

Abstract
Accumulation of misfolded proteins in the endoplasmic reticulum (ER) causes ER stress and activates inositol-requiring protein-1 (IRE1), among other ER-associated signaling proteins of the unfolded protein response (UPR) in mammalian cells. IRE1 signaling becomes attenuated under prolonged ER stress. The mechanisms by which this occurs are not well understood. An ER resident protein, Bax inhibitor-1 (BI-1), interacts with IRE1 and directly inhibits IRE1 activity. However, little is known about regulation of the BI-1 protein. We show here that bifunctional apoptosis regulator (BAR) functions as an ER-associated RING-type E3 ligase, interacts with BI-1, and promotes proteasomal degradation of BI-1. Overexpression of BAR reduced BI-1 protein levels in a RING-dependent manner. Conversely, knockdown of endogenous BAR increased BI-1 protein levels and enhanced inhibition of IRE1 signaling during ER stress. We also found that the levels of endogenous BAR were reduced under prolonged ER stress. Our findings suggest that post-translational regulation of the BI-1 protein by E3 ligase BAR contributes to the dynamic control of IRE1 signaling during ER stress.
Keywords: E3 Ubiquitin Ligase, ER Stress, Signal Transduction, Ubiquitin, Ubiquitination, Bax Inhibitor-1, Unfolded Protein Response
Introduction
The lumen of the endoplasmic reticulum (ER)2 possesses various molecular chaperones and enzymes that ensure proper folding of newly synthesized proteins destined for secretion or expression as membrane-bound proteins. Factors that compromise the folding capacity of the ER result in accumulation of unfolded/misfolded proteins, causing the phenomenon of ER stress (1). ER stress is triggered by multiple cellular disturbances, including glucose deprivation, alterations of Ca2+ concentrations in the ER, perturbations of the redox state of the ER lumen (caused by hypoxia, oxidative insults, or reactive chemicals), viral infection, accumulation of mutant proteins, and other stimuli. ER stress has been associated with a variety of diseases, including ischemia, neurodegenerative diseases, cancer, diabetes, viral infections, inflammatory bowel disease, and a variety of genetic disorders that result in mutant proteins that fail to fold properly, among other causes (2). At least three types of ER stress transducers have been identified in ER membranes in mammalian cells: IRE1, protein kinase RNA-like ER kinase (PERK), and activating transcription factor-6 (ATF6). Each initiates a distinct signaling pathway, collectively named the unfolded protein response (UPR).
Among these three known ER stress transducers, only IRE1 is highly conserved from yeast to mammals. IRE1 contains a kinase domain and a kinase-extension nuclease domain with endoribonuclease activity. During ER stress, dimerization/oligomerization and trans-autophosphorylation of IRE1 are coupled with endoribonuclease activation (3), which cleaves mRNA encoding transcriptional factor XBP1 (known as HAC1 mRNA in yeast). In parallel, IRE1 also interacts with tumor necrosis factor receptor-associated factor 2 (TRAF2) in mammals, which initiates phosphorylation cascades that include apoptotic signaling kinase-1 (ASK1) and downstream stress kinases, c-Jun N-terminal kinases (JNKs) and p38 mitogen-activated protein kinases (p38 MAPKs) (4). The role of UPR is to promote cellular adaptation to ER stress by increasing the folding capacity of the ER, reducing the unfolded protein load, and enhancing ER-associated protein degradation (ERAD). However, components of the UPR can also trigger cell death if protein homeostasis is not restored (1, 5). Although it remains poorly understood how stressed cells make decisions regarding survival versus death, a recent study provides some insights by revealing that the duration of individual UPR signal transduction events can be differentially regulated. For example, during persistent ER stress, PERK activation (which inhibits mRNA translation by phosphorylation of eukaryotic initiation factor 2α (eIF2α)) can be sustained, whereas IRE1 activation becomes attenuated (6). In this case, IRE1-mediated XBP1 mRNA splicing was shown to promote cell survival. The mechanism by which IRE1 signaling is selectively suppressed during prolonged ER stress is unknown.
BI-1, an evolutionarily conserved ER membrane protein, was recently found to negatively inhibit IRE1 activity (7). Indeed, BI-1 knock-out mice showed enhanced XBP1 mRNA splicing and JNK phosphorylation in vivo (8, 9). The inhibitory effect of BI-1 on IRE1 signaling is specific, in that eIF2α phosphorylation downstream of PERK activation is not altered in BI-1 knock-out mice (8). BI-1 forms protein complexes with IRE1 (7, 9), and the endoribonuclease activity of IRE1 was reported to be directly suppressed in vitro by BI-1 protein (7). Importantly, in contrast to time-dependent decline of IRE1 activation after prolonged ER stress, IRE1 activation and downstream XBP1 mRNA splicing were sustained in BI-1 knock-out cells (7), suggesting BI-1 plays a crucial role in the dynamic control of IRE1 activation during ER stress. However, little is known about the mechanisms by which BI-1 is regulated.
Here we studied the post-translational regulation of the BI-1 protein by BAR. We discovered that BI-1 interacts with BAR, a RING-type E3 ligase on the ER membrane (10). BAR induces BI-1 ubiquitination and promotes BI-1 proteasomal degradation, as well as catalyzing its own ubiquitination in a RING-dependent manner. BAR thus opposes BI-1, removing an inhibitory influence on IRE1 signaling. We also observed that levels of endogenous BAR protein are reduced by sustained ER stress. Taken together, our findings suggest that post-translational regulation of BI-1 by ER-associated E3 ligase BAR contributes to regulation of IRE1 signaling during ER stress.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Thapsigargin was purchased from Axxora. Tunicamycin, MG132, bafilomycin A1, cycloheximide, and doxycycline were from Sigma. Protease inhibitor mixture and phosphatase inhibitor mixture were from Roche Applied Science. Lipofectamine 2000 and Lipofectamine RNAiMAX were from Invitrogen. ECL Western blotting detection reagents were from GE Healthcare. Restore Western blot stripping buffer was from Thermo Scientific. Vectorshield mounting medium was from Vector Laboratories. We used the following antibodies: rabbit antibodies to BAR (10), ubiquitin (Lys48-specific, number 05-1307), and ubiquitin (Lys63-specific, number 05-1308, Millipore), calnexin (number SPA-860, Stressgen), active-JNK (V793, Promega), phospho-c-Jun (Ser73) (number 9164), phospho-p38 MAPK (Thr180/Tyr182) (number 9211), phospho-eIF2α (Ser51) (number 3597), JNK (number 9252), c-Jun (number 9165), and MAPK (number 9212, Cell Signaling Technology), eIF2α (sc-11386, Santa Cruz Biotechnology); mouse antibodies to glutathione S-transferase (GST) (number 554805) and Bcl-2 (number 550847, BD Biosciences), ubiquitin (P4D1) (number 3936, Cell Signaling Technology), the c-Myc epitope (Santa Cruz Biotechnology), the hemagglutinin (HA) epitope (Roche Applied Science), the FLAG epitope and α-tubulin (Sigma); and rat antibody to HA and HRP-conjugated anti-HA (Roche Applied Science). The following secondary antibodies were used: HRP-conjugated secondary antibodies, Alexa Fluor 488-conjugated and Alexa Fluor 594-conjugated secondary antibodies (Invitrogen), and TrueBlot HRP-conjugated secondary antibodies (eBioscience).
