

Abstract
Cooperation between nuclear factor of activated T cells (NFAT) and AP-1 (Fos–Jun) proteins on composite NFAT–AP-1 DNA elements constitutes a powerful mechanism for signal integration of the calcium and protein kinase C/Ras pathways in the regulation of gene expression. Here we report that NFAT can induce expression of certain genes in T cells without the need for cooperative recruitment of Fos and Jun. Using NFAT1 mutant proteins that are unable to interact with Fos–Jun dimers but are unaffected in DNA binding or transcriptional activity, we show that expression of interleukin (IL)-2, granulocyte–macrophage colony-stimulating factor (GM-CSF), IL-3, IL-4, MIP1α and Fas ligand mRNAs is absolutely dependent on cooperation between NFAT and Fos–Jun; in contrast, NFAT induces tumor necrosis factor α (TNFα) mRNA and IL-13 promoter activity without any necessity to recruit Fos and Jun. Furthermore, we show that NFAT–Fos–Jun cooperation is also essential to elicit the NFAT-dependent program of activation-induced cell death. Our results support the hypothesis that even in a single cell type, NFAT activation can evoke two distinct biological programs of gene expression, dependent or independent of NFAT–AP-1 cooperation.
Keywords: activation-induced cell death/AP-1/cytokine expression/nuclear factor of activated T cells/transcriptional regulation
Introduction
Gene expression in eukaryotes is controlled by the binding of arrays of transcription factors to regulatory promoter/enhancer sequences in DNA (Tjian and Maniatis, 1994). The cooperative and synergistic binding of transcription factors to DNA promotes the assembly of transcriptionally active ‘enhanceosome’ complexes, which interact with coactivator proteins and the core transcriptional machinery. The participation of multiple unrelated transcription factors in the enhanceosome complex permits the stringent and tissue-specific control of gene expression. Because each transcription factor is activated in response to specific signaling pathways, enhanceosome assembly ensures that optimal levels of gene expression are reached only when all the component proteins have been optimally activated.
Within the context of an enhanceosome complex, neighboring transcription factors may make cooperative binding interactions on ‘composite’ DNA elements or may bind essentially independently to adjacent binding sites on DNA. These two behaviors may be distinguished by a simple kinetic test: cooperative binding is defined by the greater stability/lower dissociation rate of the composite complex from DNA, relative to the stability/dissociation rate of the individual components alone. Among the pairs of unrelated transcription factors that have been found to bind cooperatively to composite sites in DNA, well known examples include PU1 and PIP/IRF4 (Pongubala et al., 1993), the ternary complex factor that assembles on the c-fos promoter and contains SRF and Elk (Hill et al., 1993) and the NFAT–AP-1 complex formed by the nuclear factor of activated T cells (NFAT) and Fos–Jun (AP-1) dimers (Jain et al., 1992, 1993a,b; Chen et al., 1995, 1998; Peterson et al., 1996).
The NFAT family of transcription factors contains five distinct NFAT proteins: NFAT1 (NFATp or NFATc2), NFAT2 (NFATc or NFATc1), NFAT3 (NFATc4), NFAT4 (NFATc3 or NFATx) and NFAT5 (TonEBP). These proteins show a high sequence similarity in their DNA-binding domains (DBDs), which are distantly related to the DBDs of Rel/NF-κB (Jain et al., 1995a; Wolfe et al., 1997; Chen et al., 1998). NFAT proteins are expressed in multiple cell types both within and outside the immune system, where they regulate a wide variety of putative target genes (reviewed in Rao et al., 1997; Crabtree, 1999; Kiani et al., 2000).
In cells of the immune system, cooperative NFAT–AP-1 complexes are induced by stimulation of the antigen receptors of T and B cells, the Fcγ receptors of macrophages and natural killer (NK) cells, and the Fcε receptors of mast cells and basophils (Rao et al., 1997). These receptors are coupled on the one hand to calcium mobilization and on the other hand to activation of protein kinase C (PKC)/Ras/Raf/MAP kinase pathways (van Leeuwen and Samelson, 1999). Calcium mobilization activates the phosphatase calcineurin, which dephosphorylates NFAT on multiple serine residues and causes its translocation from the cytoplasm to the nucleus; this process can be inhibited by the calcineurin inhibitors cyclosporin A (CsA) and FK506 (Rao et al., 1997; Crabtree, 1999; Kiani et al., 2000). Stimulation of the PKC/Ras pathway results in activation of AP-1 transcription complexes (Karin, 1995), which are composed of dimers of the basic region–leucine zipper (bZIP) proteins Fos (c-Fos, FosB, Fra1 and Fra2) and Jun (c-Jun, JunB and JunD). Fos and Jun proteins bind DNA through their basic region and interact with one another through their leucine zipper domains (Glover and Harrison, 1995). NFAT and AP-1 activity can be pharmacologically induced with calcium ionophores such as ionomycin and phorbol esters such as phorbol myristate acetate (PMA), respectively.
Cooperation between NFAT and AP-1 proteins thus constitutes a powerful mechanism for signal integration of the calcium and PKC/Ras pathways. Cooperative binding of NFAT and AP-1 has been demonstrated on the promoter/enhancer regions of many genes involved in the productive immune response, for instance those encoding the cytokines interleukin (IL)-2, IL-3, IL-4 and granulocyte–macrophage colony-stimulating factor (GM-CSF) (Cockerill et al., 1993, 1995; Jain et al., 1995b; Rooney et al., 1995). In particular, the distal ARRE2 elements of the human and murine IL-2 promoters are classic composite sites for cooperative binding of NFAT and AP-1. Although non-consensus binding sites for NFAT and AP-1 can be discerned within the ARRE2, NFAT and Fos–Jun heterodimers bind poorly to their individual sites within these elements in the absence of the partner protein, and a strongly cooperative complex is only formed in the presence of all three transcription factors and DNA (Jain et al., 1993a,b; Chen et al., 1995; Peterson et al., 1996). The specificity and cooperative nature of the NFAT–Fos–Jun interaction is due to the large number of contacts among the DBDs of NFAT, Fos and Jun, as well as among these DBDs and DNA (Chen et al., 1998).
In this study we have addressed the question of whether NFAT–AP-1 cooperation is mandatory for the expression of NFAT target genes. Using mutant NFAT proteins that bind DNA normally but are unable to interact with Fos–Jun or Jun–Jun dimers, we have demonstrated that certain NFAT target genes are completely dependent on NFAT–AP-1 cooperation, while others show no such requirement. Our results are consistent with the possibility that NFAT transcription factors are capable of activating distinct but partially overlapping programs of gene expression in any given cell type, dependent on whether calcium mobilization is accompanied by concomitant MAP kinase activation or not.
Results
Point mutations in critical residues of the NFAT1 DBD impair its ability to interact with Fos and Jun
Based on the crystal structure of the NFAT1–Fos–Jun–DNA complex, we identified several amino acids that are likely to be important for maintaining the interface between the NFAT1 DBD and the leucine zipper regions of c-Fos and c-Jun, respectively (Figure 1). These contacts are conserved in all members of the NFAT, Fos and Jun families (Chen et al., 1998). Arginine 468 of murine NFAT1 (Arg466 of human NFAT1) is located in the CX loop and contacts the negatively charged residues Asp170 and possibly Glu173 of Fos; similarly, Thr535 in the E′F loop of murine NFAT1 (Thr533 in human NFAT1) makes contacts within a pocket created by Arg285, Arg288 and Lys292 in Jun (Chen et al., 1998) (Figure 1). Hence, substitution of Arg468 with glutamic acid would be expected to interfere with the cooperative binding of NFAT to Fos–Jun heterodimers, while substitution of Thr535 to arginine would be expected to impair the binding of NFAT1 to Jun.

Fig. 1. Residues involved in NFAT1–Fos–Jun interaction. (A) Close-up view of the NFAT1–Fos–Jun–DNA complex (Chen et al., 1998). Ribbon diagram made using Rasmol. Side chains of NFAT1 involved in NFAT1–Fos, NFAT1–Jun and NFAT1–DNA interactions that were mutated in this study are shown. (B) Amino acid sequence and secondary structure of the NFAT1 DBD. Residues that contact Fos are shown in open boxes while residues involved in Jun contacts are shown in white inside black boxes. The residues mutated in this study are indicated by asterisks.
