

Abstract
Recent studies have shown that activation of complement and contact systems results in the generation of antibacterial peptides. Streptococcus pyogenes, a major bacterial pathogen in humans, exists in >100 different serotypes due to sequence variation in the surface-associated M protein. Cases of invasive and life-threatening S. pyogenes infections are commonly associated with isolates of the M1 serotype, and in contrast to the large majority of M serotypes, M1 isolates all secrete the SIC protein. Here, we show that SIC interferes with the activation of the contact system and blocks the activity of antibacterial peptides generated through complement and contact activation. This effect promotes the growth of S. pyogenes in human plasma, and in a mouse model of S. pyogenes sepsis, SIC enhances bacterial dissemination, results which help explain the high frequency of severe S. pyogenes infections caused by isolates of the M1 serotype.
Keywords: Antimicrobial Peptides, Bacteria, Blood Coagulation Factors, Complement, Innate Immunity, Kallikrein, Contact System, SIC, Streptococcus Pyogenes
Introduction
The innate immune system provides a rapid and nonspecific defense at biological boundaries prone to infection. Essential elements involved in this immediate host response are antimicrobial peptides (AMPs)2 and complement components (1–3). The complement system can be activated through three different pathways; the classical, alternative, and lectin pathways (4). Although the initiation of these complement cascades differs, they all converge at the level of C3 with the generation of several C3 peptide fragments including the anaphylatoxin C3a. This proinflammatory peptide has a number of biological effects, such as enhancement of vascular permeability, contraction of smooth muscle cells, and histamine release from mast cells (5). C3a also exerts a potent antimicrobial activity, a property that has been highly conserved from invertebrates to humans (6–8).
Another branch of innate immunity is represented by the human contact system, which generates AMPs upon activation (9). This system comprises three serine proteinases: factor XII (FXII), factor XI (FXI), and plasma kallikrein (PK), and the nonenzymatic cofactor high molecular weight kininogen (HK) (10–12). HK consists of six domains: the cystatin-like domains D1–D3; the bradykinin-containing domain D4; domain D5, which binds to negatively charged surfaces; and domain D6, which binds FXI and PK (see Fig. 2A, schematic). Classical consequences of contact activation are cleavage of HK leading to the release of the potent proinflammatory peptide bradykinin and the initiation of the intrinsic pathway of coagulation through FXI activation. When activated at the surface of significant human bacterial pathogens, including Streptococcus pyogenes, further processing of HK results in the generation of antibacterial fragments from domain D3 of HK, mainly involving a 26-amino acid sequence (9).
FIGURE 2.

SIC binds to the NAT26 peptide sequence of HK and inhibits its antibacterial activity. A, schematic representation of HK. S, signal sequence; D1–D3, cystatin-like domains; D4, bradykinin-containing domain; D5, domain binding to negatively charged surfaces; D6, domain binding to FXI and plasma kallikrein. Peptides derived from the D3 sequence and used in this study are shown. Numbers refer to amino acid residue positions (bars are not in scale), and the letters indicate three NH2-terminal amino acids followed by the total number of residues. The position of the peptides in the D3 domain is also indicated. B, microtiter plates were coated with 0.5 μg/ml of the synthetic peptides from domain D3 of HK: GKD25 (○), GCP28 (Δ), NAT26 (▾), KKY30 (▿), RET27 (⊕), and LDC27 (■). After incubation with SIC in a serial dilution, bound protein was detected with a specific antiserum against SIC and a peroxidase-labeled secondary antibody. C, various amounts of the D3-derived peptides were applied to a PVDF membrane. The membrane was incubated with radiolabeled SIC and autoradiographed. D, homology model of the D3 domain of HK. Peptides NAT26 (red) and LDC27 (green) are located in two separate intersecting antiparallel β-strands. E, AP1 bacteria were incubated with NAT26 (black bars) or LL-37 (gray bars) at 0.642 μm and 0.446 μm, respectively. At these concentrations, no cfu were detected. The bactericidal effect of the peptides was inhibited with indicated concentrations of SIC. Bars represent the mean ± S.E. of at least three experiments.
S. pyogenes is a common and important human bacterial pathogen causing a wide spectrum of diseases ranging from uncomplicated throat and skin infections to life-threatening invasive conditions, such as necrotizing fasciitis, septicaemia, and toxic shock syndrome (13–15). Based on antigenic differences in the M protein, a surface protein of S. pyogenes, isolates are divided into >100 serotypes (16, 17). Several studies have demonstrated that the M1 serotype is the serotype most frequently associated with invasive infections and toxic shock (18–23), and all strains of the M1 serotype secrete the SIC (streptococcal inhibitor of complement) protein that inhibits the function of the membrane attack complex of complement (24). The sic gene shows a high degree of variation among different M1 strains, and nearly 300 alleles are known (25). SIC also binds to and interferes with the activity of several antibacterial proteins and peptides such as lysozyme, LL-37, members of the defensin family, and the chemokine MIG/CXCL9 (26–29). Recently, SIC was also shown to inhibit an antibacterial peptide produced by Streptococcus salivarius, a bacteriocin-like inhibitory substance (30).
In the present work, we demonstrate that SIC interferes with innate immunity functions of the complement and contact systems. As a consequence, SIC enhances the virulence of S. pyogenes by promoting growth in human plasma and bacterial dissemination in a mouse model of sepsis.
EXPERIMENTAL PROCEDURES
Bacteria, Growth Conditions, Plasma Sources, Analysis of Bacterial Multiplication, and SIC Production during Growth
The S. pyogenes strain AP1 (40/58), was from the World Health Organization Collaborating Centre for Reference and Research on Streptococci (Prague, Czech Republic). The mutant strain SIC− has been described previously (29). Bacteria were cultivated in Todd-Hewitt broth (TH; Difco) at 37 °C. Fresh frozen plasma from healthy individuals was obtained from the blood bank at Lund University Hospital (Lund, Sweden) and kept at −80 °C until use. Human plasma depleted of HK, PK, FXII, or FXI, respectively, was purchased from George King BioMedical, Inc. (Overland Park, KS).
Bacterial cultivation in plasma was performed as follows. 10 μl of an overnight bacterial culture in TH was added to 250 μl plasma, and bacteria were left to grow at 37 °C. At various time points, including time 0, growth was monitored by plating appropriate dilutions of the bacterial solution on TH agar plates. Plates were incubated overnight at 37 °C, and the number of cfu were determined. The multiplication factor (MF) was calculated by dividing the number of cfu at the individual time points with the number of cfu at time point zero. Bacteria grown for 8 h in plasma (undiluted or diluted 1:1 in TH) were collected by centrifugation. Plasma supernatants were analyzed for SIC content by ELISA. Supernatants were also precipitated with 5% TCA for 30 min on ice followed by centrifugation at 15,000 × g (4 °C for 20 min). Precipitated material was dissolved in SDS sample buffer and subjected to SDS-PAGE and Western blot analysis. The bacterial cells were washed with PBS, and bound proteins were eluted with 0.1 m glycine-HCl, pH 2.0. The pH of the eluted material was raised to 7.5 with the addition of 1 m Tris. Eluted proteins were TCA-precipitated and analyzed with SDS-PAGE and Western blot.