DNA Constructs
Plasmids encoding GST-BAR(1–139), Myc-BAR, Myc-BARΔRING (10), BI-1-HA (11), and Myc-TRAF6 (12) have been described previously. Plasmids encoding HA-ubiquitin, HA-K48R ubiquitin, HA-JAMP, Myc-MmUbc6, Myc-MmUbc7, HA-TCRα, and HA-CD3δ were gifts from Dr. Ze'ev Ronai. The HA-IRE1α construct was a gift from Dr. Claudio Hetz. The Flag-ATF6 construct was a gift from Dr. Ron Prywes. The HA-STIM1 construct was a gift from Dr. Axel Methner. BAR and BARΔRING were subcloned into pcDNA3-HA expression vector. BI-1 was subcloned into pcDNA3-Flag vector. K48-only ubiquitin and K63-only ubiquitin were generated from pET-7xK-R ubiquitin using the site-directed mutagenesis kit (Stratagene), subcloned into pcDNA3-HA expression vector, and verified by DNA sequencing.
Cell Culture and Transfection
293T cells and HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Mediatech) with 10% fetal bovine serum (FBS), penicillin (100 IU), and streptomycin (100 μg/ml) at 37 °C in 5% CO2, 95% air. Doxycycline-inducible stable HeLa cells expressing BI-1-HA were maintained in DMEM with 10% Tet-free FBS (Omega Scientific), G418 (100 μg/ml), hygromycin B (100 μg/ml), penicillin (100 IU), and streptomycin (100 μg/ml) at 37 °C in 5% CO2, 95% air. To induce BI-1 expression, 500 ng/ml of doxycycline (unless otherwise indicated) was added to culture medium. Transient transfections were performed using Lipofectamine 2000.
In Vitro Ubiquitination Assay
GST-tagged BAR (amino acids 1–139) in pGEX vector was transformed into Escherichia coli BL21(DE3). The protein was induced with 0.1 mm isopropyl thio-β-d-galactopyranoside at room temperature for 3 h. GST-BAR was purified using glutathione-agarose (Sigma), eluted with 20 mm reduced glutathione in 50 mm Tris-Cl, pH 8.0, and dialyzed in buffer containing 50 mm sodium phosphate, pH 8.0, 200 mm NaCl, and 1 mm DTT. GST-BAR (300 nm final concentration) was used for ubiquitination reactions in buffer containing 50 mm HEPES, pH 7.6, 5 mm MgCl2, 1 mm ATP, 1 mm DTT, 1 μm ubiquitin, 50 nm E1, and 200 nm E2 (UbcH5b, UbcH5c, or Ubc13/UEV1A, as indicated). Reactions were conducted for 1 h at room temperature on a thermomixer, and terminated by adding SDS-PAGE sample buffer and boiling for 5 min. Reaction products were resolved by 15% SDS-PAGE and subjected to immunoblotting with mouse anti-ubiquitin and mouse anti-GST antibodies.
Immunoprecipitation and Protein Analysis
Cells in 10-cm plates were lysed in 1 ml of 0.2% Triton X-100 in phosphate-buffered saline (PBS), pH 7.4, containing protease and phosphatase inhibitor mixture at 4 °C for 1 h on a wheel rotor. The lysates were centrifuged at 11,000 × g for 10 min and the resulting supernatants were preincubated with protein G-Sepharose beads at 4 °C for 1 h to reduce nonspecific binding. After brief centrifugation, the supernatants were incubated with specific antibodies overnight at 4 °C followed by incubation with protein G-Sepharose beads at 4 °C for 1 h. After brief centrifugation and washing with the lysis buffer, the immunoprecipitated proteins were resolved with SDS-PAGE and subjected to immunoblotting.
For direct immunoblot analysis using cell lysates, cells were lysed with RIPA buffer (50 mm Tris-Cl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 0.1% SDS, 0.5% deoxycholic acid, 1% Triton X-100) (unless otherwise described) containing protease and phosphatase inhibitor mixture at 4 °C for 30 min on a wheel rotor. The lysates were centrifuged at 11,000 × g for 10 min. Proteins in supernatants were quantified using Bradford reagent, using aliquots of 10–30 μg of proteins for SDS-PAGE (10, 12, or 4–20% gels) analysis and immunoblotting. For sequential probing with multiple antibodies, the blots were stripped using Restore Western blot stripping buffer at room temperature for 15 to 60 min and washed with 0.1% Tween 20, PBS.
Immunofluorescent Labeling and Confocal Microscopy
Transfected HeLa cells cultured on 22-mm round coverslips were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. After washing with PBS three times, the cells were incubated in buffer containing 0.2% Triton X-100, 3% bovine serum albumin (BSA) in PBS at 4 °C for 1 h. Cells were then incubated with primary antibodies in 3% BSA in PBS overnight at 4 °C. After washing with PBS, the cells were incubated with Alexa Fluor 594 and Alexa Fluor 488-conjugated secondary antibodies in 3% BSA in PBS at 4 °C for 1 h. Alexa Fluor 488-phalloidin was used to label actin. The nuclei were stained with Hoechst at 4 °C for 5 min. The stained cells were mounted using Vectorshield mounting medium. Confocal images were acquired with a Radiance 2100 multiphoton LSCM using oil objective lens ×60 and at a resolution of 1024 × 1024.