To test these predictions, we generated the DBD mutants R468E and T535R and assessed their ability to form complexes with Fos–Jun and Jun–Jun dimers on DNA. Electrophoretic mobility shift assays (EMSAs) were performed using the ARRE2 probe from the distal NFAT site of the murine IL-2 promoter (an NFAT1–AP-1 composite site), recombinant wild-type or mutant NFAT1 DBDs, and recombinant full-length c-Fos and c-Jun proteins. As predicted, the R468E mutant NFAT1 DBD showed a highly reduced capacity, relative to its wild-type equivalent, for cooperative binding with Fos–Jun heterodimers to DNA (Figure 2B, compare first and second panels). Interestingly, this mutant protein showed an ∼3-fold increase in binding when Jun homodimers were used (Figure 2A, first, second and fourth panels), suggesting that the newly introduced Glu residue made electrostatic interactions with nearby basic residues (e.g. Lys292) of Jun. Similarly, the T535R mutant showed a dramatic decrease in its ability to bind cooperatively with either Jun homodimers or Fos–Jun heterodimers (Figure 2A and B, compare first and third panels). Similar results have been reported for the corresponding T551A mutation of human NFAT2 (Sun et al., 1997).

Fig. 2. The R468E and T535R mutations in the NFAT1 DBD strongly influence the interaction of NFAT1 with Fos and Jun. (A) Jun homodimers. (B) Fos–Jun heterodimers. EMSAs were performed with the murine IL-2 promoter ARRE2 probe and wild-type (left panels), R468E (middle panels) or T535R (right panels) recombinant NFAT1 DBDs. Right panels show results of densitometric quantification.
To engineer an NFAT1 mutant with a minimal capacity to interact with AP-1 proteins, we tested different combinations of point mutations in residues involved in maintaining the NFAT1–AP-1 complex. Arg468 and the adjacent Fos-interacting residue Ile469 were substituted with alanine, while Thr535 was substituted with glycine, to avoid potential unexpected electrostatic interactions such as those observed with the R468E mutant. In EMSAs, two of the resulting mutant DBDs, R468A/T535G (data not shown) and R468A/I469A/T535G (Figure 3A), showed an almost complete inability to bind cooperatively with Fos–Jun heterodimers on the ARRE2 probe, without any loss of binding affinity for DNA (Figure 3B).

Fig. 3. Mutations that impair the ability of NFAT to interact with Fos and Jun have no effect on its DNA-binding activity. EMSAs were performed using the murine IL-2 promoter ARRE2 probe and recombinant NFAT1 DBD proteins. Right panels show results of densitometric quantification. (A) Ability of wild-type NFAT1 DBD and two mutant DBDs, F475A and R468A/I469A/T535G, to bind cooperatively with Fos and Jun to DNA. (B) The same probe was used to measure the DNA-binding activity of wild-type NFAT1 DBD and the two mutants F475A and R468A/I469A/T535G. (C) Effect of the H427R mutation on NFAT1 DNA-binding activity.
We also tested the functional significance of Phe475 (Phe473 in human NFAT1). This residue is the center of a small hydrophobic patch that appears to be important for stabilizing the E′F loop (Chen et al., 1998). When this phenylalanine was mutated to alanine, the resulting F475A mutant showed a greatly impaired capacity to form cooperative complexes with Fos–Jun heterodimers while maintaining a normal affinity for DNA (Figure 3A and B). These results confirm the important structural role of F475 in maintaining the integrity of the E′F loop.
In the DNA-specificity loop of Rel-family transcription factors (RFRYxCEG), the second arginine residue makes an important base contact with a guanine residue in the binding site and stabilizes DNA binding (Muller et al., 1995). In the calcium-regulated NFAT proteins NFAT1–4, the corresponding DNA-specificity loop has the invariant sequence RAHYETEG, thus showing a substitution of histidine in this position. Changing this histidine back to an arginine residue in a short DNA-binding fragment of NFAT2, corresponding to the N-terminal domain of the Rel-homology region, resulted in a highly increased affinity of the mutant protein for DNA (Zhou et al., 1998). We asked whether, by making a similar change in the NFAT1 DBD, we could create a protein with increased DNA-binding affinity that might bind stably to DNA in the absence of AP-1 proteins. However, contrary to the results described for the short NFAT2 DBD fragment (175 amino acids), the H425R mutant NFAT1 DBD (297 amino acids) did not show greater DNA-binding affinity than the wild-type protein (Figure 3C). This indicates that residues at the very end of the N-terminal domain of the Rel-homology region (which are missing in the NFAT2 fragment) contribute strongly to stabilizing the NFAT–DNA complex.
Taken together, the results in this section identify several mutations in the NFAT1 DBD that impair its ability to bind cooperatively with Fos and Jun to DNA. The doubly mutated protein R468A/T535G and the triply mutated protein R468A/I469A/T535G showed very striking impairments, and were used to ask whether NFAT–AP-1 complexes elicit transcription of a different spectrum of target genes from NFAT alone.
NFAT1 AP-1-interaction mutants fail to transactivate from NFAT–AP-1 composite sites but are fully active on AP-1-independent NFAT sites
To test the transcriptional functions of full-length NFAT1 proteins bearing DBD mutations that impaired cooperative interactions with AP-1, we performed transient transfection experiments in Jurkat cells using a reporter plasmid (NFAT/AP-1 3×Luc) carrying three tandem copies of the murine IL-2 promoter ARRE2 site. All DBD mutants tested showed less transactivation than the wild-type protein (Figure 4A). This decrease showed a clear correlation with the reduction in cooperative binding to AP-1 in EMSAs of the recombinant DBDs (Figures 1 and 2 and data not shown). In particular, R468A/T535G showed very pronounced impairment, while the R468A/I469A/T535G mutant showed no significant transcriptional activity in the NFAT/AP-1 3×Luc reporter system (Figure 4A). Based on these results, we chose the R468A/T535G and R468A/I469A/T535G mutants for subsequent detailed study.

Fig. 4. Mutations that impair the interaction of NFAT1 with AP-1 also selectively impair its transcriptional activity on a composite NFAT–AP-1 site. (A) Jurkat cells were cotransfected with the NFAT/AP-1 3×-luciferase reporter plasmid and expression plasmids encoding wild-type NFAT1 or different AP-1 interaction mutants. Activity was measured after 8 h of stimulation with PMA and ionomycin. (B) Jurkat cells were cotransfected with expression plasmids encoding wild-type NFAT1 or R468A/T535G mutant NFAT1 and either a luciferase reporter plasmid driven by three copies of the IL-2 promoter ARRE-2 site (NFAT/AP-1 3×Luc; left panel) or a CAT reporter plasmid carrying five copies of the κ3 element of the TNFα promoter (κ3-5×CAT; right panel). Where indicated, the cells were stimulated with PMA and ionomycin for 8 h. Values are mean + SE of three independent experiments. A slight (2-fold) enhancement was seen with the mutant relative to the wild-type protein in the κ3-5×CAT reporter experiments, but this did not reach significant levels. (C) HEK293 cells were cotransfected with expression plasmids encoding wild-type NFAT1 or R468A/I469A/T535G mutant NFAT1 and with the NFAT/AP-1 3× (left panel) or TNFα promoter (right panel) luciferase reporter plasmids. Reporter activity was measured after 8 h of stimulation with PMA and ionomycin. Lower panels show western blots using an anti-NFAT1 antibody to detect expression of wild-type and mutant NFAT1.
We tested whether these mutants were active in driving transcription from AP-1-independent NFAT sites. Jurkat cells were cotransfected with the R468A/T535G mutant and with reporter plasmids carrying tandem copies of either the murine ARRE2 NFAT–AP-1 composite site (NFAT/AP-1 3×Luc) or an AP-1-independent NFAT site (κ3) from the tumor necrosis factor α (TNFα) promoter (κ3-5×CAT). To provide the most stringent test of the need for NFAT–AP-1 cooperation, the cells were stimulated with both PMA and ionomycin to achieve maximal activation of AP-1, other inducible transcription factors (e.g. NF-κB) and any nuclear proteins (e.g. basal transcription machinery, coactivator proteins) that might contribute to gene transcription and need full stimulation for their activity. The NFAT mutant showed practically no transactivation on the NFAT–AP-1 composite site even under these optimal conditions, but it strongly transactivated from the AP-1-independent κ3 NFAT site (Figure 4B). To extend this result under conditions where we could demonstrate equivalent expression of the wild-type and mutant NFAT1 proteins, experiments were performed in HEK293 cells, which do not express NFAT1, using similar reporters and the R468A/I469A/T535G mutant. Expression of wild-type and mutant NFAT1 in HEK293 cells was equivalent as judged by western blotting (Figure 4C, lower panels). However, the triply mutated protein was unable to drive transcription from the NFAT–AP-1 composite site, but was unimpaired in its ability to activate transcription driven by the full TNFα promoter (Figure 4C).