Proteins, Antibodies, and Iodination
Human HK was purchased from Kordia. The synthetic peptides based on sequences in domain D3 of HK were described previously (31) and are shown in Fig. 2A. LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) and peptide HKH20 (479HKHGHGHGKHKNKGKKNGKH498 in domain D5 of HK) were synthesized by Innovagen AB (Lund, Sweden). The complement peptide C3a was from Calbiochem, and rabbit anti-C3 was from Serotec. Protein SIC was purified from the S. pyogenes strain AP1 as described (24). Polyclonal antisera to protein SIC and NAT26 were raised in rabbits. HRP-conjugated goat anti-rabbit IgG was purchased from Pierce, and HRP-conjugated protein A was from Sigma. FITC-conjugated anti-rabbit IgG F(ab′)2 was purchased from Sigma, and anti-NAT26 F(ab′)2 fragments were prepared as described (9). Polyclonal anti-SIC IgG was affinity-purified using protein A-Sepharose (GE Healthcare), and the FabRICATOR® Kit (Genovis) was then utilized for preparation and purification of F(ab′)2 antibody fragments. Proteins were radiolabeled with 125I using iodobeads (Pierce) as described by the manufacturer. Binding of radiolabeled protein to bacteria was performed as described (32).
ELISA
Microtiter plates (Maxisorb, NUNC, Denmark) were coated with peptides spanning the HK domain D3 (0.5 μg/ml). After washing with PBST (PBS containing 0.05% Tween 20), the plates were incubated with protein SIC in a serial dilution (2n; starting concentration, 5 μg/ml). Bound protein was detected with a polyclonal rabbit antibody against protein SIC (1:500), followed by HRP-conjugated goat anti-rabbit IgG (1:5000). All incubations were done at 37 °C for 1 h and followed by a washing step. Substrate solution, 0.1% diammonium-2,2-azino-bis-(3-ethyl-2,3-dihydrobenzthiazoline)-6-sulfonate, 0.012% H2O2 in 100 mm citric acid, 100 mm NaH2PO4, pH 4.5, was added and the change in absorbance at 405 nm was determined after 30 min. Plates were also coated with plasma supernatants from bacterial growth, and the content of SIC in the samples was determined as above.
SDS-PAGE, Western Blot, and Slot-binding Analysis
SDS-PAGE was performed as described by Laemmli (33) using a polyacrylamide concentration of 10% and 3.3% cross-linking or a Tricine-SDS/PAGE system was used (34). Separated proteins were transferred to PVDF membranes (Amersham Biosciences). Membranes were blocked with PBST containing 5% dry milk powder (blocking buffer), incubated with primary antibody (rabbit anti-SIC, 1:1000) in blocking buffer for 30 min at 37 °C. Following a washing step with PBST, the membranes were incubated with secondary antibodies (HRP-conjugated protein A, 1:5000) in blocking buffer for 30 min at 37 °C. The membranes were washed, and bound antibodies were detected by chemiluminescence. When radiolabeled SIC was analyzed, gels were dried, and radioactivity was visualized by autoradiography using Kodak x-Omat AR films and regular intensifying screens. Peptides were also directly applied to PVDF membranes using a Milliblot-D system (Millipore). Membranes were blocked in TBS (0.05 m Tris-HCl, pH 7.5, 0.15 m NaCl) containing 3% bovine serum albumin, incubated with 125I-labeled protein SIC for 3 h, and washed with TBS containing 0.05% Tween 20. Autoradiography was carried out as described above.
Antimicrobial Assay
AP1 bacteria were grown to mid-log phase in TH, washed, and diluted in 10 mm Tris-HCl, pH 7.5, containing 5 mm glucose. 50 μl of bacterial solution (2 × 106 cfu/ml) was incubated with NAT26, LL-37, HKH20, or C3a at various concentrations for 1 h at 37 °C. In subsequent experiments, bacteria were incubated with bactericidal concentrations of the peptides together with various concentrations of protein SIC. The bactericidal activity was quantified by plating serial dilutions of the incubation mixtures on TH agar, incubated overnight at 37 °C, and the number of cfu were determined.
Flow Cytometry
S. pyogenes AP1 bacteria were cultivated overnight at 37 °C, washed, and resuspended in PBS (final concentration of 2 × 108 cfu/ml). 400 μl of the bacterial solution (8 × 107 cfu) was incubated with 400 μl PBS or plasma together with various concentrations of protein SIC for 1 h at 37 °C. The bacterial cells were collected, washed with PBS, and resuspended in 75 μl PBS. Then, 25 μl bacterial solution (2.6 × 107 cfu) was incubated with rabbit anti-NAT26 F(ab′)2 fragments (0.2 mg/ml) or rabbit anti-C3 (0.1 mg/ml) for 30 min at room temperature. Following a washing step with PBS, the bacterial cells were incubated with FITC-conjugated goat anti-rabbit IgG F(ab′)2 fragments (11 μg/ml) for 30 min at room temperature. After washing with PBS, bacteria were resuspended in PBS, and samples were analyzed on a FACSCalibur (Becton Dickinson), and 100,000 events were acquired using logarithmic settings. Data were analyzed using histogram plots of FITC fluorescence intensity acquired in Cell Quest Pro software.
Animal Experiments and Clotting Assay
AP1 bacteria were grown to early logarithmic phase (A620 nm ∼ 0.5), washed, and diluted in PBS to 1 × 108 cfu/ml. Female Balb/c mice (10 weeks old) were anesthetized with isofluorane and injected subcutaneously in an air pouch on the neck with 200 μl of the bacterial solution (2 × 107 cfu/animal) or with 200 μl PBS (control group). At 8 and 20 h post-infection, 200 μl protein SIC (200 μg/dose) or PBS was injected intraperitoneally in infected animals. (Mice in the control group were injected with PBS.) At 30 h post-infection, animals were terminally anesthetized with isofluorane, and blood was drawn by cardiac puncture into polypropylene tubes containing one-tenth volume of 3.8% trisodium citrate. Following centrifugation, the separated plasma was collected. The concentration of IL-6 in the plasma was determined using an ELISA kit (Invitrogen), and clotting times were measured in a coagulometer (Amelung). For the activated partial thromboplastin time (aPTT), 50 μl of plasma was prewarmed at 37 °C for 60 s, incubated with 50 μl DAPTTIN (Technoclone) for 60 s, followed by the addition of 50 μl of 30 mm CaCl2, and the time to form a clot was measured. For determination of bacterial dissemination, the spleens were removed, kept on ice, and homogenized in PBS, and the materials were plated on TH agar and incubated overnight at 37 °C, and the number of cfu were determined. p values were determined by using the Mann-Whitney U test. The animal experiments were approved by the regional ethic committee for animal experimentation (permit M220-08).
