochemical evidence available in yeast (Frand and Kaiser, 1999) and with studies performed with the Dsb proteins of the bacterial periplasm (Bader et al., 1999). Unlike PDI, Ero1-Lα contains 12 additional cysteine residues whose functions are unknown. Interestingly, the multimembrane-spanning bacterial periplasmic protein DsbD (also a CXXC protein) has a disulfide-rich amino acid sequence similar to Ero1-Lα (Missiakas et al., 1995). DsbD is involved in providing reducing (rather than oxidizing) equivalents to the periplasmic space in order to maintain DsbC in a reduced state (Joly and Swartz, 1997). To function, DsbD has been postulated to shuttle electrons from the CXXC motif along its length by a cascade of intramolecular disulfide exchange reactions that alter the protein’s conformation and allow the periplasm to communicate with thioredoxin in the cytosol (Stewart et al., 1999). Although Ero1-Lα does not pass the membrane (Pagani et al., 2000), one might speculate that a disulfide-mediated cascade along the length of the protein, accompanied by conformational changes, could re-oxidize Ero1-Lα. In vitro reconstitution experiments using purified mammalian ER components should allow us to address this issue fully. From an evolutionary viewpoint, it is intriguing that prokaryotes and eukaryotes may have found convergent solutions to the problem of protein folding in an oxidizing environment.
Materials and methods
Cell lines and virus
The Chinese hamster ovary-derived cell line DUKX was a kind gift from Drs A.Dorner and R.J.Kaufman, then at Genetics Institute, Cambridge, MA, and was maintained in α minimal essential medium (MEM) supplemented with nucleosides and 8% fetal calf serum (FCS) (Dorner et al., 1990). The human fibrosarcoma cell line HT1080 was maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FCS, and the human cervical carcinoma cell line HeLa was maintained in MEM supplemented with non-essential amino acids and 10% FCS. All cell lines were kept at 37°C under 5% CO2.
The recombinant vaccinia virus expressing the T7 polymerase (VVT7) was a kind gift from Dr J.Rose, Yale University, New Haven, CT (Fuerst et al., 1986; Rose et al., 1991) and was propagated in HeLa cells. Infections were performed with a multiplicity of infection (m.o.i.) of ∼5 infectious virus particles per cell.
Antibodies
The polyclonal A29 anti-peptide serum was generated as follows: a peptide was synthesized corresponding to the 20 C-terminal residues of the Ero1-Lα protein, incorporating an additional N-terminal cysteine residue for cross-linking purposes (CRISTSVKELENFRNLLQNIH). Peptide purity was verified by HPLC and mass spectrometry and the peptide was cross-linked to keyhole limpet hemocyanin (KLH) using the immunoject system (Pierce). New Zealand White rabbits were immunized with 100 µg of conjugated peptide emulsified in montanide per injection. Plasma was collected at regular intervals by plasmaphoresis.
The polyclonal anti-PDI antiserum was generated by challenging New Zealand White rabbits with 100 µg of PDI in montanide per immunization. A fraction of the PDI protein used for immunization was a kind gift from Dr P.Klappa, University of Canterbury, UK. It had been purified as described previously (Lambert and Freedman, 1983).
The polyclonal anti-Erp57 serum was a kind gift from Dr S.High (University of Manchester, UK). The anti-myc murine monoclonal antibody 9E10 was used to detect myc-tagged Ero1-Lα complexes (Chan et al., 1987).
DNA constructs
The construction and sequencing of the Ero1-Lα cDNA behind the T7 promoter in pcDNA3.1 and the generation of the C391A, C394A and C397A mutations has been described (Cabibbo et al., 2000). All mutations were confirmed by DNA sequencing. The gp120 cDNA sequence expressed from pBluescript (pBS-gp120) was derived from an HIV LAI genomic SalI–XhoI fragment (a kind gift from Dr B.Berkhout). The gp120 stop codon was introduced by PCR, and tat and vpu coding sequences were removed.
Infections/transfections
HeLa cells cultured in 6 cm dishes were infected with an m.o.i. of ∼5. One hour after infection, cells were transiently transfected with 2 µg of pcDNA3.1-Ero1-Lα (or mutant cDNAs), mixed with 16 µl of lipofectace (Gibco-BRL) according to the manufacturer’s instructions and analyzed for protein expression 4 h later. Transfections/infections using lipofectAMINE (Gibco-BRL) or CV1 cells gave identical results.
The C3 cell line, which constitutively overexpresses Ero1-Lα, was generated by transfection of the pcDNA3.1-Ero1-Lα vector into HT1080 cells, using the calcium phosphate precipitation method. Cells were maintained in the presence of 500 µg/ml G418, which was omitted during experimental procedures. The overexpression of Ero1-Lα in the C3 line was verified by immunofluorescence before use.