We examined several different cytokine promoters to see whether they depended on the NFAT–AP-1 interaction or could be transactivated by a mutant NFAT1 (R468A/I469A/T535G) that was incapable of cooperatively recruiting Fos and Jun. Jurkat cells were cotransfected with expression plasmids for wild-type or mutant NFAT1 and with reporter plasmids driven by the full promoters of the IL-2, TNFα and GM-CSF genes and the Th2 cytokines IL-4 and IL-13 (Figure 5). Wild-type NFAT1 increased transcription of the IL-2 promoter 4-fold, while the mutant protein did not increase reporter activity, showing as expected that the IL-2 promoter was strongly dependent on NFAT/AP-1 cooperation (Figure 5A). In contrast, both wild-type and mutant NFAT1 proteins greatly enhanced transcription of the TNFα promoter, confirming that this promoter can be efficiently activated in the absence of NFAT–AP-1 cooperation both in Jurkat cells (Figure 5B) and in HEK293 cells (Figure 4C). The GM-CSF promoter was intermediate, retaining substantial activity when tested with the mutant NFAT1 (Figure 5C). A similar range was seen in the case of the Th2 cytokines: activity of the IL-4 promoter was significantly dependent on NFAT1–AP-1 cooperation while the IL-13 promoter was not (Figure 5D and E).

Fig. 5. Different NFAT-dependent cytokine gene promoters show differing requirements for cooperation of NFAT with AP-1. Jurkat cells were cotransfected with expression plasmids encoding wild-type NFAT1 or R468A/I469A/T535G mutant NFAT1 and luciferase reporter plasmids bearing the promoter regions of the (A) IL-2, (B) TNFα, (C) GM-CSF, (D) IL-4 and (E) IL-13 genes. Reporter activity was measured in cells activated with PMA and ionomycin for 8 h. (–) indicates the level of endogenous reporter activity in cells transfected with a control empty plasmid. Results are mean + SE of three independent experiments.
Differing requirements for NFAT–AP-1 cooperation in transcription of endogenous cytokine genes
We used the mutant proteins to identify endogenous NFAT target genes whose expression was dependent on, or independent of, the cooperative interaction between NFAT and AP-1 (Figure 6). These studies are important because the behavior of a proximal promoter does not always predict the behavior of an endogenous gene, especially if distal regulatory elements play an important role (Cockerill et al., 1993; Agarwal et al., 2000). Jurkat cells were transiently transfected with plasmids expressing mouse CD4 and either wild-type NFAT1 or the R468A/T535G mutant protein. Transfected cells were selected for CD4 expression with magnetic beads to enrich for cells coexpressing NFAT1, then stimulated with PMA and ionomycin. The pattern of cytokine expression was analyzed by RNase protection assay after 4 h of stimulation. Cells transfected with wild-type NFAT1 showed a pronounced increase in IL-2 mRNA induction, while cells transfected with the R468A/T535G mutant had levels of IL-2 transcripts similar to those of the untransfected cells (Figure 6A). In contrast, in a parallel experiment, both wild-type and mutant NFAT1 proteins produced a marked increase in TNFα mRNA production (Figure 6B; note differences in RNA loading as assessed by signal from L32 and GAPDH). These data show clearly that IL-2 expression requires NFAT–AP-1 cooperation, while TNFα expression is NFAT dependent but can occur in the absence of physical cooperation between NFAT and AP-1.
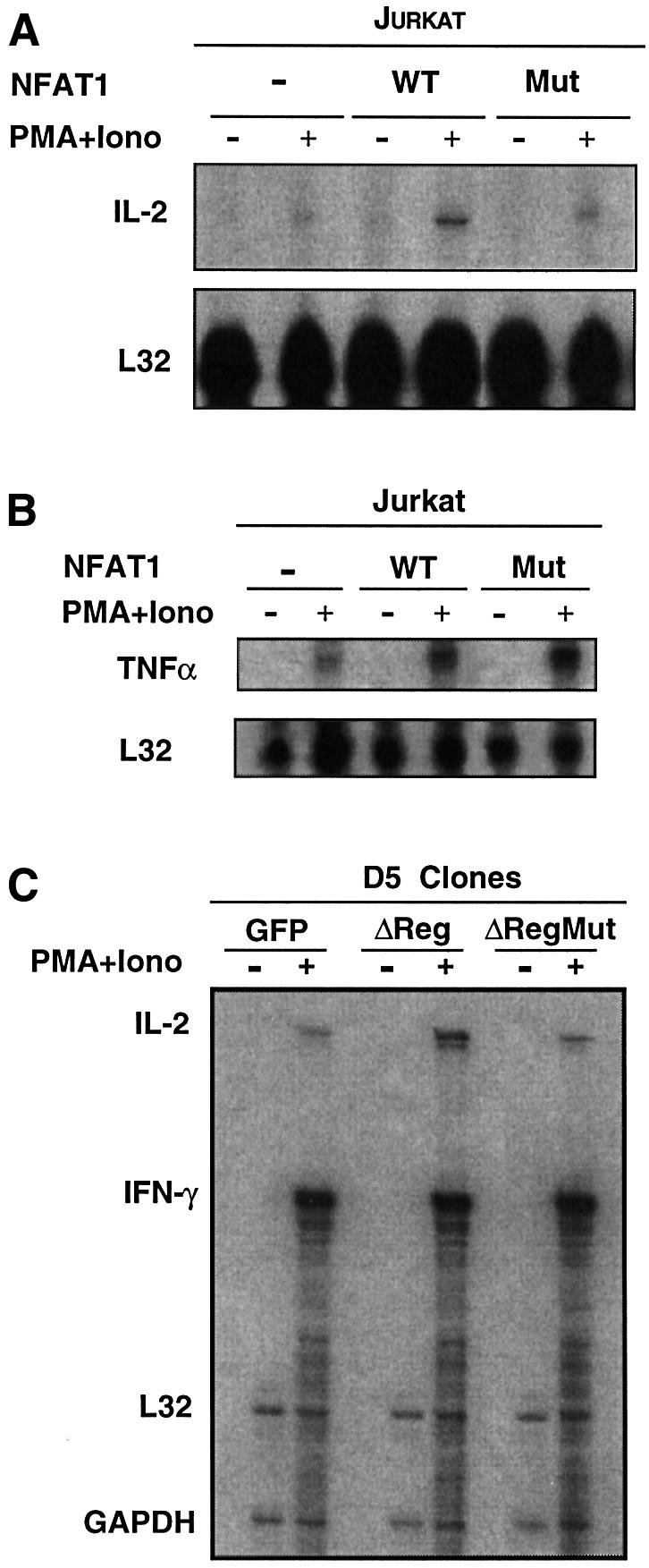
Fig. 6. AP-1-dependent and -independent expression patterns of endogenous NFAT target genes. (A) Jurkat cells were cotransfected with expression plasmids encoding murine CD4 and wild-type or R468A/I469A/T535G mutant NFAT1 (1 µg of each expression plasmid/106 cells). After 24 h, transfected cells were selected with CD4–Dynabeads and left unstimulated or stimulated with PMA and ionomycin for 4 h. Levels of expression of IL-2 (A) and TNFα (B) were determined by RNase protection assay. Levels of protected L32 transcripts indicate mRNA loading in each lane. (C) RNA from D5 clones stably expressing GFP, wild-type NFAT1ΔReg or mutant NFAT1ΔReg[R468A/T535G] were analyzed by RNase protection assay using a multi-set probe that allowed detection of IL-2 and IFNγ mRNAs. Where indicated, cells were stimulated with PMA and ionomycin for 4 h. Levels of L32 and GAPDH mRNAs show similar loading in all lanes.