RNAi Experiments
HeLa cells (2 × 105) with inducible BI-1-HA protein expression were transfected with 15 nm scrambled siRNA (siCtrl, Ambion) or siRNA targeting human BAR (Ambion) using the reverse transfection procedure with Lipofectamine RNAiMAX according to the manufacturer's instructions (Invitrogen). The transfected cells were cultured in 6-well plates for 24 h before 500 ng/ml of doxycycline was added to the culture medium to induce BI-1-HA protein expression. After 24 h, cells were subjected to ER stress stimuli as described. 293T cells (2 × 105) were transfected with 15 nm scrambled siRNA or siRNA targeting human BAR as described above for 24 h. Cells were then transfected with 1 μg of pcDNA3-Myc-BAR and 1 μg of pcDNA3-BI-1-HA with Lipofectamine 2000 and cultured for an additional 48 h before collecting the cells. The two targeting sequences for BAR were 5′-GCTAGAACGTGTCAAAGCA-3′ (BAR siRNA-1) and 5′-GGGATGCCATTGAAAAGTT-3′ (BAR siRNA-2).
XBP1 Splicing Analysis
Total RNA was extracted from cells using the RNeasy mini kit (Qiagen) along with on-column DNase treatment. RT-PCR analysis was performed using the SuperScript one-step RT-PCR system (Invitrogen) employing conditions previously described with minor modifications (6). Briefly, primers used for XBP1 were 5′-TTACGAGAGAAAACTCATGGC-3′ and 5′-GGGTCCAAGTTGTCCAGAATGC-3′. Primers used for GAPDH were 5′-GGTGAAGGTCGGAGTCAACGGA-3′ and 5′-GAGGGATCTCGCTCCTGGAAGA-3′. The following RT-PCR conditions were employed: 50 °C for 30 min; 94 °C for 2 min; 35 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s; 72 °C for 10 min. The PCR products were resolved on a 3% agarose gel. For quantitative real time PCR analysis of XBP1 splicing, cDNA was generated using the StrataScript first-strand synthesis system (Stratagene). Two sets of primers were used. 5′-TGCTGAGTCCGCAGCAGG-3′ and 5′-GGGTCCAAGTTGTCCAGAAT-3′ were employed for spliced XBP1 amplification; 5′-CCGCAGCACTCAGACTACG and 5′-GGGTCCAAGTTGTCCAGAAT-3′ were utilized for unspliced XBP1 amplification. Thermal DNA melting experiments showed single melting peaks for the products generated with each primer set. Quantitative PCR was performed using the Mx3000P Q-PCR system (Stratagene) with SYBR Green PCR master mixture (Qiagen) and analyzed with the MxPro software (Stratagene).
RT-PCR Analysis of BAR mRNA Level
Total RNA was extracted from cells. RT-PCR analysis was performed using the SuperScript one-step RT-PCR system. Primers used for BAR were 5′-AGAAGTGAACTGAAGACCGTGCCT-3′ and 5′-CGGAGTTCCTTGACCACAAGATCA-3′. We used the following RT-PCR conditions: 50 °C for 30 min; 94 °C for 2 min; 32 cycles of 94 °C for 30 s, 50 °C for 30 s, and 72 °C for 45 s; 72 °C for 10 min. A 182-bp RT-PCR product was generated.
Statistical Analysis
Statistical significance (p < 0.05) was assessed using Student's t test or one-way analysis of variance and Tukey post test. Data were analyzed and calculated with Image J and GraphPad Prism 4 software.
RESULTS
BAR Has E3 Ligase Activity in Vitro
Protein ubiquitination is achieved through sequential action of an ubiquitin-activating enzyme (E1), an ubiquitin-conjugating enzyme (E2), and an ubiquitin ligase (E3). The RING domain defines the largest group of E3 ligases in eukaryotes (13). Because BAR contains a RING domain at the N terminus, we studied whether BAR was able to promote substrate-independent ubiquitin-protein ligation in vitro, as documented for other E3 ligases (14). The first 139 amino acids of BAR, which contains the RING domain, was fused in-frame with GST, expressed in bacteria, and purified to near homogeneity (supplemental Fig. S1). GST-BAR(1–139) or GST alone was incubated with purified human E1, E2 (UbcH5b or UbcH5c), ubiquitin, and ATP at room temperature for 1 h. The reaction was stopped and ubiquitination was monitored by immunoblot analysis with an anti-ubiquitin antibody (Fig. 1, upper panel). The presence of GST or GST-BAR(1–139) in the reaction was confirmed with an anti-GST antibody (Fig. 1, lower panel). Polyubiquitin (poly-Ub) chains were formed in reactions containing both E2 and GST-BAR(1–139) (Fig. 1, upper panel). On the contrary, no poly-Ub chains were formed in reactions containing either E2 alone or E2 and control GST protein (Fig. 1, upper panel). We did not observe polyubiquitination of GST-BAR(1–139) itself (Fig. 1, lower panel), possibly due to the short fragment we used for the experiment, which may lack the internal sites of autoubiquitination found in the full-length BAR protein. These data indicate that BAR has E2-dependent E3 ligase activity in vitro.
FIGURE 1.