Immunofluorescence
HT1080 and C3 cells were grown on coverslips, fixed for 5 min with methanol at –20°C and blocked in 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) containing calcium and magnesium, prior to incubation with a 1:1000 dilution of the A29 antiserum. The antigen was visualized using Cy3-conjugated goat anti-rabbit antibodies (Americium) at 1:100 dilution after washing with 0.2% BSA in PBS with calcium and magnesium.
Semi-permeabilized cell assays
The pcDNA3.1–Ero1-Lα construct was first linearized with EagI. pBS–gp120 was linearized with XhoI. T7 RNA polymerase-driven transcription reactions were performed using the Promega riboprobe system in accordance with the manufacturer’s instructions. In vitro translations using semi-permeabilized cells as an ER source were performed essentially as described (Wilson et al., 1995). Briefly, the cells in a 90% confluent T75 flask were permeabilized selectively with 6 µg of digitonin in 6 ml of KHM buffer (110 mM KOAc, 2 mM MgOAc, 20 mM HEPES pH 7.2) and washed with buffer H (90 mM KOAc, 50 mM HEPES pH 7.2). Endogenous mRNA was destroyed using micrococcal nuclease, which was activated with 1 mM CaCl2. Nuclease activity was inactivated subsequently by quenching calcium with EGTA. Washed cells were reconstituted with rabbit reticulocyte lysate (Promega) supplemented with 50 mM KCl, 0.04 mM amino acids and 10 µCi of in vitro 35S-labeling mix (Amersham, UK) per 50 µl reaction (four samples). Translation was initiated at 30°C upon introduction of the relevant mRNA in the complete absence of reducing agents and exogenous glutathione. Translation was stopped by quenching aliquots with 20 mM NEM on ice, to trap free sulfhydryl groups. Proteins were taken up into sample buffer and analyzed non-reduced and reduced (with 50 mM DTT) on 7.5% SDS–PAGE gels.
Metabolic labeling and pulse–chase analysis
Subconfluent cells in 6 cm dishes were starved with MEM lacking cysteine and methionine (Gibco-BRL) for 30 min, pulse-labeled for the times stated with 50 µCi of in vitro 35S-labeling mix (Amersham, UK) per dish and subsequently chased when necessary with complete medium supplemented with 5% FCS, 10 mM HEPES pH 7.4, 5 mM methionine, 5 mM cysteine and 1 mM cycloheximide. At given time intervals, the chase was stopped by flooding the cells with ice-cold HBSS (Gibco-BRL) supplemented with 20 mM NEM to trap folding intermediates. The cells were lysed in 600 µl of lysis buffer [110 mM KOAc, 2 mM MgOAc, 20 mM HEPES pH 7.2, containing 2% CHAPS, 10 µg/ml each of chymostatin, leupeptin, antipain and pepstatin, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 20 mM NEM] and subjected to specific immunoprecipitations after pelleting the nuclei for 10 min at 4°C and 13 000 r.p.m.
Immunoprecipitations
Immunoprecipitations were performed at 4°C for 2 h or overnight using antibodies immobilized on 30 µl of a 10% suspension of protein A–Sepharose beads (Amersham Pharmacia). Collected complexes were washed twice at room temperature in wash buffer (300 mM NaCl, 10 mM Tris–HCl, 0.05% Triton X-100 and 0.05% SDS, pH 8.6) prior to uptake in sample buffer. After a 5 min incubation at 95°C, half the samples were reduced with 50 mM DTT, and the proteins were analyzed by 7.5% SDS–PAGE.
Acknowledgments
Acknowledgements
We wish to thank Professor H.Tabak for support, Dr B.Berkhout and E.Van Anken for the HIV LAI envelope vector construct, A.Land for sharing unpublished data on HIV envelope protein, Dr M.Farmery for assistance with semi-permeabilized cell techniques, and members of the Braakman, Bulleid and Sitia groups for useful discussions. This work was funded by NWO/CW (A.B. and I.B.), by the Royal Netherlands Academy of Arts and Sciences (I.B.) and by Utrecht University (A.B.). A.C., A.F. and R.S. were supported in part through grants from Associazione per la Ricerca sul Cancro, Consiglio Nazionale delle Ricerche (Target project on Biotechnology grant CNR 97.01296.PF49; 5% Biotechnology grant CNR 98.00393.PF31) and Ministero della Sanita (RF9853). N.B. was supported by the Royal Society.