Immunofluorescence Microscopy
The S. pyogenes strains AP1 and SIC− were grown overnight at 37 °C. Ten μl from these cultures were inoculated into 250 μl of citrated plasma (1:1 in TH), and bacteria were allowed to grow at 37 °C for 20 h in Eppendorf tubes. The bacterial cells were washed twice in PBS supplemented with goat serum to a final concentration of 1% (wash buffer). Bacteria were then incubated for 30 min on ice with polyclonal rabbit anti-SIC IgG F(ab′)2 fragments (0.1 mg/ml), washed with wash buffer, and incubated with secondary fluorochrome-conjugated goat anti-rabbit F(ab′)2 fragments (Alexa Fluor 594; Invitrogen) (1:600), for 30 min on ice. The bacterial cells were washed with wash buffer, and bound antibodies were visualized in a fluorescence microscope (Nikon Eclipse TE300 inverted fluorescence microscope equipped with a Hamamatsu C4742–95 cooled CCD camera, using a Plan Apochromat 100× objective with a numerical aperture of 1.4 (Nikon)). The software used for acquisition was NIS-Elements (version 3.0; Nikon). The same exposure time was used for all images, and during post-processing, all images were treated identically.
Negative Staining and Transmission Electron Microscopy
Negative staining of AP1 bacteria incubated with PBS, plasma, or plasma with protein SIC (see above) was performed with 0.75% uranyl formate as described (35). Specimens were examined in a Jeol JEM 1200 EX transmission electron microscope operated at 60 kV accelerating voltage. Digital images were recorded with a Gatan Multiscan 791 CCD camera.
Homology Modeling of D3 Domain of HK
Amino acids 261–370 in the D3 domain of HK was aligned with human cystatin F (36) and subjected to a Swiss model (37) automated protein homology modeling using cystatin F (Protein Data Bank code 2CH9, chain A) as the template. The model was edited and visualized using VMD (version 1.8.7) (38) and Tachyon Ray tracer (39).
RESULTS
Multiplication of S. pyogenes in Human Plasma Is Controlled by Complement and Contact Systems
To investigate the importance of contact activation in controlling bacterial growth, the S. pyogenes isolate AP1 (a strain of the M1 serotype used throughout this study) was cultivated in human plasma deficient of the various contact factors. As compared with normal plasma, the growth rate was enhanced in plasma that was deficient of PK, FXII, or HK (Fig. 1A). After 8 h of growth, the bacterial numbers were significantly higher in plasma lacking either of these contact factors (Fig. 1A, inset), suggesting that the generation of antibacterial peptide fragments from HK was affected. Factor XI triggers the intrinsic pathway of coagulation but does not participate in the cleavage of HK, and the growth of AP1 bacteria in FXI deficient plasma was the same as in normal plasma (Fig. 1A).
FIGURE 1.

Growth of S. pyogenes in human plasma is controlled by the contact and complement systems. A, AP1 bacteria were grown in human citrated plasma samples: normal plasma (●), PK-deficient plasma (♢), FXII-deficient plasma (△), HK-deficient plasma (○), or FXI-deficient plasma (□). At indicated time points, bacteria were plated on TH agar plates. Plates were incubated overnight at 37 °C; the number of cfu was determined, and MF was calculated by dividing the number of cfu at each time point with the number of cfu at time 0. Mean values of at least four experiments are shown. The bars in the inset represent the mean values ± S.E. after 8 h of growth. p values were determined by using the Mann-Whitney U test. B, AP1 bacteria were cultivated in citrated plasma (black bars), serum (gray bars), and heat-inactivated serum (white bars). Bacterial growth was followed as above. Mean values of three experiments ± S.E. are shown.
The complement cascade is another proteolytic system that is activated at sites of infection. Therefore, the contribution of this system in reducing the multiplication of S. pyogenes was examined by performing growth experiments in human serum. Compared with plasma, AP1 bacteria grew better in serum, and in complement-inactivated serum (heated to 56 °C), a further increase of bacterial multiplication was recorded (Fig. 1B). Taken together, the results suggest that the contact and complement systems both interfere with S. pyogenes growth in human blood.
SIC Interferes with Antibacterial Activity of HK-derived Peptides
Following assembly of the contact factors on the bacterial surface, activation of the contact system results in the generation of antibacterial peptides derived from the D3 domain of HK (9). SIC is secreted by some S. pyogenes strains, including all isolates of the M1 serotype, and an interaction between SIC and HK mediated through domains D3 and D5 was demonstrated recently (40). Here, synthetic peptides spanning the D3 domain (Fig. 2A) were analyzed by indirect ELISA for binding to SIC. As shown in Fig. 2B, a strong binding was obtained with the NAT26 peptide. Furthermore, peptide LDC27 in the COOH terminus of D3 interacted with SIC, whereas the other peptides showed no affinity (Fig. 2B). The results were in full agreement with experiments where the peptides were applied to PVDF filters and probed with 125I-SIC (Fig. 2C). A homology model of the cystatin-like D3 domain of HK was generated by aligning amino acids 261–370 of HK with human cystatin F (E-value 4.33 × 10−10) (36). The Swiss-model automated homology modeling server (37) revealed a typical cystatin-like structure with one α-helix and four antiparallel β-strands (Fig. 2D). Interestingly, the SIC-binding peptides NAT26 (red) and LDC27 (green) are predicted to be located in two separate intersecting antiparallel β-strands forming surface-exposed epitopes within the D3 domain (Fig. 2D). Intact domain D3 is antibacterial, and this activity is mainly located in the NAT26 sequence (9). When tested at concentrations of 12–13 μm, the peptide LDC27 reduced the bacterial number with ∼8% as compared with a 100% cfu reduction obtained with the peptide NAT26 (9), suggesting that the antibacterial activity of the NAT26 peptide could be blocked by SIC. Indeed, at a bactericidal concentration of NAT26 (0.642 μm), SIC blocked the killing of AP1 bacteria. In these experiments, the inhibitory effect of SIC on the classical antibacterial peptide LL-37 (0.446 μm) was included as a positive control (Fig. 2E). Fragments of domain D5 of HK, including the sequence HKH20, are also antibacterial (41). Such fragments are, however, not generated as a result of contact activation (9), but at the site of infection, they are produced through proteolytic cleavage of HK by enzymes released by activated neutrophils (41). Also, the antibacterial activity of HKH20 against AP1 was blocked by SIC (data not shown).
SIC Blocks Antibacterial Activity of Complement C3a
Complement activation leads to cleavage of C3 and generation of the inflammatory mediator C3a. Previous studies have demonstrated that this peptide is capable of killing various microorganisms (6–8). Here, C3a also was found to kill AP1 bacteria (Fig. 3A), and at an antibacterial concentration of the peptide (0.22 μm), SIC inhibited this activity and promoted bacterial growth (Fig. 3B). However, higher molar concentrations of SIC were required to efficiently block C3a activity as compared with peptides NAT26 and LL-37. Whereas SIC at a concentration of 60 nm inhibited the activity of these peptides to 80–90% (Fig. 2E), C3a still killed ∼60% of the bacteria (Fig. 3B).