The subline of Jurkat cells that we used does not express several cytokines at significant levels upon stimulation, and so we turned to murine Th1 and Th2 T cell clones (D5 and D10, respectively) to examine the effects of wild-type and mutant NFAT1 proteins on the expression of other inducible genes. These IL-2-dependent clones are not as highly transfectable as Jurkat T cells and so we needed to make stably transfected cell lines expressing wild-type and AP-1-interaction-deficient mutant NFAT1 (a bonus of making stably expressing cell lines is that the proteins are not overexpressed relative to endogenous NFAT1; see Figure 7C). To generate the stable transfectants, we used a truncated form of NFAT1, NFAT1ΔReg, which lacks the calcineurin-binding regulatory domain of NFAT1 (amino acids 143–398). In resting cells, this protein shows combined cytoplasmic–nuclear distribution that remains unchanged after stimulation and is insensitive to CsA; however, it becomes fully transcriptionally active only after stimulation and this transcriptional activation is also insensitive to CsA (F.Macián and C.García-Rodríguez, unpublished results).

Fig. 7. The NFAT1ΔReg[R468A/T535G] mutant protein shows selective dominant-negative activity on composite NFAT–AP-1 sites. (A) The ability of the mutant R468A/T535G NFAT1 DBD (DBDMut) to compete with wild-type NFAT1 DBD (DBDWT) for binding to an NFAT–AP-1 composite site was evaluated by EMSA using the murine ARRE-2 probe. Fixed concentrations of Jun (20 nM), Fos (20 nM) and recombinant DBDWT (4 nM) were used in all binding reactions and competition by increasing amounts of DBDMut was monitored by the decrease in intensity of the band corresponding to the wild-type Fos–Jun–NFAT1 DBD complex (NFAT1:Fos:Jun). The migration positions of DNA–protein complexes containing NFAT1 DBDWT and DBDMut are indicated. (B) Jurkat cells were transiently transfected with the NFAT/AP-1 3×luciferase reporter plasmid with or without an expression plasmid encoding mutant NFAT1ΔReg[R468A/T535G], and luciferase activity was determined in cells activated with PMA and ionomycin for 8 h. A western blot showing the levels of expression of full-length endogenous NFAT1 and the mutant NFAT1ΔReg protein in transfected cells is shown (inset). (C) A D10 clone stably expressing NFAT1ΔReg[R468A/T535G] was generated by retroviral infection of D10 cells and subsequent selection. The level of protein expression achieved in this clone was determined by western blotting using an anti-NFAT1 antibody. (D) The pattern of cytokine expression of a D10 clone that expressed R468A/T535G NFAT1 ΔReg was analyzed by RNase protection assay using a multi-set cytokine probe. Where indicated, cells were stimulated with PMA and ionomycin for 4 h in the presence or absence of CsA. Levels of protected L32 transcripts show that loading was similar in all lanes.
Three different retroviral (RV) constructs, RV-IRES-GFP, RV-NFAT1ΔReg-IRES-GFP and RV-NFAT1ΔReg [R468A/T535G]-IRES-GFP, were used to infect the murine Th1 clone D5. After infection, cells were selected for G418 resistance and clones were isolated. The expression of green fluorescent protein (GFP) was verified by immunocytochemistry, and western blotting was used to confirm NFAT1 expression (data not shown). Three different clones were picked; their pattern of cytokine expression after stimulation was analyzed by RNase protection assay. Once again, only D5 cells expressing wild-type NFAT1ΔReg showed enhanced IL-2 transcription, while cells expressing R468A/T535G mutant NFAT1ΔReg showed levels of IL-2 transcripts similar to or even lower than the control GFP-expressing clone (Figure 6C). Note that there is no change in the level of interferon γ (IFNγ) transcripts in cells expressing either the wild-type or the mutant NFAT1ΔReg protein. While this could be attributed to a lack of NFAT target sites in the IFNγ gene, we believe that it is more likely to reflect the high levels of IFNγ expressed by D5 cells, combined with the difficulty of achieving sufficient overexpression of NFAT1ΔReg in stably transfected cells.
The NFAT1ΔReg[R468A/T535G] mutant protein inhibits transcription of genes whose expression is dependent on NFAT–AP-1 cooperation
As mentioned before, NFAT1ΔReg is a truncated form of NFAT1 that shows both cytoplasmic and nuclear localization in resting cells. Nevertheless, stimulation with PMA and ionomycin is still needed to render this protein fully transcriptionally active. Since NFAT1ΔReg has the advantage of being already in the nucleus and presumably bound to NFAT sites in resting cells, it should be able to compete for DNA binding with endogenous NFAT proteins that translocate into the nucleus after stimulation. Thus, NFAT1ΔReg with mutations in its AP-1-interacting residues would be predicted to bind non-productively to NFAT–AP-1 composite sites, thereby behaving as a dominant-negative protein that selectively diminishes expression of NFAT–AP-1-dependent genes.
To test this prediction, we first evaluated the ability of the R468A/T535G mutant DBD to inhibit binding of wild-type NFAT1 DBD to the murine ARRE2 composite site in the presence of Fos and Jun. A 4-fold excess of mutant DBD (16 nM) competed >70% of DNA binding by the wild-type DBD, while a 16-fold excess (64 nM) competed binding by >95% (Figure 7A). These results indicated that NFAT1ΔReg[R468A/T535G] would have a dominant-negative effect at composite NFAT–AP-1 sites if expressed at appropriate levels. Transient reporter assays in Jurkat T cells (Figure 7B) confirmed that the cells expressing mutant NFAT1ΔReg[R468A/T535G] showed decreased activity of an NFAT–AP-1-3×luciferase reporter, relative to the activity of this reporter plasmid in cells transfected with a control expression vector (Figure 7B). Jurkat cells are highly transfectable and hence were used for these reporter assays in preference to D5 or D10 T cell clones. Western blotting (inset) indicated that the mutant protein was expressed in the whole cell population at similar levels to endogenous NFAT1, indicating higher expression levels in the fraction of productively transfected cells. Thus the decrease in reporter activity is consistent with a dominant-negative effect of the mutant protein due to its competition with endogenous NFAT proteins for the NFAT–AP-1 sites in the reporter plasmid. In similar experiments, wild-type NFAT1ΔReg produced at least a 2-fold increase in the expression of the reporter gene (data not shown).
Based on these results, we compared gene expression in uninfected D10 cells and in the D10 cell line stably expressing the NFAT1ΔReg[R468A/T535G] mutant protein (to a level ∼2-fold higher than endogenous NFAT1; Figure 7C). The cells were stimulated with PMA and ionomycin for 4 h and their pattern of cytokine expression was analyzed by RNase protection assay. Compared with uninfected D10 cells, the clone expressing the mutant NFAT1ΔReg protein showed a detectable, CsA-resistant increase in TNFα mRNA expression, but produced significantly less fasL, MIP-1α and IL-3 mRNA upon stimulation (Figure 7D). These results identify new genes (fasL, MIP-1α, IL-3) whose expression is dependent on NFAT–AP-1 cooperation, and confirm that the NFAT1ΔReg[R468A/T535G] mutant protein exerts a selective dominant-negative effect at NFAT–AP-1 composite sites as well as driving transcription of TNFα mRNA by binding to AP-1-independent NFAT sites.
NFAT1 enhances activation-induced cell death in T cells in a manner that requires cooperation with AP-1
Activation through the T-cell receptor (TCR) leads to a characteristic form of Fas-mediated apoptosis in T cells termed activation-induced cell death (AICD) (Brunner et al., 1995; Dhein et al., 1995; Ju et al., 1995). This process depends on activation-induced transcription of the fasL gene; interaction of FasL with Fas at the cell surface triggers apoptosis. Multiple transcription factors, including NF-κB, NFAT, EGR and Forkhead, have been proposed to mediate transcription of the fasL gene (Latinis et al., 1997; Mittelstadt and Ashwell, 1998; Brunet et al., 1999; Kasibhatla et al., 1999; Rengarajan et al., 2000). As the experiment of Figure 7D suggested that cooperating NFAT–AP-1 complexes were involved in regulating fasL gene transcription, we asked whether AICD in T cells would be enhanced by overexpressing NFAT1 and, if so, whether AP-1 interaction would be needed. D5 clones that expressed GFP, NFAT1ΔReg or NFAT1ΔReg[R468A/T535G] were stimulated with immobilized anti-CD3. After 48 h, cells were collected and stained with propidium iodide to detect apoptotic cells. As expected, stimulation with anti-CD3 caused an increase in cell death in the control D5 clone expressing GFP. D5 cells expressing wild-type NFAT1ΔReg showed a 4- to 5-fold increase in cell death compared with the control GFP-expressing clone, directly implicating NFAT proteins in the process of AICD. In contrast, no enhancement of AICD was observed in the clone expressing the mutant NFAT1ΔReg[R468A/T535G] (Figure 8), indicating strongly that NFAT–AP-1 cooperation is needed.