References
- Aslund F. and Beckwith,J. (1999) The thioredoxin superfamily: redundancy, specificity and gray-area genomics. J. Bacteriol., 181, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader M., Muse,W., Ballou,D.P., Gassner,C. and Bardwell,J.C. (1999) Oxidative protein folding is driven by the electron transport system. Cell, 98, 217–227. [DOI] [PubMed] [Google Scholar]
- Bardwell J.C., McGovern,K. and Beckwith,J. (1991) Identification of a protein required for disulphide bond formation in vivo. Cell, 67, 581–589. [DOI] [PubMed] [Google Scholar]
- Beinert H. and Kiley,P.J. (1999) Fe–S proteins in sensing and regulatory functions. Curr. Opin. Chem. Biol., 3, 152–157. [DOI] [PubMed] [Google Scholar]
- Braakman I., Hoover-Litty,H., Wagner,K.R. and Helenius,A. (1991) Folding of influenza hemagglutinin in the endoplasmic reticulum. J. Cell Biol., 114, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I., Helenius,J. and Helenius,A. (1992) Role of ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature, 356, 260–262. [DOI] [PubMed] [Google Scholar]
- Bulleid N.J. and Freedman,R.B. (1988) Defective co-translational formation of disulphide bonds in protein disulphide-isomerase-deficient microsomes. Nature, 335, 649–651. [DOI] [PubMed] [Google Scholar]
- Bullough P.A., Hughson,F.M., Skehel,J.J. and Wiley,D.C. (1994) Structure of influenza haemagglutinin at the pH of membrane fusion. Nature, 371, 37–43. [DOI] [PubMed] [Google Scholar]
- Cabibbo A., Pagani,M., Fabbri,M., Rocchi,M., Farmery,M.R., Bulleid,N.J. and Sitia,R. (2000) ERO1-L, a human protein that favors disulphide bond formation in the endoplasmic reticulum. J. Biol. Chem., 275, 4827–4833. [DOI] [PubMed] [Google Scholar]
- Chan S., Gabra,H., Hill,F., Evan,G. and Sikora,K. (1987) A novel tumour marker related to the c-myc oncogene product. Mol. Cell Probes, 1, 73–82. [DOI] [PubMed] [Google Scholar]
- Chen J., Lee,K.H., Steinhauer,D.A., Stevens,D.J., Skehel,J.J. and Wiley,D.C. (1998) Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell, 95, 409–417. [DOI] [PubMed] [Google Scholar]
- Cuozzo J.W. and Kaiser,C.A. (1999) Competition between glutathione and protein thiols for disulphide-bond formation. Nature Cell Biol., 1, 130–135. [DOI] [PubMed] [Google Scholar]
- Dorner A.J., Wasley,L.C., Raney,P., Haugejorden,S., Green,M. and Kaufman,R.J. (1990) The stress response in Chinese hamster ovary cells. Regulation of ERp72 and protein disulphide isomerase expression and secretion. J. Biol. Chem., 265, 22029–22034. [PubMed] [Google Scholar]
- Ferrari D.M. and Söling,H.D. (1999) The protein disulphide-isomerase family: unravelling a string of folds. Biochem. J., 339, 1–10. [PMC free article] [PubMed] [Google Scholar]
- Frand A.R. and Kaiser,C.A. (1998) The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell, 1, 161–170. [DOI] [PubMed] [Google Scholar]
- Frand A.R. and Kaiser,C.A. (1999) Ero1p oxidizes protein disulphide isomerase in a pathway for disulphide bond formation in the endoplasmic reticulum. Mol. Cell, 4, 469–477. [DOI] [PubMed] [Google Scholar]
- Frand A.R., Cuozzo,J.W. and Kaiser,C.A. (2000) Pathways for protein disulphide bond formation. Trends Cell Biol., 10, 203–210. [DOI] [PubMed] [Google Scholar]
- Fuerst T.R., Niles,E.G., Studier,F.W. and Moss,B. (1986) Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl Acad. Sci. USA, 83, 8122–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillece P., Luz,J.M., Lennarz,W.J., Javier de la Cruz,F. and Römisch,K. (1999) Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol., 147, 1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B., Tachibana,C. and Winther,J.R. (1997) Active site mutations in yeast protein disulphide isomerase cause dithiothreitol sensitivity and a reduced rate of protein folding in the endoplasmic reticulum. J. Cell Biol., 138, 1229–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes E.A. and Cresswell,P. (1998) The thiol oxidoreductase ERp57 is a component of the MHC class I peptide-loading complex. Curr. Biol., 8, 709–712. [DOI] [PubMed] [Google Scholar]
- Hwang C., Sinskey,A.J. and Lodish,H.F. (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science, 257, 1496–1502. [DOI] [PubMed] [Google Scholar]
- Joly J.C. and Swartz,J.R. (1997) In vitro and in vivo redox states of the Escherichia coli periplasmic oxidoreductases DsbA and DsbC. Biochemistry, 36, 10067–10072. [DOI] [PubMed] [Google Scholar]