FIGURE 3.

SIC inhibits the bactericidal activity of C3a. A, S. pyogenes strain AP1 (2 × 106 cfu/ml) was incubated with C3a at indicated concentrations and cfu were determined. B, the bactericidal effect of C3a at 0.22 μm was inhibited with various concentrations of SIC. Experiments were repeated at least three times, and mean values ± S.E. are shown.
Antibacterial Action of Contact System Is Inhibited by SIC
The activation of the contact system at bacterial surfaces requires initial binding of the contact factors. HK has been demonstrated to bind to the surface of S. pyogenes (42), and therefore, we tested whether SIC could interfere with this binding. Binding of radiolabeled HK to AP1 bacteria (Fig. 4A) was efficiently blocked by SIC (Fig. 4B), indicating that SIC not only blocks the action of antibacterial peptides generated through contact activation but also interferes with the activation process itself. We therefore investigated whether SIC influences bacterial growth in human plasma, and exogenously added SIC was found to enhance the multiplication of AP1 bacteria (Fig. 5A). This finding suggests that antibacterial HK fragments are bound and inactivated by SIC, and/or that SIC blocks binding of HK to the bacterial surface and thereby reduces the generation of antibacterial D3 fragments.
FIGURE 4.
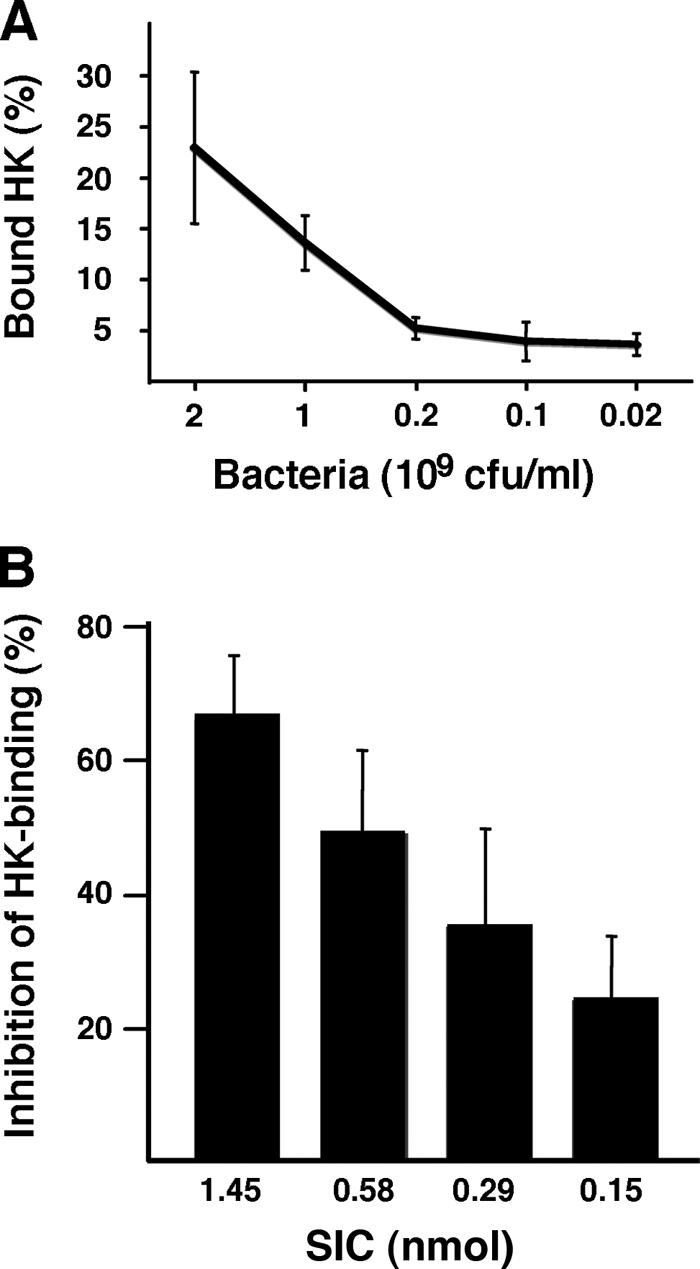
Binding of HK to S. pyogenes is inhibited by SIC. A, AP1 bacteria at indicated concentrations were tested for binding of radiolabeled HK. B, the binding of radiolabeled HK to AP1 bacteria (2 × 109 cfu/ml) was inhibited with various amounts of SIC. The bars represent the mean ± S.E. of at least three experiments.
FIGURE 5.

SIC protects S. pyogenes bacteria from killing in plasma. A, AP1 bacteria were cultivated in human citrated plasma with various concentrations of SIC: plasma alone (●), plasma+SIC 7.65 μm (□), plasma+SIC 15.3 μm (△), and plasma+SIC 23 μm (○). At indicated time points, bacteria were plated on TH agar plates. Plates were incubated overnight at 37 °C, the number of cfu was determined, and the MF was calculated by dividing the number of cfu at each time point with the number of cfu at time 0. The bars in the inset represent the mean values ± S.E. of at least three experiments following 8 h of growth. B, AP1 bacteria (8 × 107 cfu) were incubated with human citrated plasma at 37 °C for 1 h ± SIC. Bacteria were washed with PBS, and the binding of HK or HK fragments was analyzed by flow cytometry. Rabbit anti-NAT26 F(ab′)2-fragments were used as primary antibodies, and FITC-conjugated goat anti-rabbit F(ab′)2-fragments were employed in the secondary step. Blue line, background fluorescence obtained with secondary antibodies; green line, detection of HK/D3-containing fragments with primary and secondary antibodies; red line, detection of HK/D3-containing fragments in the presence of SIC (13.5 μm). C–E, an aliquot of AP1 bacteria incubated with human plasma (B) was analyzed by electron microscopy following negative staining. C, AP1 bacteria incubated with PBS. D, AP1 bacteria incubated with plasma. E, AP1 bacteria incubated with plasma in the presence of SIC (13.5 μm). Scale bar, 2 μm.
Next, AP1 bacteria were incubated with human plasma, extensively washed, and then incubated with antibodies against NAT26, followed by FITC-labeled secondary antibodies. Flow cytometry was then used to analyze bacterial cells interacting with anti-NAT26. The background signal was defined as the fluorescence signal in the presence of secondary antibodies alone (Fig. 5B, blue line). The increase in the fluorescence signal obtained in the presence of both primary and secondary antibodies (Fig. 5B, green line) represents that proportion of the cell population that have specifically bound the primary antibodies. In Fig. 5B, binding of HK or NAT26-containing HK fragments were detected in 35% of the population as these cells have a fluorescent intensity greater than background as calculated using Cell Quest software. For bacteria preincubated with SIC (Fig. 5B, red line), only 13% of the population have a fluorescent intensity greater than background, corresponding to ∼60% inhibition. The bacterial cells were also analyzed by electron microscopy following negative staining. Contrary to bacteria incubated with PBS (Fig. 5C), the cell walls of bacteria exposed to plasma were disintegrated, resulting in the leakage of cytoplasmic material (Fig. 5D). This effect was much less pronounced when excess amount of SIC was present during plasma incubation (Fig. 5E), suggesting that SIC interferes with the antibacterial activity of peptides generated through contact activation.