Fig. 8. Activation-induced cell death of the D5 T cell clone is enhanced by overexpression of wild-type NFAT1ΔReg but not the AP-1-interaction mutant NFAT1ΔReg[R468A/T535G]. D5 clones that expressed GFP, wild-type NFAT1ΔReg or the AP-1-interaction mutant NFAT1ΔReg[R468A/T535G] were either left unstimulated or stimulated with plate-bound anti-CD3 (1 µg/ml). After 48 h, cells were stained with propidium iodide and the proportion of dead cells was determined.
Discussion
In this report we have explored the role of specific residues located in the contact surfaces of the NFAT–Fos–Jun–DNA complex, focusing on the functional importance of these contacts in regulating the expression of NFAT-dependent genes. Based on our results, we have been able to classify selected NFAT target genes into distinct categories: those dependent and those not dependent on NFAT–AP-1 cooperation. We have confirmed that NFAT– AP-1 cooperation is required for the induction of a large number of cytokine genes; however, there is clearly a subset of NFAT-dependent genes that can be turned on by mutant NFAT proteins unable to cooperate with AP-1. Using these mutant proteins as tools, we have shown that AICD, a complex biological process in which the role of NFAT–AP-1 cooperation had not previously been formally established, is also dependent on cooperative occupancy of composite NFAT–AP-1 sites.
Identification of mutations that disrupt the NFAT1–Fos–Jun interaction without affecting DNA binding by NFAT1
The crystal structure of the NFAT1–Fos–Jun–DNA complex shows an extended contact surface between NFAT1, Fos and Jun (Chen et al., 1998). In NFAT1, the contacts involve mainly residues in the N-terminal ‘specificity’ domain of the Rel-homology region of NFAT1, although some contacts with Fos occur within the cc′ loop of the C-terminal domain. Formation of the extended contact interface is facilitated by bending of both the Fos–Jun dimer and DNA. The binding of NFAT to DNA is markedly stabilized in the presence of Fos and Jun (Jain et al., 1992); conversely, the presence of NFAT on composite sites increases by ∼10-fold the affinity of Jun dimers for DNA (Peterson et al., 1996).
Despite this extended network of contacts, NFAT1–AP-1 cooperation was easily disrupted by limited substitutions in the NFAT1–Fos–Jun interface. Specifically, combined mutation of one or two residues in the CX loop, which contacts Fos (Arg468 with or without Ile469), and one in the E′F loop, which contacts Jun (Thr535), essentially eliminated all cooperative interactions between NFAT1 and AP-1 without affecting DNA binding in any way (Figure 3). Arg468 and Thr535 are completely conserved in NFAT1–4 (Chen et al., 1998), leading to the prediction that the corresponding substitutions in NFAT2, NFAT3 and NFAT4 would have the same effect. The E′F loop has two interesting features. First, it is unstructured in solution in the context of a small DNA-binding fragment of NFAT2, but acquires an α-helical structure upon binding to DNA (Zhou et al., 1998). This DNA-induced folding may require the formation of backbone contacts between protein and DNA (Chen et al., 1998; Zhou et al., 1998); however, it is clear that the T535G mutation in the E′F loop does not compromise DNA binding directly (Figure 3). Secondly, the E′F loop is supported by a small hydrophobic patch centered on Phe475 (Chen et al., 1998; Zhou et al., 1998). Substitution of this residue with alanine yielded a mutant protein (F475A) that bound DNA with comparable affinity to the wild type but was greatly impaired in its ability to interact with Fos–Jun heterodimers on composite DNA sites (Figures 2 and 3).
Taken together, our data confirm and extend previous observations showing that the CX and E′F loops are critical for Fos–Jun binding (Sun et al., 1997; Chen et al., 1998), and form the basis for generation of the R468A/T535G and R468A/I469A/T535G mutant NFAT1 proteins that are unable to interact with AP-1.
Categories of NFAT-dependent gene expression: role of physical cooperation between NFAT and AP-1
In this initial study with AP-1 interaction mutants of NFAT, we limited our analysis to genes induced during the productive immune response. Our results establish that expression of some genes and promoters (IL-2, GM-CSF, IL-3, MIP-1α, IL-4, fasL) can only be achieved through coordinate activation of both NFAT and AP-1 proteins, while other genes and promoters (TNFα, IL-13) respond to NFAT1 independently of NFAT–AP-1 cooperation. These findings are consistent with previous data showing that the promoter/enhancer regions of the IL-2, IL-3, IL-4 and GM-CSF genes contain composite NFAT–AP-1 sites (Cockerill et al., 1993, 1995; Jain et al., 1995b; Rooney et al., 1995; reviewed in Rao et al., 1997; Kiani et al., 2000).
The crux of this paper, however, is that we can use the mutants to classify endogenous NFAT-dependent genes into categories requiring or not requiring cooperation between NFAT and AP-1. In this context, our finding that TNFα falls into the NFAT–AP-1-independent category was not predictable from earlier work in our own and other laboratories (Goldfeld et al., 1993; Tsai et al., 1996a,b). These studies examined only the proximal region of the TNFα promoter. While we have confirmed that the activity of the promoter region in reporter assays does not require a physical interaction between NFAT and AP-1, we have also shown that the endogenous TNFα gene is similarly independent of NFAT–AP-1 cooperation. This is important because the behavior of the proximal promoter does not necessarily predict the behavior of the endogenous gene: a priori, the TNFα gene might have contained distal enhancer elements, potentially containing NFAT–AP-1 composite sites, such as those identified in the GM-CSF, IL-3 and IL-4 cytokine genes (Cockerill et al., 1993, 1995; Jain et al., 1995b; Rooney et al., 1995).
It has been known for some time that, while most NFAT-dependent cytokine genes are maximally induced only upon combined stimulation of T cells with PMA and ionomycin, a few (e.g. TNFα) are substantially induced in response to ionomycin alone (Goldfeld et al., 1993; Jain et al., 1995b). Since NFAT is activated by calcium and calcineurin while Fos and Jun are synthesized and activated in response to PMA, the question arises as to whether we could have predicted the requirement for NFAT–AP-1 cooperation based on whether or not PMA was required for maximal activation of a given gene. However, activation of a gene with ionomycin alone does not rule out the possibility of NFAT cooperation with AP-1 proteins such as c-Jun and JunD, which are present in unstimulated T cells (Jain et al., 1993b). Conversely, stimulation with both ionomycin and PMA does not automatically imply a physical cooperation between NFAT and AP-1, since PMA might be needed for maximal activation of the basal transcription machinery, coactivator proteins or other transcription factors (including NF-κB and AP-1) that bind to specific target sites independently of NFAT. Indeed, in transient reporter assays, maximal activation of both the TNFα and IL-13 promoters required combined stimulation with PMA and ionomycin, despite the fact that both promoters were independent of strict NFAT–AP-1 cooperation since they could be efficiently activated by NFAT mutants unable to cooperate physically with AP-1 (Figure 5B and E). Thus the use of AP-1 interaction mutants of NFAT provides a far more stringent criterion to assess the dependence of gene expression on NFAT–AP-1 cooperation than the response to stimulation with PMA and ionomycin versus ionomycin alone.
Which are the features of the NFAT sites that do not require NFAT–AP-1 cooperation? Can common features be recognized in all such sites? Potentially, they could resemble NF-κB sites, to which NFAT proteins bind as dimers (Rao et al., 1997); this unusual architecture of NFAT sites is found in the κ3 NFAT site of the TNFα promoter, in a handful of other cytokine promoters and in the HIV-1 long terminal repeat (McCaffrey et al., 1994; Kinoshita et al., 1997; Rao et al., 1997; Macián and Rao, 1999). An interesting possibility is that it is characteristic of genes on which NFAT acts independently of any physical cooperation with AP-1. In this context we note that, in the TNFα promoter, the κ3 NFAT site is located immediately adjacent to a CRE element that binds pre-existing ATF2–Jun dimers and shows functional although not physical cooperation with the κ3 site (Tsai et al., 1996a,b). An alternative possibility is that in the ‘NFAT–AP-1-independent’ category of NFAT-dependent genes, NFAT binds to composite sites where it utilizes another nuclear partner distinct from AP-1. A precedent is provided by skeletal and cardiac muscle, where there is evidence that NFAT utilizes nuclear partners of the GATA family: NFAT2–GATA2 and NFAT3–GATA4 complexes have been implicated in skeletal muscle and cardiac hypertrophy, respectively (Molkentin et al., 1998; Musaro et al., 1999).