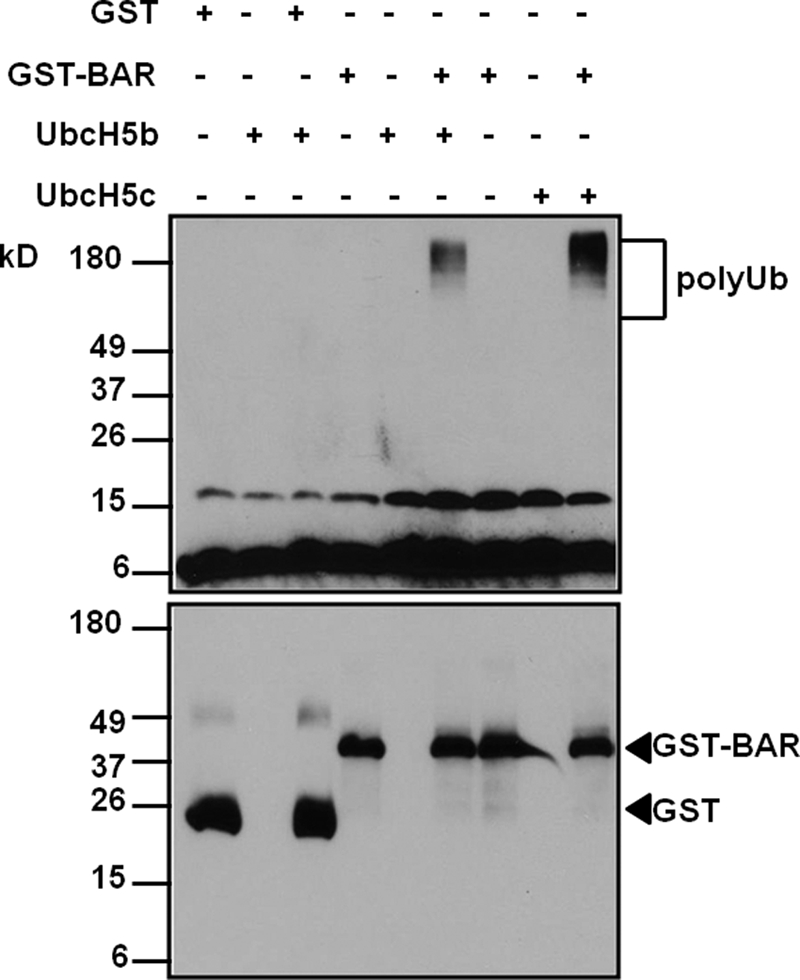
BAR has E3 ligase activity in vitro. Purified GST or GST BAR(1–139) was incubated with E1, E2 (UbcH5b or UbcH5c), ubiquitin, and ATP at room temperature for 1 h. The reaction was stopped by adding 5× SDS sample buffer, boiled for 5 min, and resolved with 15% SDS-PAGE. The blot was probed with mouse anti-ubiquitin and anti-GST antibodies. Note that poly-Ub chains were detected only in reactions containing both GST-BAR and E2s (sixth and ninth lanes). No poly-Ub chains were formed in reactions containing either E2 alone (second, fifth, and eighth lanes) or E2 and GST protein (third lane).
BAR Mediates RING-dependent Self-ubiquitination and Proteasomal Degradation
Our observation that BAR has E3 ligase activity in vitro prompted us to examine whether BAR functions as an E3 ligase in vivo and mediates its autodegradation. We expressed Myc-tagged wild-type BAR (Myc-BAR) or RING-deletion mutant (Myc-BARΔRING) in 293T cells. Cells were lysed 24 h later and BAR protein levels were analyzed by immunoblotting. Consistent with previous observations (10), BAR protein levels were significantly higher when the RING domain was deleted (Fig. 2A). Treatment with the proteasome inhibitor MG132 also significantly increased BAR protein levels (Fig. 2A). Thus the RING domain appears to mediate BAR protein autodegradation by proteasomes.
FIGURE 2.

BAR mediates RING-dependent self-ubiquitination and proteasomal degradation. A, 293T cells were singly transfected with pcDNA3 vector (first, second, seventh, and eighth lanes), or plasmids encoding Myc-BAR (third, fourth, ninth, and tenth lanes) or Myc-BARΔRING (fifth, sixth, eleventh, and twelfth lanes) for 24 h. 5 h before collecting the cells, proteasome inhibitor MG132 (20 μm) was added (+), as indicated. Cells lysates were analyzed by immunoblotting using rabbit anti-BAR antibody. α-Tubulin served as a loading control. Compared with BAR (third and fourth lanes), deletion of the RING domain stabilized the BAR protein level (fifth and sixth lanes). Treatment with MG132 also increased the BAR protein level (ninth and tenth lanes). B, 293T cells were co-transfected with HA-ubiquitin and pcDNA3 vector (first, second, seventh, and eighth lanes) or plasmids encoding BAR (third, fourth, ninth, and tenth lanes) or BARΔRING (fifth, sixth, eleventh, and twelfth lanes) for 24 h. MG132 was added (+) 5 h before collecting the cells, as indicated. Cell lysates were immunoprecipitated (IP) using mouse anti-Myc antibody. The inputs (1/20 of lysates used for immunoprecipitation) and the immunoprecipitated proteins were analyzed by immunoblotting using rabbit anti-HA to detect ubiquitination and anti-BAR antibody. Poly-Ub chains were detected in BAR immunoprecipitates (ninth lane), which were dramatically increased when cells were treated with MG132 (tenth lane). In contrast, deletion of the RING domain significantly reduced ubiquitination of BAR (eleventh and twelfth lanes). C, equivalent portions of the immunoprecipitated proteins described in B were analyzed by immunoblotting using rabbit anti-Lys48-specific ubiquitin antibody (upper) and anti-BAR antibody (lower). D, 293T cells were co-transfected with Myc-BAR and various HA-tagged ubiquitin plasmids (WT, K48-only, K48R, and K63-only) for 24 h. Cell lysates were immunoprecipitated with mouse anti-Myc antibody. Non-transfected cell lysate (NT) was used as a negative control for immunoprecipitation. The inputs (1/20 of lysates used for immunoprecipitation) and immunoprecipitates were analyzed by immunoblotting using rabbit anti-HA to detect ubiquitination and anti-BAR antibodies. Note that BAR was able to form poly-Ub chains joined through sites other than K48 (ninth and tenth lanes, upper panel).
A major signal for proteasomal degradation is lysine 48 (K48)-linked poly-Ub chains on target proteins (15). In addition, poly-Ub chains formed at other lysine residues in ubiquitin may also target proteins for degradation (16). We thus studied whether BAR mediates poly-Ub chain formation on itself. Myc-BAR or Myc-BARΔRING was co-expressed with HA-ubiquitin in 293T cells for 24 h. The cells were lysed and BAR protein was immunoprecipitated with anti-Myc antibody. BAR protein levels in the precipitates were analyzed by immunoblotting with anti-BAR antibody and ubiquitination was analyzed with anti-HA antibody. BAR was polyubiquitinated as detected by anti-HA antibody (Fig. 2B) and the polyubiquitinated BAR protein accumulated to much higher levels when MG132 was added to cultures for 5 h before collecting the cells (Fig. 2B). In contrast, ubiquitination of BARΔRING was minimal and levels of this protein were not impacted by MG132 treatment. Thus, BAR undergoes RING-dependent self-ubiquitination. Ubiquitination can occur on all seven lysine residues within the ubiquitin molecule, with K48- and K63-linked chains among the most abundant linkages in vivo (17). We examined whether K48-linked polyubiquitination occurred on BAR protein by using an antibody that specifically recognizes poly-Ub chains linked through K48 (18). Indeed, BAR was able to form K48-linked poly-Ub chains, which were significantly increased when cells were treated with MG132 (Fig. 2C, upper panel). Deletion of the RING domain from the BAR protein markedly reduced K48-linked poly-Ub chain formation (Fig. 2C, upper panel). Using an antibody that specifically recognizes poly-Ub chains linked through K63 (18), we found BAR could also form K63-linked poly-Ub chains, although the signal was weak compared with another RING-type E3 ligase TRAF6 (supplemental Fig. S2), which preferentially forms K63-linked chains (19). Treatment of cells with MG132 increased the K63-linked poly-Ub signal, possibly suggesting that mixed chains of K48 and K63 are present on BAR in cells.