Multiplication of S. pyogenes in Plasma Is Enhanced by SIC
When AP1 bacteria were grown in human plasma, only trace amounts of SIC could be detected in the plasma by Western blot or ELISA following 8 h of growth. This observation indicated that the SIC secreted by the bacteria in plasma environment could be bound to the bacterial surface where it blocks binding of HK or the activity of generated antibacterial HK fragments. The SIC-deficient isogenic mutant strain SIC− derived from AP1 (29) was included as a negative control in these experiments. Due to the poor growth of S. pyogenes in 100% plasma, the bacteria were cultivated in 50% plasma to enhance the number of bacterial cells. Following growth, bacteria were carefully washed, and proteins associated with the surface were released by low pH and subjected to Western blot analysis using antibodies against SIC. A prominent band of 35 kDa, released from AP1 bacteria, reacted with the SIC antibody (Fig. 6A, lane 1). No such band was identified in the material eluted from the SIC− mutant (Fig. 6A, lane 2). SIC eluted from AP1 bacteria appeared slightly smaller than the SIC control isolated from AP1 growth medium (Fig. 6A, lane 3), indicating that the surface-bound SIC represents a processed form of the protein. In addition, immunoreactive bands of ∼31, 50, and 55 kDa, respectively, were present in the material eluted from both strains. AP1 bacteria bind several abundant plasma proteins, including albumin, fibrinogen, and IgG, via the surface proteins H and M1 (43, 44). Thus, the band migrating at 55 kDa corresponds to IgG heavy chains interacting with the HRP-conjugated protein A used in the assay. M1 protein is released from the bacterial surface (44), and the 50-kDa band represents M1 protein interacting with the rabbit antibody against SIC through nonimmune IgGFc binding (44). AP1 bacteria secretes IdeS, a proteolytic enzyme that cleaves IgG heavy chains in the hinge region (45). Surface-bound IgG is cleaved by IdeS, and under reducing conditions, heavy chain fragments of 31 kDa are generated, which bind the secondary protein A reagent in the Western blot assay. NH2-terminal sequencing of this band confirmed it to be the 31-kDa IgG fragment mentioned above.
FIGURE 6.
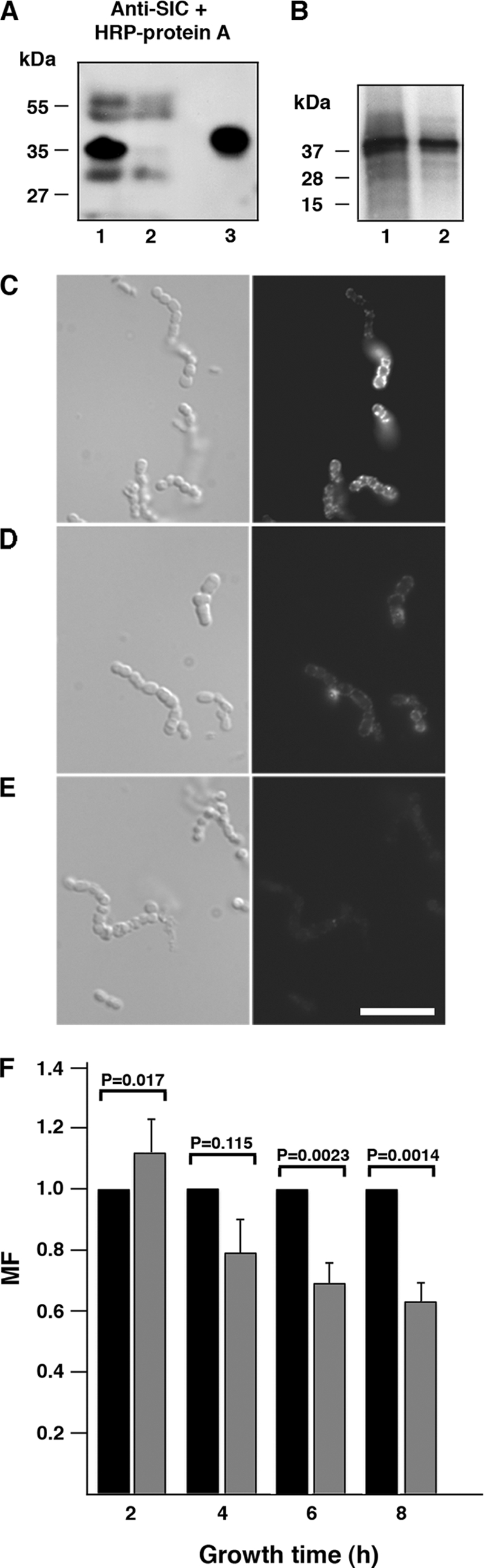
SIC associated with the surface of S. pyogenes enhances the bacterial multiplication in human plasma. A, the AP1 strain and the isogenic AP1 SIC-deficient mutant strain SIC− were cultivated in human plasma (diluted in TH (1:1)) for 8 h at 37 °C. Bacteria were washed with PBS, and bound proteins were eluted at pH 2.0 and subjected to SDS-PAGE followed by Western blotting. Lane 1, proteins eluted from AP1 bacteria; lane 2, proteins eluted from the AP1 mutant SIC−; and lane 3, SIC, 0.5 μg. B, AP1 bacteria (1 × 108 cfu) were incubated with radiolabeled protein SIC (750,000 cpm) in TH or in plasma (diluted 1:5 with TH) at 37 °C for 1 h. The bacteria were washed with PBS and resuspended in SDS sample buffer. Following separation by Tricine-SDS-PAGE, the gel was dried and subjected to autoradiography. Lane 1, AP1 in TH; lane 2, AP1 in plasma (1:5 with TH). C–E, wild-type AP1 bacteria and the isogenic mutant SIC− were grown in human plasma (diluted in TH (1:1)) overnight at 37 °C. The presence of SIC at the bacterial surface was demonstrated by fluorescence microscopy (right panel) using rabbit anti-SIC F(ab′)2 fragments, followed by Alexa Fluor 594-conjugated goat anti-rabbit F(ab′)2 fragments. Scale bar represents 10 μm. C, AP1 bacteria. D, SIC− bacteria. E, AP1 background fluorescence with the secondary goat F(ab′)2 fragments only. F, wild-type AP1 bacteria (black bars) and the isogenic mutant SIC− (gray bars) were grown in human plasma. At indicated time points, bacteria were plated on TH agar plates. Plates were incubated overnight at 37 °C, the number of cfu was determined, and the MF was calculated. The MF of AP1 was set to 1 at each time point in each individual experiment, and the MF of the mutant SIC− was related to this. Mean values ± S.E. of 10 experiments are shown. p values were determined by using the Mann-Whitney U test.