Might NFAT elicit different genetic programs in the presence and absence of Fos and Jun?
In immune cells, potential genetic programs regulated by NFAT–AP-1 cooperation include developmental processes such as positive and negative selection in the thymus, as well as cell proliferation, cytokine production, tolerance and cell death in the periphery. We have shown that the differential expression of cytokine genes by Th1 and Th2 cells is not related to subtype-specific differences in their requirement for AP-1. Th2 cells coordinately express IL-4, IL-5, IL-10 and IL-13, but while transcription of the IL-4 promoter was clearly AP-1 dependent, confirming previous reports (Jain et al., 1993a; Rooney et al., 1995), transcription of the IL-13 promoter was not. In contrast, the NFAT/FasL-dependent program of AICD requires cooperation between NFAT and AP-1: expression of a wild-type NFAT in D5 T cells potentiated cell death in response to stimulation with anti-CD3, but no potentiation of AICD was observed when the AP-1 interaction mutant of NFAT was expressed (Figure 8). This finding was not predictable from earlier work. The NFAT site in the fasL promoter does not contain an obvious adjacent AP-1 site, and the effect of NFAT on FasL expression has been suggested to be indirectly mediated through EGR proteins (Rengarajan et al., 2000).
An interesting implication of our results is that positive and negative selection in the thymus might have differing requirements for NFAT–AP-1 cooperation. Negative selection in the thymus is the process of clonal deletion whereby autoreactive T cell clones enter into apoptosis and are eliminated. This process, which is analogous to AICD, requires TCR stimulation of double-positive thymocytes and stimulation of CD28, which induces activation of Jnk (Rincon et al., 1998). CD28 knockout mice have an impaired negative selection and in vivo blockade of B7 proteins results in decreased thymocyte apoptosis (Samoilova et al., 1997; Noel et al., 1998). Both positive and negative selection occur at a similar stage of thymic development, the double-positive stage, and positive selection is blocked by CsA (Gao et al., 1988). NFAT4–/– mice show a moderate impairment in positive selection, providing further evidence for NFAT involvement in this process (Oukka et al., 1998). However, positive and negative selection differ in that positive selection occurs on thymic epithelial antigen-presenting cells under conditions of low antigen stimulation, while negative selection occurs on bone marrow-derived dendritic cells and requires a high threshold of antigen stimulation (Anderson et al., 1996). Thus it is plausible that positive selection, unlike negative selection, involves greater activation of NFAT relative to AP-1, initiating a program of thymocyte gene expression that dictates cell survival rather than cell death.
Finally, in non-immune cells such as skeletal and cardiac muscle, selection of GATA versus AP-1 proteins as partners would also be expected to result in differing patterns of NFAT-dependent gene expression (Molkentin et al., 1998; Musaro et al., 1999). Gene expression patterns dependent on NFAT–AP-1 versus NFAT–GATA cooperation might be mutually exclusive rather than overlapping, and might underlie the implementation of widely differing genetic programs.
Therapeutic implications
Our data also suggest that it might be useful to identify inhibitors that selectively disrupt the NFAT–AP-1 interaction without affecting the ability of NFAT proteins to interact with DNA. Disruption of the NFAT–AP-1 interaction in T cells should inhibit AICD as well as the production of many cytokines required for the productive immune response. Compounds that bind the CX loop or E′F loops of NFAT or the corresponding interacting regions of Fos and Jun, affecting their structure or blocking their accessibility for protein–protein interactions, would constitute a promising new class of immunosuppressive agents, distinct from either the calcineurin inhibitors CsA and FK506 or the specific peptide inhibitor of NFAT, VIVIT, which functions by disrupting the docking interaction between calcineurin and NFAT (Aramburu et al., 1999). The clinical usefulness of inhibiting NFAT activation is demonstrated by the widespread use of CsA and FK506 to prevent transplant rejection; unfortunately, these drugs have numerous adverse effects, most likely because they inhibit other signaling pathways that are also regulated by calcineurin (Kiani et al., 2000). Potentially, compounds that targeted the NFAT–AP-1 contact region would be immunosuppressive while lacking the adverse effects of CsA and FK506.
Materials and methods
Mutagenesis
Mutated versions of pNFATpXS(1–297) (Jain et al., 1995a), a bacterial expression plasmid encoding a His6-tagged form of the NFAT1 DBD, were constructed by site-directed PCR-mediated mutagenesis with Pfu polymerase (Promega). Introduction of the mutation was confirmed by DNA sequencing. The sequenced DNA fragment containing the mutation was subcloned back into pNFATpXS(1–297).
EMSAs
Recombinant wild-type NFAT1 DBD and its mutant derivatives, and full-length c-Fos and c-Jun proteins were expressed as His6-tagged proteins and purified as previously described (Jain et al., 1993a). Binding reactions were performed in a buffer containing 10 mM HEPES pH 7.5, 120 mM NaCl, 10% glycerol, 20 µg/ml poly(dI)–poly(dC), 0.8 mg/ml bovine serum albumin and 0.25 mM dithiothreitol, in a total volume of 15 µl. Approximately 10 000 c.p.m. of 32P-end-labeled ARRE2 probe (Jain et al., 1993a) (0.1–0.4 ng) were used in each binding reaction. After 20 min of incubation at room temperature, DNA–protein complexes were separated from free probe by electrophoresis in a 5% polyacrylamide gel. Dried gels were exposed to autoradiography film and/or quantified using a Molecular Dynamics Storm System.
Plasmids
The murine NFAT1 expression plasmids pLGP3-mNFAT1C, encoding isoform C of NFAT1, and pLGP3-mNFAT1ΔReg, which carries a deletion from residues 143 to 398 in the NFAT1 regulatory domain, have been described (Luo et al., 1996; Garcia-Rodriguez and Rao, 1998). pLGP3-mNFAT1 plasmids carrying mutations in the DBD were constructed by subcloning BglII–MluI fragments from mutated derivatives of the pNFATpXS(1–297) bacterial expression plasmid encoding the NFAT1 DBD into either pLGP3-mNFAT1C or pLGP3-mNFAT1ΔReg. The luciferase reporter plasmid NFAT/AP-1 3×Luc, which contains three copies of the distal NFAT–AP-1 site of the murine IL-2 promoter, and pIL2(–585)-Luc, which contains the murine IL-2 promoter, were kindly provided by D.J.McKean (Hedin et al., 1997). The chloramphenicol acetyl-transferase (CAT) reporter plasmid κ3 5×CAT (Goldfeld et al., 1993), which contains five copies of the κ3 site of the TNFα promoter, was a gift from A.Goldfeld. TNFα-Luc, in which the expression of the luciferase gene is regulated by the human TNFα promoter, was a gift from S.L.McKnight. pHGM (Cockerill et al., 1995), which contains the human GM-CSF promoter driving the expression of the CAT gene, was kindly provided by P.N.Cockerill. Luciferase reporter plasmids containing the murine IL-4 promoter (Szabo et al., 1993) and the human IL-13 promoter (Dolganov et al., 1996) were gifts from K.M.Murphy and D.B.Lewis, respectively.
Cell culture and transfections
Jurkat cells and HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 10 mM HEPES and 2 mM l-glutamine. D5 (Ar-5) (Rao et al., 1984) and D10 (D10.G4.1) (Kaye et al., 1983) murine T cell clones were cultured in DMEM supplemented with 10% fetal calf serum, l-glutamine, penicillin, streptomycin, non-essential amino acids, sodium pyruvate, vitamins, HEPES, 2-mercaptoethanol and 10 U/ml purified rat IL-2 (Collaborative Biomedical Products). D10 cell cultures were also supplemented with 25 U/ml recombinant IL-4. T cell clones were restimulated with antigen and irradiated antigen-presenting cells every 4 weeks.