- Kamitani S., Akiyama,Y. and Ito,K. (1992) Identification and characterization of an Escherichia coli gene required for the formation of correctly folded alkaline phosphatase, a periplasmic enzyme. EMBO J., 11, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert N. and Freedman,R.B. (1983) Structural properties of homogeneous protein disulphide-isomerase from bovine liver purified by a rapid high-yielding procedure. Biochem. J., 213, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist J.A., Jensen,O.N., Mann,M. and Hämmerling,G.J. (1998) ER-60, a chaperone with thiol-dependent reductase activity involved in MHC class I assembly. EMBO J., 17, 2186–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J.L. (1995) Thioredoxin—a fold for all reasons. Structure, 3, 245–250. [DOI] [PubMed] [Google Scholar]
- Martin J.L., Bardwell,J.C. and Kuriyan,J. (1993) Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature, 365, 464–468. [DOI] [PubMed] [Google Scholar]
- Missiakas D., Schwager,F. and Raina,S. (1995) Identification and characterization of a new disulphide isomerase-like protein (DsbD) in Escherichia coli. EMBO J., 14, 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari M. and Helenius,A. (1999) Glycoproteins form mixed disulphides with oxidoreductases during folding in living cells. Nature, 402, 90–93. [DOI] [PubMed] [Google Scholar]
- Morrice N.A. and Powis,S.J. (1998) A role for the thiol-dependent reductase ERp57 in the assembly of MHC class I molecules. Curr. Biol., 8, 713–716. [DOI] [PubMed] [Google Scholar]
- Oliver J.D., Roderick,H.L., Llewellyn,D.H. and High,S. (1999) ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol. Biol. Cell, 10, 2573–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani M., Fabbri,M., Benedetti,C., Fassio,A., Pilati,S., Bulleid,N.J., Cabibbo,A. and Sitia,R. (2000) Endoplasmic reticulum oxidoreductin 1-Lβ (ERO1-Lβ) a human gene induced in the course of the unfolded protein response. J. Biol. Chem., 275, 23685–23692. [DOI] [PubMed] [Google Scholar]
- Pind S., Davidson,H., Schwaninger,R., Beckers,C.J., Plutner,H., Schmid,S.L. and Balch,W.E. (1993) Preparation of semi-intact cells for study of vesicular trafficking in vitro. Methods Enzymol., 221, 222–234. [DOI] [PubMed] [Google Scholar]
- Plutner H., Davidson,H.W., Saraste,J. and Balch,W.E. (1992) Morphological analysis of protein transport from the ER to Golgi membranes in digitonin-permeabilized cells: role of the P58 containing compartment. J. Cell Biol., 119, 1097–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard M.G., Travers,K.J. and Weissman,J.S. (1998) Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell, 1, 171–182. [DOI] [PubMed] [Google Scholar]
- Raina S. and Missiakas,D. (1997) Making and breaking disulphide bonds. Annu. Rev. Microbiol., 51, 179–202. [DOI] [PubMed] [Google Scholar]
- Reddy P., Sparvoli,A., Fagioli,C., Fassina,G. and Sitia,R. (1996) Formation of reversible disulfide bonds with the protein matrix of the endoplasmic reticulum correlates with the retention of unassembled Ig light chains. EMBO J., 15, 2077–2085. [PMC free article] [PubMed] [Google Scholar]
- Rietsch A. and Beckwith,J. (1998) The genetics of disulphide bond metabolism. Annu. Rev. Genet., 32, 163–184. [DOI] [PubMed] [Google Scholar]
- Rose J.K., Buonocore,L. and Whitt,M.A. (1991) A new cationic liposome reagent mediating nearly quantitative transfection of animal cells. Biotechniques, 10, 520–525. [PubMed] [Google Scholar]
- Roth R.A. and Pierce,S.B. (1987) In vivo cross-linking of protein disulphide isomerase to immunoglobulins. Biochemistry, 26, 4179–4182. [DOI] [PubMed] [Google Scholar]
- Stewart E.J., Katzen,F. and Beckwith,J. (1999) Six conserved cysteines of the membrane protein DsbD are required for the transfer of electrons from the cytoplasm to the periplasm of Escherichia coli. EMBO J., 18, 5963–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traenckner E.B., Wilk,S. and Baeuerle,P.A. (1994) A proteasome inhibitor prevents activation of NF-κB and stabilizes a newly phosphorylated form of I κB-α that is still bound to NF-κB. EMBO J., 13, 5433–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson I.A., Skehel,J.J. and Wiley,D.C. (1981) Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature, 289, 366–373. [DOI] [PubMed] [Google Scholar]
- Wilson R., Allen,A.J., Oliver,J., Brookman,J.L., High,S. and Bulleid,N.J. (1995) The translocation, folding, assembly and redox-dependent degradation of secretory and membrane proteins in semi-permeabilized mammalian cells. Biochem. J., 307, 679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]