To further study the nature of BAR-mediated autoubiquitination, we next co-expressed BAR with HA-tagged wild-type (WT) or mutant variants of ubiquitin (K48-only and K63-only, which have 6 of 7 lysines mutated to arginine, and K48R, which has only 1 of 7 lysines mutated), then examined poly-Ub chain formation and BAR protein stability. These experiments suggest that BAR ubiquitination involves chains encompassing K48, K63, and probably other types of poly-Ub linkages (Fig. 2D). Treatment with MG132 stabilized BAR protein levels in all cases (supplemental Fig. S3). Note that we cannot exclude the possibility that endogenous ubiquitin targeted BAR for proteasomal degradation, which was blocked by MG132 treatment in the experiment. We also found that treatment with the lysosomal inhibitor bafilomycin A1 modestly increased BAR protein levels when cells were co-transfected with K63-only ubiquitin, but not K48-only ubiquitin (supplemental Fig. S4), consistent with a previous report suggesting that K63-linked poly-Ub chains target protein for lysosomal degradation (20). In contrast to MG132 treatment, bafilomycin A1 treatment did not change BAR protein levels in cells (supplemental Fig. S4) suggesting that under normal conditions, BAR is mainly degraded via proteasomes rather than lysosomes.
BAR Co-localizes and Interacts with BI-1 in ER Membranes
BAR was reported previously to localize mainly to the ER, and it has at least one predicted transmembrane domain typical of integral membrane proteins (21). We performed co-localization studies by immunofluoresence confocal microscopy and compared the intracellular localizations of BAR with another ER resident protein BI-1 (Fig. 3A). MG132 treatment stabilized the BAR protein and increased its levels (detected using anti-Myc antibody). Both Myc-BAR and BI-1-HA (detected using anti-HA antibody) demonstrated perinuclear reticular staining patterns (first and second panels). Significant co-localization was observed between Myc-BAR and BI-1-HA (fourth panel), as well as ER chaperone protein calnexin (third panel), which served as an ER marker. Similar subcellular localization results were obtained using a ΔRING mutant of BAR, which accumulates to easily detectable levels without requirement for MG132 treatment (supplemental Fig. S5). To determine whether BI-1 interacts with BAR, we performed co-immunoprecipitation experiments. BI-1-HA was co-expressed with Myc-BAR or Myc-BARΔRING. BAR and its associated proteins were immunoprecipitated from cell lysates with anti-Myc antibody and analyzed by SDS-PAGE/immunoblotting. Using anti-HA antibody, we found the BI-1-HA protein was present in the BAR immunoprecipitates (Fig. 3B). MG132 treatment and RING deletion of BAR both resulted in increased levels of BI-1 protein, as well as BAR protein (Fig. 3B and supplemental Fig. S6). In contrast, little or no BAR or BI-1 was immunoprecipitated by normal IgG, confirming the specificity of BAR and BI-1 co-immunoprecipitation results. As an additional control for specificity, we also co-expressed BI-1-HA with another RING-type E3 ligase, Myc-TRAF6. BI-1 did not co-immunoprecipitate with TRAF6 (supplemental Fig. S7), further confirming specific interactions between BAR and BI-1.
FIGURE 3.

BAR co-localizes and interacts with BI-1 in ER. A, HeLa cells were singly transfected with Myc-BAR, BI-1-HA, or co-transfected with both plasmids (normalizing total DNA transfected with pcDNA3). After 24 h, cells transfected with Myc-BAR were treated with 20 μm MG132 for 2 h. Cells were fixed and labeled with actin, Myc epitope, calnexin, or HA epitope antibodies as indicated, and analyzed by immunofluorescence confocal microscopy. B, 293T cells were co-transfected with BI-1-HA and Myc-BAR (first to sixth lanes) or Myc-BARΔRING (seventh to ninth lanes) for 48 h. MG132 was added 6 h before collecting cells, as indicated (fourth to sixth lanes). BAR or BARΔRING protein in cell lysates was immunoprecipitated with mouse anti-Myc antibody (third, sixth, and ninth lanes). Normal mouse IgG was used as a negative control for immunoprecipitation (second, fifth, and eighth lanes). The precipitates were analyzed by immunoblotting using rabbit anti-HA and anti-BAR antibodies. BI-1 co-immunoprecipitated with BAR (third lane), which was enhanced when treated with MG132 (sixth lane) or co-transfected with BARΔRING (ninth lane). C, inducible BI-1-HA-expressing HeLa cells were treated with doxycycline (Dox) at increasing concentrations for 24 h. Dose-dependent induction of BI-1-HA was detected by immunoblot. α-Tubulin served as a loading control. D, HeLa cells were induced to express BI-1-HA with low concentrations of Dox for 24 h. Cell lysates were immunoprecipitated with anti-HA antibody and subjected to immunoblotting with anti-BAR antibody. Cell lysates without Dox treatment were used as a negative control for immunoprecipitation (fourth lane). Note that BAR protein was detected in BI-1-HA immunoprecipitates (upper panel, fifth, and sixth lanes). The levels of precipitated BI-1-HA protein were also shown (lower panel).