To further demonstrate an association of SIC with the bacterial surface, AP1 bacteria were incubated with radiolabeled SIC. As shown in Fig. 6B, SIC is bound to AP1 also when there are no plasma proteins present. Next, we used fluorescence microscopy to investigate whether some of the SIC produced by AP1 is retained at the bacterial surface. In these experiments, the AP1 and SIC− strains were grown in plasma overnight. Bacteria were then carefully washed and incubated with anti-SIC followed by a secondary fluorophore-conjugated antibody. The results show SIC associated with AP1 bacteria and a low background binding of the antibody to the SIC− mutant (Fig. 6, C–E). This finding is in agreement with the Western blot data above, and the combined data suggest that a fraction of secreted SIC associates with the bacterial surface during growth. Moreover, analysis of the S. pyogenes surface proteome identified SIC at the surface of the M1-SF370 strain, further supporting our finding (46).
To examine whether SIC provides a selective advantage, AP1 bacteria and the mutant strain SIC− were grown in human citrated plasma. At various time points, bacteria were plated, cfu was determined, and the MF was calculated. For comparison, the MF of AP1 was set to 1 in each individual experiment (10 in total), and the MF of the mutant SIC− analyzed in parallel experiments was related to this (Fig. 6F). After 6 and 8 h, the Mann-Whitney U test showed a highly significant growth advantage for wild-type AP1 as compared with the SIC mutant, data supporting the notion that SIC promotes bacterial survival in human plasma.
SIC Promotes Bacterial Growth in Vivo
The AP1 strain is of the M1 serotype and produces SIC, and this strain is also highly virulent to mice. Previous studies have demonstrated that compared with the wild-type AP1 strain, the isogenic mutant strain SIC− is attenuated in virulence (29). To study whether blocking of the antibacterial activity of the contact system in vivo by SIC influences the virulence of AP1 bacteria, we used a mouse model of S. pyogenes infection (47). Mice were injected subcutaneously with AP1 bacteria (2 × 107 cfu/mouse), followed by an intraperitoneal injection of PBS or SIC (200 μg/mouse) after 8 and 20 h. Mice injected subcutaneously and intraperitoneally with PBS served as a healthy control group. After 30 h, the dissemination of bacteria to the spleen and the concentration of the proinflammatory marker IL-6 in plasma were determined. Statistical analysis revealed that significantly higher numbers of bacteria were detected in animals treated with SIC as compared with the animals treated with PBS (p = 0.042) (Fig. 7). Also, the concentration of IL-6 was higher in the group given SIC, but this was not statistically significant (p = 0.055). In plasma from the control group, the concentration of IL-6 was below the detection level, and no bacteria were found in the spleens.
FIGURE 7.

Inhibition of the antibacterial activity of the contact system by SIC promotes bacterial growth in vivo. Female Balb/c mice were injected subcutaneously with AP1 bacteria (2 × 107 cfu/animal), and at 8 and 20 h post-infection, PBS or SIC was injected intraperitoneal (200 μg SIC/dose). Thirty hours after infection, blood was drawn by cardiac puncture, and the concentration of IL-6 in the plasma was determined. Activation of the intrinsic pathway of coagulation was measured as the aPTT in these samples, and the number of cfu in the spleens of the sacrificed mice was determined. In the group treated with SIC (n = 8), the number of cfu/ml spleen homogenate was significantly higher as compared with the group treated with PBS alone (n = 7; p = 0.042). The increased concentration of IL-6 in the SIC-treated group was not statistically significant (p = 0.055). Compared with the healthy control group (n = 12), the increase in aPTT was significantly prolonged in the groups treated with PBS (p = 0.022) or SIC (p = 0.0073). There was no statistical difference between the PBS- and SIC-treated groups (p = 0.61). The Mann-Whitney U test was used for the determination of p values. Bars represent the median value in each group.
To determine whether the contact system is activated in S. pyogenes-infected animals, clotting assays were performed. In plasma from infected animals, activation of the intrinsic pathway of coagulation was impaired as judged by the prolonged aPTT (Fig. 7). This increase of aPTT was statistically significant compared with the healthy control group, demonstrating that activation of the contact system in infected animals results in a consumption of contact factors. There was no statistical difference of the aPTT between the groups of infected animals treated with PBS or SIC, which does not exclude that SIC interferes with HK binding and contact activation at the bacterial surface in vivo. These results demonstrate that invading AP1 bacteria activate the contact system, whereas the enhanced dissemination of bacteria to the spleen in mice treated with SIC suggests that antibacterial peptides generated through contact activation are inhibited by SIC.
DISCUSSION
The contact and complement systems constitute important components of innate immunity that are activated locally at the site of infection to prevent bacterial invasion and dissemination. Upon activation of these proteolytic cascades, antibacterial peptide fragments are generated, which efficiently kill several bacterial pathogens, including S. pyogenes. This common and significant human pathogen is responsible for a number of diseases, including both localized and systemic infections (13–15). In the present investigation, the SIC protein, secreted by S. pyogenes of the M1 serotype, was found to interfere with the antibacterial function of the contact and the complement systems, both in vitro and in vivo.
S. pyogenes is known to induce powerful local inflammatory responses causing vascular leakage and the emergence of plasma proteins at the infectious site, leading to the activation of the contact and the complement systems and the generation of antibacterial peptides. The importance of a functional contact system in bacterial surveillance was previously demonstrated in an in vivo mouse model, where blocking of the contact system promoted bacterial dissemination and growth (9). The finding here that the growth of S. pyogenes is enhanced in human plasma deficient of the contact components FXII, PK, or HK (see Fig. 1A) further emphasizes the role of contact activation in controlling bacterial numbers. In addition, both constitutively expressed AMPs as well as AMPs produced in response to inflammation at the site of infection contribute to a rapid and unspecific defense against invasive bacteria (1). In vitro, the sic gene is expressed already during early growth phase (24), suggesting that SIC is produced immediately when S. pyogenes starts to multiply after colonization. At this early and crucial stage of infection, S. pyogenes strains expressing SIC that inactivates AMPs should have a selective advantage. Previous work has demonstrated that isogenic M1 mutant strains lacking SIC are significantly attenuated in virulence (29, 48). The significance of SIC as a virulence determinant is further supported by the present study, where SIC enhanced the bacterial dissemination in a mouse model of infection. Moreover, in a recent report by Pence and co-workers (49), it was shown that SIC bound both human and murine cathelicidins, thereby promoting resistance to LL-37 and virulence in mice. In relation to the present study, their finding that SIC enhanced S. pyogenes growth in human whole blood and serum is of particular interest.