Jurkat cells were transfected by electroporation in serum-free medium with pulses of 250 V and 960 µF. Typically, 107 cells were transfected with 2 µg of reporter plasmid and 5 µg of an NFAT1 expression plasmid or a control plasmid. Twenty-four hours after transfection, cells were stimulated with 2 µM ionomycin (Calbiochem) and 20 nM PMA (Calbiochem). Eight to 14 h after stimulation, cells were harvested and cell extracts were assayed for CAT or luciferase activity as described previously (Luo et al., 1996). One million HEK293 cells were transfected with 0.3 µg of reporter plasmid and 0.5 µg of NFAT1 expression plasmid by calcium phosphate DNA precipitation. In all cases, cotransfection of a renilla-expressing plasmid (Promega) or a human growth hormone-expressing plasmid (Luo et al., 1996) was used to normalize assays. NFAT1 expression was detected by western blotting using anti-67.1 antibody (Ho et al., 1994).
Retroviral infections to generate stable cell lines
Three different retroviral vectors were used: (i) RV-IRES-GFP, constructed by subcloning a pgk–neo cassette 3′ to the GFP gene in the RV-GFP vector (a gift from W.Sha); (ii) RV-NFAT1ΔReg-IRES-GFP; and (iii) RV-NFAT1 R468A/T535G ΔReg-IRES-GFP. The last two vectors were constructed by subcloning the DNAs encoding murine wild type and NFAT1ΔReg[R468A/T535G] in RV-IRES-GFP, respectively. Forty-eight hours after stimulation with antigen and irradiated antigen-presenting cells in medium supplemented with 40 U/ml IL-2, D5 and D10 cells were infected by spin infection with retrovirus-containing supernatants derived from the Phoenix Ecotropic packaging cell line (kindly provided by G.P.Nolan). Seventy-two hours after infection, cells were selected by GFP expression and G418 resistance. Individual clones were selected for appropriate levels of expression of NFAT1ΔReg.
RNase protection assay
Total cellular RNA was purified from resting cells or cells stimulated with 20 nM PMA and 2 µM ionomycin for 4 h, using Ultraspec reagent (Biotecx) and analyzed using the RiboQuant multiprobe RNase protection kit (Pharmingen) according to the manufacturer’s instructions. Where indicated, 1 µM CsA was added 15 min prior to stimulation. RNA from transiently transfected Jurkat cells was obtained after selecting transfected cells for expression of a cotransfected murine CD4 plasmid as previously described (Aramburu et al., 1999).
Activation-induced cell death
AICD was induced in D5 clones by stimulation with immobilized anti-CD3 (1 µg/ml 145-2C11, Pharmingen). After 48 h, cells were harvested, fixed in 70% cold ethanol, treated with RNase and stained with propidium iodide. The cell cycle of stained cells was analyzed by flow cytometry. The proportion of dead cells was determined from the pre-G1 peak.
Acknowledgments
Acknowledgements
We thank Drs P.N.Cockerill, A.Goldfeld, D.B.Lewis, D.J.McKean, S.L.McKnight, K.M.Murphy, G.P.Nolan and W.Sha for their generous gifts of reagents. This work was supported by NIH grant CA42471 and a Leukemia Society of America Stohlman Scholar Award (to A.R.). F.M. was supported by a postdoctoral fellowship from the Ministry of Education and Culture of Spain. C.G.-R. was the recipient of a Lady Tata Memorial Trust Fellowship.
References
- Agarwal S., Avni,O. and Rao,A. (2000) Cell type-restricted binding of the transcription factor NFAT to a distal IL-4 enhancer in vivo. Immunity, 12, 1–20. [DOI] [PubMed] [Google Scholar]
- Anderson G., Moore,N.C., Owen,J.J. and Jenkinson,E.J. (1996) Cellular interactions in thymocyte development. Annu. Rev. Immunol., 14, 73–99. [DOI] [PubMed] [Google Scholar]
- Aramburu J., Yaffe,M.B., Lopez-Rodriguez,C., Cantley,L.C., Hogan,P.G. and Rao,A. (1999) Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science, 285, 2129–2133. [DOI] [PubMed] [Google Scholar]
- Brunet A. et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell, 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Brunner T. et al. (1995) Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature, 373, 441–444. [DOI] [PubMed] [Google Scholar]
- Chen L., Oakley,M.G., Glover,J.N., Jain,J., Dervan,P.B., Hogan,P.G., Rao,A. and Verdine,G.L. (1995) Only one of the two DNA-bound orientations of AP-1 found in solution cooperates with NFATp. Curr. Biol., 5, 882–889. [DOI] [PubMed] [Google Scholar]
- Chen L., Glover,J.N., Hogan,P.G., Rao,A. and Harrison,S.C. (1998) Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature, 392, 42–48. [DOI] [PubMed] [Google Scholar]
- Cockerill P.N., Shannon,M.F., Bert,A.G., Ryan,G.R. and Vadas,M.A. (1993) The granulocyte–macrophage colony-stimulating factor/interleukin 3 locus is regulated by an inducible cyclosporin A-sensitive enhancer. Proc. Natl Acad. Sci. USA, 90, 2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerill P.N., Bert,A.G., Jenkins,F., Ryan,G.R., Shannon,M.F. and Vadas,M.A. (1995) Human granulocyte-macrophage colony-stimulating factor enhancer function is associated with cooperative interactions between AP-1 and NFATp/c. Mol. Cell. Biol., 15, 2071–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G.R. (1999) Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell, 96, 611–614. [DOI] [PubMed] [Google Scholar]
- Dhein J., Walczak,H., Baumler,C., Debatin,K.M. and Krammer,P.H. (1995) Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature, 373, 438–441. [DOI] [PubMed] [Google Scholar]
- Dolganov G., Bort,S., Lovett,M., Burr,J., Schubert,L., Short,D., McGurn,M., Gibson,C. and Lewis,D.B. (1996) Coexpression of the interleukin-13 and interleukin-4 genes correlates with their physical linkage in the cytokine gene cluster on human chromosome 5q23–31. Blood, 87, 3316–3326. [PubMed] [Google Scholar]
- Gao E.K., Lo,D., Cheney,R., Kanagawa,O. and Sprent,J. (1988) Abnormal differentiation of thymocytes in mice treated with cyclosporin A. Nature, 336, 176–179. [DOI] [PubMed] [Google Scholar]
- Garcia-Rodriguez C. and Rao,A. (1998) Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP). J. Exp. Med., 187, 2031–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J.N. and Harrison,S.C. (1995) Crystal structure of the heterodimeric bZIP transcription factor c-Fos–c-Jun bound to DNA. Nature, 373, 257–261. [DOI] [PubMed] [Google Scholar]
- Goldfeld A.E., McCaffrey,P.G., Strominger,J.L. and Rao,A. (1993) Identification of a novel cyclosporin-sensitive element in the human tumor necrosis factor α gene promoter. J. Exp. Med., 178, 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedin K.E., Bell,M.P., Kalli,K.R., Huntoon,C.J., Sharp,B.M. and McKean,D.J. (1997) Delta-opioid receptors expressed by Jurkat T cells enhance IL-2 secretion by increasing AP-1 complexes and activity of the NF-AT/AP-1-binding promoter element. J. Immunol., 159, 5431–5440. [PubMed] [Google Scholar]
- Hill C.S., Marais,R., John,S., Wynne,J., Dalton,S. and Treisman,R. (1993) Functional analysis of a growth factor-responsive transcription factor complex. Cell, 73, 395–406. [DOI] [PubMed] [Google Scholar]
- Ho A.M., Jain,J., Rao,A. and Hogan,P.G. (1994) Expression of the transcription factor NFATp in a neuronal cell line and in the murine nervous system. J. Biol. Chem., 269, 28181–28186. [PubMed] [Google Scholar]
- Jain J., McCaffrey,P.G., Valge-Archer,V.E. and Rao,A. (1992) Nuclear factor of activated T cells contains Fos and Jun. Nature, 356, 801–804. [DOI] [PubMed] [Google Scholar]
- Jain J., McCaffrey,P.G., Miner,Z., Kerppola,T.K., Lambert,J.N., Verdine,G.L., Curran,T. and Rao,A. (1993a) The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature, 365, 352–355. [DOI] [PubMed] [Google Scholar]