We also employed a HeLa cell line that has detectable endogenous BAR protein expression and was previously engineered to express BI-1-HA under control of a tetracycline/doxycyclin-inducible promoter (22). (Note that antibodies to endogenous BI-1 protein are unavailable despite a variety of attempts to produce them, possibly due to the highly conserved amino acid sequences among mammalian BI-1 orthologs that may limit inter-species immunogenicity.) Dose-dependent increases in the BI-1-HA protein were observed upon addition of doxycycline to the culture medium of these engineered HeLa cells (Fig. 3C). We found that endogenous BAR was able to co-immunoprecipitate with induced BI-1 protein in these cells, which we treated with low concentrations of doxycycline to avoid excessive overexpression in an effort to attain physiological levels of BI-1-HA protein production (Fig. 3D).
BAR Overexpression Promotes BI-1 Ubiquitination and Proteasomal Degradation
The specific interaction between BAR and BI-1 promoted us to study whether BAR functions as an E3 ligase for BI-1. For these experiments, BI-1-FLAG protein was co-expressed with HA-ubiquitin and Myc-BAR. After 24 h, the cells were lysed and the BI-1 protein in the lysates was recovered by immunoprecipitation using anti-FLAG antibody. BI-1 protein levels in the immunoprecipitates were analyzed by immunoblotting with anti-FLAG antibody and ubiquitination was monitored with anti-HA antibody (Fig. 4A). Less BI-1 protein was present in cell lysates when BAR was overexpressed (Fig. 4A, upper panel). Moreover, ubiquitination of BI-1 was detected when BAR was overexpressed (Fig. 4A, lower panel). In contrast, co-expression of BARΔRING significantly reduced BI-1 ubiquitination (Fig. 4A, lower panel) and increased BI-1 protein levels (Fig. 4A, upper panel).
FIGURE 4.

BAR overexpression promotes BI-1 ubiquitination and proteasomal degradation. A, 293T cells were transfected with plasmids encoding BI-1-FLAG, HA-ubiquitin, and pcDNA3 vector (first, fourth, and fifth lanes), Myc-BAR (second and sixth lanes) or Myc-BARΔRING (third and seventh lanes). After 24 h, BI-1 protein in cell lysates was immunoprecipitated with mouse anti-FLAG antibody, and the resulting immune complexes were analyzed by immunoblotting with mouse anti-FLAG and rabbit anti-HA antibodies to assess BI-1 protein levels and BI-1 ubiquitination (fifth to seventh lanes). The inputs were 1/20 of cell lysates used for immunoprecipitations. Normal mouse IgG was used as a negative control for immunoprecipitations (fourth lane). B, HA-tagged BI-1, STIM1, IRE1α, JAMP, and FLAG-tagged ATF6 were co-transfected with pcDNA3 vector or plasmids encoding either Myc-BAR or Myc-BARΔRING at 1:1 ratio in 293T cells for 48 h. Cell lysates were analyzed by immunoblotting with antibodies to HA, FLAG, and BAR. α-Tubulin served as a loading control. C, levels of BI-1 protein in B were quantified by scanning densitometry. Vector groups were adjusted to 1. Statistical significance (mean ± S.E.; n = 3) was determined by one-way analysis of variance and Tukey post test, and is denoted by asterisks (*, p < 0.05; **, p < 0.01). D, BI-1-HA was co-transfected with pcDNA3 vector, or plasmids encoding either Myc-BAR or Myc-BARΔRING at 1:1 ratio in 293T cells for 48 h. Cells were lysed either with 2% SDS or RIPA buffer and subjected to immunoblotting with mouse anti-HA. α-Tubulin served as a loading control.
To determine the specificity of the effect of BAR on BI-1 protein stability, we performed experiments where BAR was co-expressed with BI-1 or a variety of other ER resident integral membrane proteins, including ER calcium sensor STIM1, ER stress sensors IRE1α and ATF6, and JAMP, a seven-transmembrane ER-anchored protein with comparable topology to that of BI-1 (23), analyzing the steady-state levels of these ER proteins. We found that BAR specifically regulated stability of the BI-1 protein in a RING-dependent manner, but not other ER membrane proteins studied (Fig. 4, B and C, and supplemental Fig. S8).
To exclude the possibility that BAR altered the BI-1 location to the Triton-insoluble fraction instead of changing its protein level, we used 2% SDS as the lysis buffer in contrast to Triton-based lysis buffer that we used in previous experiments. Similar reduction of the BI-1 protein level was detected in BAR overexpressing cells (Fig. 4D). Furthermore, the BI-1 protein level was slightly increased when cells were co-transfected with BARΔRING compared with vector control (Fig. 4, B–D), suggesting BARΔRING may compete with endogenous BAR to prevent BI-1 degradation.
To further explore whether BAR regulates BI-1 protein levels post-translationally, we treated cells with cycloheximide (protein synthesis inhibitor) and monitored BI-1 protein levels for up to 6 h post-inhibiting protein synthesis. Compared with co-transfection with vector control and BARΔRING, co-transfection of BAR caused a significantly faster rate of BI-1 protein degradation (Fig. 5, A and B). In addition, we also observed rapid BAR protein reductions (Fig. 5A), attesting to the short half-life of the BAR protein. Consistent with our previous results (Fig. 2, A and B), RING deletion greatly stabilized the BAR protein (Fig. 5A).
FIGURE 5.

BAR modulates BI-1 protein stability. A, 293T cells were co-transfected with BI-1-HA and pcDNA3 vector (Vector), Myc-BAR (BAR), or Myc-BARΔRING (BARΔRING) for 24 h. Cells were then treated with cycloheximide (CHX, 20 μg/ml) for the indicated times to inhibit protein synthesis. Cell lysates were analyzed by immunoblotting using mouse anti-HA and rabbit anti-BAR antibodies. α-Tubulin served as a loading control within each group. The amount of total protein used in each group was adjusted and at time 0, a similar densitometric signal was achieved. B, percentage of the remaining BI-1 protein at each time point after inhibiting protein synthesis using cycloheximide in A was quantified by densitometry (mean ± S.E.; n = 2).
To confirm these observations based on BAR overexpression, we also transfected synthetic BAR siRNAs (targeting BAR coding sequences) before co-transfecting cells with plasmids encoding BAR and BI-1, asking whether BAR-mediated reductions in steady-state BI-1 protein levels could be reversed. Indeed, plasmid-derived BAR protein was readily reduced by BAR siRNAs and correspondingly the levels of transfected BI-1 were significantly increased (supplemental Fig. S9, A and B). Taken together, these results demonstrate that BAR overexpression induces BI-1 ubiquitination and promotes proteasomal degradation of BI-1 in a RING-dependent manner.