Another host defense mechanism considered as part of innate immunity is the formation of neutrophil extracellular traps (NETs) containing bactericidal molecules like AMPs, proteinases, and histones (50). Interestingly, the contact system was demonstrated recently to be activated on NETs (51), but it was not shown whether HK-derived antibacterial peptides are generated. However, it can be hypothesized that HK fragments constitute an additional factor promoting bacterial killing by NETs. In addition, M1 protein has been identified as a strong inducer of NET formation, but surprisingly, it is also important for promoting S. pyogenes survival through inhibition of LL-37 (52). The results of this study suggest that SIC bound to the bacterial surface, or secreted into the microenvironment and associated with NETs, contribute to bacterial survival.
Originally, the sic gene was identified in S. pyogenes strains of the M1 and M57 serotypes (24). Subsequent work demonstrated that distantly related sic variants (drs) are also present in strains of the M12 and M55 serotypes (53). SIC binds to and interferes with the function of the membrane attack complex of complement (24, 54), and subsequently, a number of investigations have demonstrated the ability of SIC and SIC variants to block the activity of several antibacterial proteins and peptides (26–29, 55). In this context, it is interesting that Histidine-rich glycoprotein, a plasma protein that interacts with SIC (24), and the membrane attack complex (56) exerts antibacterial activity against various bacterial species (57). Indeed, SIC was recently found to block the antibacterial activity of Histidine-rich glycoprotein against AP1 bacteria (58). When S. pyogenes bacteria of the M1 serotype are exposed to saliva or blood, sic is up-regulated (59, 60). Surprisingly, we could not detect SIC in the supernatant but only at the bacterial surface when AP1 bacteria were grown in human plasma. This could be explained if secreted SIC is bound to SIC-binding plasma proteins (Histidine-rich glycoprotein, clusterin, and/or HK). Thus, in ELISA, the antibodies may not detect SIC epitopes hidden in these protein complexes.
There is a remarkable variation among sic genes from different isolates of the M1 serotype (25) due to natural selection on mucosal surfaces (61), which may facilitate adaptation to changes in the antibacterial activity at the local site of infection and promote bacterial survival. Strains of the M1 serotype are common and are frequently associated with invasive infections (18–23), and the interference of SIC with antibacterial activities of the contact and complement systems may facilitate S. pyogenes invasion. Virulence is of course a polygenic property, but the results of the present work could in part explain the evolutionary success of the M1 serotype and the severity of infections caused by M1 strains.
Acknowledgments
We thank Ulla Johannesson and Maria Baumgarten for expert technical assistance. Rita Wallén (Cell and Organism Biology) is acknowledged for help with electron microscopy, and Pontus Nordenfelt is acknowledged for help with the immunofluorescence microscopy experiment.
This work was supported by the Swedish Research Council (Project 7480), the Foundations of Crafoord, Kock, Bergvall, and Österlund, the Royal Physiographic Society, and Hansa Medical AB.
- AMP
- antimicrobial peptide
- FXII
- factor XII
- FXI
- factor XI
- PK
- plasma kallikrein
- HK
- high molecular weight kininogen
- TH
- Todd-Hewitt broth
- MF
- multiplication factor
- NET
- neutrophil extracellular trap
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- aPTT
- activated partial thromboplastin time.
REFERENCES
- 1. Lai Y., Gallo R. L. (2009) Trends Immunol. 30, 131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morgan B. P., Marchbank K. J., Longhi M. P., Harris C. L., Gallimore A. M. (2005) Immunol. Lett. 97, 171–179 [DOI] [PubMed] [Google Scholar]
- 3. Zasloff M. (2002) Nature 415, 389–395 [DOI] [PubMed] [Google Scholar]
- 4. Rus H., Cudrici C., Niculescu F. (2005) Immunol. Res. 33, 103–112 [DOI] [PubMed] [Google Scholar]
- 5. Haas P. J., van Strijp J. (2007) Immunol. Res. 37, 161–175 [DOI] [PubMed] [Google Scholar]
- 6. Nordahl E. A., Rydengård V., Nyberg P., Nitsche D. P., Mörgelin M., Malmsten M., Björck L., Schmidtchen A. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 16879–16884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pasupuleti M., Walse B., Nordahl E. A., Mörgelin M., Malmsten M., Schmidtchen A. (2007) J. Biol. Chem. 282, 2520–2528 [DOI] [PubMed] [Google Scholar]
- 8. Sonesson A., Ringstad L., Nordahl E. A., Malmsten M., Mörgelin M., Schmidtchen A. (2007) Biochim. Biophys. Acta 1768, 346–353 [DOI] [PubMed] [Google Scholar]
- 9. Frick I. M., Åkesson P., Herwald H., Mörgelin M., Malmsten M., Nägler D. K., Björck L. (2006) EMBO J. 25, 5569–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhoola K. D., Figueroa C. D., Worthy K. (1992) Pharmacol. Rev. 44, 1–80 [PubMed] [Google Scholar]
- 11. Colman R. W., Schmaier A. H. (1997) Blood 90, 3819–3843 [PubMed] [Google Scholar]
- 12. Joseph K., Kaplan A. P. (2005) Adv. Immunol. 86, 159–208 [DOI] [PubMed] [Google Scholar]
- 13. Bisno A. L., Brito M. O., Collins C. M. (2003) Lancet Infect. Dis. 3, 191–200 [DOI] [PubMed] [Google Scholar]
- 14. Cunningham M. W. (2000) Clin. Microbiol. Rev. 13, 470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olsen R. J., Shelburne S. A., Musser J. M. (2009) Cell. Microbiol. 11, 1–12 [DOI] [PubMed] [Google Scholar]
- 16. Facklam R. F., Martin D. R., Lovgren M., Johnson D. R., Efstratiou A., Thompson T. A., Gowan S., Kriz P., Tyrrell G. J., Kaplan E., Beall B. (2002) Clin. Infect. Dis. 34, 28–38 [DOI] [PubMed] [Google Scholar]
- 17. Fischetti V. A. (1989) Clin. Microbiol. Rev. 2, 285–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aziz R. K., Kotb M. (2008) Emerg. Infect. Dis. 14, 1511–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imöhl M., Reinert R. R., Ocklenburg C., van der Linden M. (2010) FEMS Immunol. Med. Microbiol. 58, 389–396 [DOI] [PubMed] [Google Scholar]