- Jain J., Miner,Z. and Rao,A. (1993b) Analysis of the preexisting and nuclear forms of nuclear factor of activated T cells. J. Immunol., 151, 837–848. [PubMed] [Google Scholar]
- Jain J., Burgeon,E., Badalian,T.M., Hogan,P.G. and Rao,A. (1995a) A similar DNA-binding motif in NFAT family proteins and the Rel homology region. J. Biol. Chem., 270, 4138–4145. [PubMed] [Google Scholar]
- Jain J., Loh,C. and Rao,A. (1995b) Transcriptional regulation of the IL-2 gene. Curr. Opin. Immunol., 7, 333–342. [DOI] [PubMed] [Google Scholar]
- Ju S.T., Panka,D.J., Cui,H., Ettinger,R., el-Khatib,M., Sherr,D.H., Stanger,B.Z. and Marshak-Rothstein,A. (1995) Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature, 373, 444–448. [DOI] [PubMed] [Google Scholar]
- Karin M. (1995) The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem., 270, 16483–16486. [DOI] [PubMed] [Google Scholar]
- Kasibhatla S., Genestier,L. and Green,D.R. (1999) Regulation of fas-ligand expression during activation-induced cell death in T lymphocytes via nuclear factor κB. J. Biol. Chem., 274, 987–992. [DOI] [PubMed] [Google Scholar]
- Kaye J., Porcelli,S., Tite,J., Jones,B. and Janeway,C.A.,Jr (1983) Both a monoclonal antibody and antisera specific for determinants unique to individual cloned helper T cell lines can substitute for antigen and antigen-presenting cells in the activation of T cells. J. Exp. Med., 158, 836–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani A., Rao,A. and Aramburu,J. (2000) Manipulating immune responses with immunosuppressive agents that target NFAT. Immunity, 12, 359–372. [DOI] [PubMed] [Google Scholar]
- Kinoshita S., Su,L.S., Amano,M., Timmerman,L.A., Kaneshima,H. and Nolan,G.P. (1997) The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity, 6, 235–244. [DOI] [PubMed] [Google Scholar]
- Latinis K.M., Norian,L.A., Eliason,S.L. and Koretzky,G.A. (1997) Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J. Biol. Chem., 272, 31427–31434. [DOI] [PubMed] [Google Scholar]
- Luo C., Burgeon,E., Carew,J.A., McCaffrey,P.G., Badalian,T.M., Lane,W.S., Hogan,P.G. and Rao,A. (1996) Recombinant NFAT1 (NFATp) is regulated by calcineurin in T cells and mediates transcription of several cytokine genes. Mol. Cell. Biol., 16, 3955–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macián F. and Rao,A. (1999) Reciprocal modulatory interaction between human immunodeficiency virus type 1 Tat and transcription factor NFAT1. Mol. Cell. Biol., 19, 3645–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey P.G., Goldfeld,A.E. and Rao,A. (1994) The role of NFATp in cyclosporin A-sensitive tumor necrosis factor-α gene transcription. J. Biol. Chem., 269, 30445–30450. [PubMed] [Google Scholar]
- Mittelstadt P.R. and Ashwell,J.D. (1998) Cyclosporin A-sensitive transcription factor Egr-3 regulates Fas ligand expression. Mol. Cell. Biol., 18, 3744–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin J.D., Lu,J.R., Antos,C.L., Markham,B., Richardson,J., Robbins,J., Grant,S.R. and Olson,E.N. (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell, 93, 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller C.W., Rey,F.A., Sodeoka,M., Verdine,G.L. and Harrison,S.C. (1995) Structure of the NF-κB p50 homodimer bound to DNA. Nature, 373, 311–317. [DOI] [PubMed] [Google Scholar]
- Musaro A., McCullagh,K.J., Naya,F.J., Olson,E.N. and Rosenthal,N. (1999) IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature, 400, 581–585. [DOI] [PubMed] [Google Scholar]
- Noel P.J., Alegre,M.L., Reiner,S.L. and Thompson,C.B. (1998) Impaired negative selection in CD28-deficient mice. Cell. Immunol., 187, 131–138. [DOI] [PubMed] [Google Scholar]
- Oukka M., Ho,I.C., de la Brousse,F.C., Hoey,T., Grusby,M.J. and Glimcher,L.H. (1998) The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity, 9, 295–304. [DOI] [PubMed] [Google Scholar]
- Peterson B.R., Sun,L.J. and Verdine,G.L. (1996) A critical arginine residue mediates cooperativity in the contact interface between transcription factors NFAT and AP-1. Proc. Natl Acad. Sci. USA, 93, 13671–13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongubala J.M., Van Beveren,C., Nagulapalli,S., Klemsz,M.J., McKercher,S.R., Maki,R.A. and Atchison,M.L. (1993) Effect of PU.1 phosphorylation on interaction with NF-EM5 and transcriptional activation. Science, 259, 1622–1625. [DOI] [PubMed] [Google Scholar]
- Rao A., Faas,S.J. and Cantor,H. (1984) Activation specificity of arsonate-reactive T cell clones. Structural requirements for hapten recognition and comparison with monoclonal antibodies. J. Exp. Med., 159, 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A., Luo,C. and Hogan,P.G. (1997) Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol., 15, 707–747. [DOI] [PubMed] [Google Scholar]
- Rengarajan J., Mittelstadt,P.R., Mages,H.W., Gerth,A.J., Kroczek,R.A., Ashwell,J.D. and Glimcher,L.H. (2000) Sequential involvement of NFAT and EGR transcription factors in FasL regulation. Immunity, 12, 293–300. [DOI] [PubMed] [Google Scholar]
- Rincon M., Whitmarsh,A., Yang,D.D., Weiss,L., Derijard,B., Jayaraj,P., Davis,R.J. and Flavell,R.A. (1998) The JNK pathway regulates the in vivo deletion of immature CD4(+)CD8(+) thymocytes. J. Exp. Med., 188, 1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney J.W., Hoey,T. and Glimcher,L.H. (1995) Coordinate and cooperative roles for NF-AT and AP-1 in the regulation of the murine IL-4 gene. Immunity, 2, 473–483. [DOI] [PubMed] [Google Scholar]
- Samoilova E.B., Horton,J.L., Bassiri,H., Zhang,H., Linsley,P.S., Carding,S.R. and Chen,Y. (1997) B7 blockade prevents activation-induced cell death of thymocytes. Int. Immunol., 9, 1663–1668. [DOI] [PubMed] [Google Scholar]
- Sun L.J., Peterson,B.R. and Verdine,G.L. (1997) Dual role of the nuclear factor of activated T cells insert region in DNA recognition and cooperative contacts to activator protein 1. Proc. Natl Acad. Sci. USA, 94, 4919–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo S.J., Gold,J.S., Murphy,T.L. and Murphy,K.M. (1993) Identification of cis-acting regulatory elements controlling interleukin-4 gene expression in T cells: roles for NF-Y and NF-ATc Mol. Cell. Biol., 13, 4793–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjian R. and Maniatis,T. (1994) Transcriptional activation: a complex puzzle with few easy pieces. Cell, 77, 5–8. [DOI] [PubMed] [Google Scholar]
- Tsai E.Y., Jain,J., Pesavento,P.A., Rao,A. and Goldfeld,A.E. (1996a) Tumor necrosis factor α gene regulation in activated T cells involves Atf-2/Jun and NFATp. Mol. Cell. Biol., 16, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai E.Y., Yie,J.M., Thanos,D. and Goldfeld,A.E. (1996b) Cell-type-specific regulation of the human tumor necrosis factor α gene in B cells and T cells by NFATp, and ATF-2/Jun. Mol. Cell. Biol., 16, 5232–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen J.E. and Samelson,L.E. (1999) T cell antigen-receptor signal transduction. Curr. Opin. Immunol., 11, 242–248. [DOI] [PubMed] [Google Scholar]
- Wolfe S.A., Zhou,P., Dotsch,V., Chen,L., You,A., Ho,S.N., Crabtree,G.R., Wagner,G. and Verdine,G.L. (1997) Unusual Rel-like architecture in the DNA-binding domain of the transcription factor NFATc. Nature, 385, 172–176. [DOI] [PubMed] [Google Scholar]
- Zhou P., Sun,L.J., Dotsch,V., Wagner,G. and Verdine,G.L. (1998) Solution structure of the core NFATC1/DNA complex. Cell, 92, 687–696. [DOI] [PubMed] [Google Scholar]