BI-1 Protein Levels Are Increased by Knockdown of Endogenous BAR Expression
To complement the observations based on BAR overexpression, we next studied whether endogenous BAR has E3 ligase activity toward BI-1, using HeLa cells with inducible BI-1 expression. First, we employed synthetic BAR siRNAs to reduce endogenous BAR expression in HeLa cells and then induced BI-1 expression by adding doxycycline to the culture medium. The steady-state levels of BAR and BI-1 proteins were subsequently determined by immunoblot analysis. BAR siRNAs dramatically reduced endogenous BAR protein expression (Fig. 6A, middle panel). Consistent with previous observations that overexpressed BAR promotes BI-1 degradation, we observed increased BI-1 protein levels in cells with reduced BAR expression (Fig. 6, A, upper panel, and B). These findings further support the hypothesis that endogenous BAR operates as an E3 ligase for BI-1.
FIGURE 6.

BI-1 protein levels are increased in BAR-deficient cells. A, BI-1-expressing HeLa cells were transfected with scrambled siRNA or BAR siRNA (BAR siRNA-1 or BARsiRNA-2). After 24 h, cells were treated with 500 ng/ml of doxycycline for 24 h to induce BI-1 expression. Cell lysates were analyzed by immunoblotting with antibodies to HA epitope and BAR. α-Tubulin served as a loading control. B, quantification of A was performed by scanning densitometry. The scrambled siRNA group was adjusted to 1. Statistical significance (mean ± S.E.; n = 3) was determined by one-way analysis of variance and Tukey post test, and is denoted by asterisks (**, p < 0.01).
BAR Knockdown Suppresses ER Stress-induced IRE1 Signaling
BI-1 was reported to regulate ER stress responses by directly interacting with IRE1 and inhibiting its activation (7, 9). Based on the results that BAR modulates BI-1 protein levels via enhancing its proteasomal degradation, we reasoned that knocking down endogenous BAR expression would cause a secondary reduction in IRE1 signaling in cells undergoing ER stress. IRE1 has both endoribonuclease and protein kinase activities and initiates XBP1 splicing and protein kinase cascades during ER stress (1, 5). In this regard, we assessed the effects of endogenous BAR knockdown on both pathways. BI-1-expressing HeLa cells were transfected with BAR siRNA and treated with the ER stress inducer tunicamycin (inhibitor of N-linked glycosylation in ER) at increasing concentrations for 2.5 h, and the levels of XBP1 mRNA splicing were evaluated with both RT-PCR and real time PCR (Fig. 7, A and B). A dose-dependent increase in XBP1 mRNA splicing was observed with tunicamycin treatment in HeLa cells. Compared with scrambled siRNA-transfected cells, cells transfected with BAR siRNA showed less up-regulation of XBP1 splicing during ER stress (Fig. 7, A and B). Less XBP1 splicing was also observed in BAR siRNA-transfected cells when treated with thapsigargin (inhibitor of ER Ca2+-ATPase) and DTT (supplemental Fig. S10A), thus extending these observations to additional ER stress stimuli.
FIGURE 7.

BAR knockdown modulates ER stress-induced IRE1 signaling. A, BI-1-expressing HeLa cells were transfected with scrambled siRNA or BAR siRNA-1. After 24 h, cells were treated with 500 ng/ml of doxycycline for 24 h to induce BI-1 expression. Cells were then treated with increasing concentrations of tunicamycin as indicated for 2.5 h. The levels of XBP1 mRNA splicing were determined by RT-PCR. Spliced XBP1 (XBP1 S) and unspliced XBP1 (XBP1 U) were indicated. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA level was determined by RT-PCR. B, the levels of spliced XBP1 and unspliced XBP1 after tunicamycin treatment were also determined by real time PCR. The ratios of spliced XBP1/unspliced XBP1 were calculated. Statistical significance (mean ± S.E.; n = 3) was determined by Student's t test, and is denoted by asterisks (**, p < 0.01). C, BI-1-expressing HeLa cells were transfected with scrambled siRNA (Sc) or BAR siRNA-1. After 24 h, cells were treated with 500 ng/ml of doxycycline for an additional 24 h to induce BI-1 expression. Cells were then treated with 5 μm thapsigargin (TG) for 1 h. Cell lysates were analyzed by immunoblotting with antibodies to various ER stress markers, HA epitope, and α-tubulin. The blot was first probed with antibody to the phosphoprotein, followed by antibody stripping and probing with phospho-independent antibody directed to the same protein. Additionally, the blot was re-probed with anti-BAR antibody confirming the results shown in Fig. 6A (not shown). D, scanning densitometry was performed and the ratios were determined of phosphoprotein:total protein corresponding to various ER stress markers. The ratios in the scrambled siRNA group were adjusted to 1. Statistical significance (mean ± S.E.; n = 3) was determined by Student's t test, and is denoted by asterisks (**, p < 0.01).
To determine the effect of BAR knockdown on the IRE1-activated protein kinase cascade, BI-1-expressing HeLa cells were transfected with BAR siRNA and treated with the ER stress inducer thapsigargin for 1 h. The levels of total and activated (phosphorylated) JNK and its direct substrate c-Jun were monitored with immunoblotting. Compared with scrambled siRNA, transfection of BAR siRNA correlated with suppressed JNK and c-Jun activation after treatment with thapsigargin (Fig. 7, C and D). Reduction of JNK phosphorylation was also detected in BAR siRNA-transfected cells when treated with DTT and tunicamycin (supplemental Fig. S10B), thus extending these observations to additional ER stress stimuli. Although activation of p38 MAPK is also a downstream signaling event associated with IRE1 activation (5), no significant change was observed in phosphorylation of this stress kinase in HeLa cells treated with thapsigargin (Fig. 7, C and D). Importantly, phosphorylation of eIF2α, downstream of the ER stress transducer PERK, was not altered by BAR siRNA transfection (Fig. 7, C and D), thus showing a specific effect of BAR siRNA on IRE1 signaling. Taken together, these experiments demonstrate that knocking down expression of endogenous BAR specifically suppresses IRE1, but not PERK, signaling during ER stress.