- 20. Luca-Harari B., Darenberg J., Neal S., Siljander T., Strakova L., Tanna A., Creti R., Ekelund K., Koliou M., Tassios P. T., van der Linden M., Straut M., Vuopio-Varkila J., Bouvet A., Efstratiou A., Schalén C., Henriques-Normark B., Jasir A. (2009) J. Clin. Microbiol. 47, 1155–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Musser J. M., Krause R. M. (1998) in Emerging Infections (Krause R. M. ed.) pp. 185–218, Academic Press, New York [Google Scholar]
- 22. Steer A. C., Law I., Matatolu L., Beall B. W., Carapetis J. R. (2009) Lancet Infect. Dis. 9, 611–616 [DOI] [PubMed] [Google Scholar]
- 23. Vikerfors A., Haggar A., Darenberg J., Low A., Melhus A., Hedlund J., Sylvan S., Norrby-Teglund A., Eriksson B. M. (2009) Scand. J. Infect. Dis. 41, 823–830 [DOI] [PubMed] [Google Scholar]
- 24. Åkesson P., Sjöholm A. G., Björck L. (1996) J. Biol. Chem. 271, 1081–1088 [DOI] [PubMed] [Google Scholar]
- 25. Stockbauer K. E., Grigsby D., Pan X., Fu Y. X., Mejia L. M., Cravioto A., Musser J. M. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 3128–3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Egesten A., Eliasson M., Johansson H. M., Olin A. I., Mörgelin M., Mueller A., Pease J. E., Frick I. M., Bjorck L. (2007) J. Infect. Dis. 195, 684–693 [DOI] [PubMed] [Google Scholar]
- 27. Fernie-King B. A., Seilly D. J., Binks M. J., Sriprakash K. S., Lachmann P. J. (2007) Microbes Infect. 9, 300–307 [DOI] [PubMed] [Google Scholar]
- 28. Fernie-King B. A., Seilly D. J., Davies A., Lachmann P. J. (2002) Infect. Immun. 70, 4908–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frick I. M., Åkesson P., Rasmussen M., Schmidtchen A., Björck L. (2003) J. Biol. Chem. 278, 16561–16566 [DOI] [PubMed] [Google Scholar]
- 30. Minami M., Ohmori D., Tatsuno I., Isaka M., Kawamura Y., Ohta M., Hasegawa T. (2009) FEMS Microbiol. Lett. 298, 67–73 [DOI] [PubMed] [Google Scholar]
- 31. Herwald H., Hasan A. A., Godovac-Zimmermann J., Schmaier A. H., Müller-Esterl W. (1995) J. Biol. Chem. 270, 14634–14642 [PubMed] [Google Scholar]
- 32. Björck L., Kronvall G. (1984) J. Immunol. 133, 969–974 [PubMed] [Google Scholar]
- 33. Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 34. Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 35. Roth J., Bendayan M., Orci L. (1978) J. Histochem. Cytochem. 26, 1074–1081 [DOI] [PubMed] [Google Scholar]
- 36. Schüttelkopf A. W., Hamilton G., Watts C., van Aalten D. M. (2006) J. Biol. Chem. 281, 16570–16575 [DOI] [PubMed] [Google Scholar]
- 37. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Humphrey W., Dalke A., Schulten K. (1996) J. Mol. Graphics 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 39. Stone J. E. (1998) An Efficient Library for Parallel Ray Tracing and Animation. M.Sc. thesis, Missouri University of Science and Technology, Rolla, MO [Google Scholar]
- 40. Åkesson P., Herwald H., Rasmussen M., Håkansson K., Abrahamson M., Hasan A. A. K., Schmaier A., Müller-Esterl W., Björck L. (August 12, 2010) Microbiology 10.1099/mic.0.039578-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nordahl E. A., Rydengård V., Mörgelin M., Schmidtchen A. (2005) J. Biol. Chem. 280, 34832–34839 [DOI] [PubMed] [Google Scholar]
- 42. Ben Nasr A. B., Herwald H., Müller-Esterl W., Björck L. (1995) Biochem. J. 305, 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frick I. M., Åkesson P., Cooney J., Sjöbring U., Schmidt K. H., Gomi H., Hattori S., Tagawa C., Kishimoto F., Björck L. (1994) Mol. Microbiol. 12, 143–151 [DOI] [PubMed] [Google Scholar]
- 44. Åkesson P., Schmidt K. H., Cooney J., Björck L. (1994) Biochem. J. 300, 877–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. von Pawel-Rammingen U., Johansson B. P., Björck L. (2002) EMBO J. 21, 1607–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rodríguez-Ortega M. J., Norais N., Bensi G., Liberatori S., Capo S., Mora M., Scarselli M., Doro F., Ferrari G., Garaguso I., Maggi T., Neumann A., Covre A., Telford J. L., Grandi G. (2006) Nat. Biotechnol. 24, 191–197 [DOI] [PubMed] [Google Scholar]
- 47. Oehmcke S., Shannon O., von Köckritz-Blickwede M., Mörgelin M., Linder A., Olin A. I., Björck L., Herwald H. (2009) Blood 114, 444–451 [DOI] [PubMed] [Google Scholar]
- 48. Lukomski S., Hoe N. P., Abdi I., Rurangirwa J., Kordari P., Liu M., Dou S. J., Adams G. G., Musser J. M. (2000) Infect. Immun. 68, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pence M. A., Rooijakkers S. H., Cogen A. L., Cole J. N., Hollands A., Gallo R. L., Nizet V. (2010) J. Innate Immun. 2, 587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D. S., Weinrauch Y., Zychlinsky A. (2004) Science 303, 1532–1535 [DOI] [PubMed] [Google Scholar]
- 51. Oehmcke S., Mörgelin M., Herwald H. (2009) J. Innate Immun. 1, 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lauth X., von Köckritz-Blickwede M., McNamara C. W., Myskowski S., Zinkernagel A. S., Beall B., Ghosh P., Gallo R. L., Nizet V. (2009) J. Innate Immun. 1, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hartas J., Sriprakash K. S. (1999) Microb. Pathog. 26, 25–33 [DOI] [PubMed] [Google Scholar]
- 54. Fernie-King B. A., Seilly D. J., Willers C., Würzner R., Davies A., Lachmann P. J. (2001) Immunology 103, 390–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fernie-King B. A., Seilly D. J., Lachmann P. J. (2004) Immunology 111, 444–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chang N. S., Leu R. W., Rummage J. A., Anderson J. K., Mole J. E. (1992) Blood 79, 2973–2980 [PubMed] [Google Scholar]
- 57. Rydengård V., Olsson A. K., Mörgelin M., Schmidtchen A. (2007) FEBS J. 274, 377–389 [DOI] [PubMed] [Google Scholar]
- 58. Shannon O., Rydengård V., Schmidtchen A., Mörgelin M., Alm P., Sørensen O. E., Björck L. (2010) Blood 116, 2365–2372 [DOI] [PubMed] [Google Scholar]
- 59. Graham M. R., Virtaneva K., Porcella S. F., Barry W. T., Gowen B. B., Johnson C. R., Wright F. A., Musser J. M. (2005) Am. J. Pathol. 166, 455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shelburne S. A., 3rd, Granville C., Tokuyama M., Sitkiewicz I., Patel P., Musser J. M. (2005) Infect. Immun. 73, 4723–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hoe N. P., Nakashima K., Lukomski S., Grigsby D., Liu M., Kordari P., Dou S. J., Pan X., Vuopio-Varkila J., Salmelinna S., McGeer A., Low D. E., Schwartz B., Schuchat A., Naidich S., De Lorenzo D., Fu Y. X., Musser J. M. (1999) Nat. Med. 5, 924–929 [DOI] [PubMed] [Google Scholar]