BAR Protein Levels Are Reduced by Prolonged ER Stress
The observation that BAR is a short-lived protein, with significant degradation occurring within 2 h after inhibition of protein synthesis using cycloheximide (Fig. 5, A and B), promoted us to assess whether dynamic changes of BAR protein levels occur with prolonged ER stress. HeLa cells with or without BI-1 protein induction were treated with the ER stress inducer thapsigargin for up to 6 h, and levels of endogenous BAR protein were analyzed by immunoblotting. BAR protein levels began to decrease as early as 1 h after thapsigargin treatment, with striking reductions observed by 6 h (Fig. 8, A and B). Similar results were obtained when cells were treated with another ER stress inducer tunicamycin (supplemental Fig. S11). These effects on BAR protein stability were not due to a general decline of cell viability (not shown). By contrast, ER stress inducers did not stimulate significant changes in the levels of anti-apoptotic protein Bcl-2 (supplemental Fig. S11), which localizes in part to ER membranes (24), thus serving as a specificity control. No significant change in BAR mRNA levels was detected during the 6-h thapsigargin treatment (measured by RT-PCR analysis) (Fig. 8, C and D). We conclude that persistent ER stress reduces BAR protein expression through post-transcriptional mechanisms.
FIGURE 8.

BAR protein levels are reduced by prolonged ER stress induced by thapsigargin. A and B, HeLa cells without BI-1-HA induction (A) or induced with 500 ng/ml of doxycycline for 24 h (B) were treated with 1 μm thapsigagin (TG) for various durations as indicated. Levels of endogenous BAR and tubulin were determined by immunoblot analysis of cell lysates (normalized for total protein content) with antibodies to BAR and tubulin. Relative levels of endogenous BAR protein were quantified by scanning densitometry (mean ± S.E.; n = 3). BAR protein levels prior to TG treatment (0 h) were adjusted to 1. C and D, HeLa cells without BI-1-HA induction (C) or induced with 500 ng/ml of doxycycline for 24 h (D) were treated with 1 μm thapsigagin (TG) for various durations as indicated. Total RNA was extracted from cells. RT-PCR analysis of BAR mRNA was performed. RT-PCR for GAPDH served as a control. No significant change in BAR mRNA levels was detected.
DISCUSSION
BI-1 is an evolutionarily conserved ER resident protein that functions to modulate the UPR in plants and animals (7, 25–27). Three parallel ER stress transducers in mammals have been characterized, namely IRE1, PERK, and ATF6. It has been reported that IRE1 signaling can become selectively attenuated under prolonged ER stress relative to other UPR signaling components such as PERK, suggesting that specific modulators of IRE1 exist (6). Recently, BI-1 was found to directly interact with IRE1 and negatively regulate IRE1/XBP1 signaling (7), making BI-1 a candidate regulator of IRE1 signaling. Little is known about how BI-1 is regulated. Numerous efforts to generate BI-1 antibodies have been unsuccessful making tracking of the endogenous BI-1 protein a challenge, and leaving only mRNA studies to provide guidance. The levels of BI-1 mRNA vary among normal tissues, and have been reported to be modulated by hypoxia and ER stress (8, 9, 27, 28). Our findings indicate that BAR is an E3 ligase of the ER that plays a role in regulating BI-1 protein levels. The rapid decline of BAR protein levels during ER stress suggests that post-translational regulation of the BI-1 protein is dynamic and could contribute to the selective control of IRE1 signaling in cells undergoing prolonged ER stress. Indeed, we observed increased BI-1 expression in cells lacking endogenous BAR expression and selective inhibition of IRE1 signaling with intact PERK activation when cells with siRNA-mediated reductions in BAR were treated with ER stress inducers.
Although we focused on IRE1 signaling as an end point for inferring cellular activity of BI-1, the mechanisms by which BI-1 modulates ER stress responses appear to be multifaceted. Although BI-1 directly interacts with IRE1 and inhibits IRE1 activation (7), BI-1 also inhibits cell death induced by pro-apoptotic protein Bax (11, 25, 29, 30), which regulates mitochondrial outer membrane permeability (31), but also binds and activates IRE1 (32). In addition, BI-1 interacts with anti-apoptotic proteins Bcl-2 and Bcl-XL in ER membranes and regulates ER Ca2+ homeostasis, which is essential for normal ER function (33, 34). Finally, BI-1 is reported to activate redox-sensitive transcription factor Nrf-2 and protect cells from oxidative stress (26). It remains to be determined how BI-1 coordinates multiple regulatory responses when confronted with ER stress-associated disturbances.
Regulation of BI-1 signaling could occur at multiple levels. Here, we studied post-translational regulation of BI-1 protein stability and found that BAR has E3 ligase activity toward BI-1. Rapid changes of BAR protein levels in cells undergoing ER stress suggest that this level of regulation is dynamic and relevant to ER stress signaling. Regulation of BI-1 could occur at other levels, including transcription, translation, and post-translational modification. Transcriptional regulation of BI-1 has been studied previously (7, 8, 27, 28). Whereas one study found no change of BI-1 mRNA levels in cells treated with the ER stress inducer tunicamycin (7), other studies reported induction of BI-1 mRNA when confronted with ER stress stimuli (8, 27, 28). The results have implied that different types of cells might tailor BI-1 levels, depending on the duration and severity of ER stress. Moreover, BI-1 expression at the mRNA level becomes altered with disease, as shown by decreased expression in the liver of obese mice (9) and increased BI-1 mRNA expression in human cancers (35–37). The unavailability of antibodies recognizing BI-1 has hampered studies of BI-1 protein regulation.
E3 ligases may interact with different E2s and catalyze distinct types of ubiquitin chain polymerization (38). K48-linked poly-Ub chains typically target proteins for degradation by the 26 S proteasome, whereas K63-linked poly-Ub chains act as a scaffolds for assembling signaling complexes involved in inflammation, DNA repair, and protein trafficking (15), as well as possibly serving as a tag for lysosomal destruction (20). We show here that BAR-mediated K48-linked poly-Ub chain formation on BI-1 and induced proteasomal degradation of BI-1 protein. On the other hand, we also found that BAR was able to collaborate with the K63-specific E2 complex Ubc13-Uev1A in vitro (supplemental Fig. S12), which specifically forms K63-linked poly-Ub chains (15). Furthermore, by using both a K63-specific ubiquitin antibody and by overexpressing K63-only mutant ubiquitin protein, we detected evidence of K63-linked poly-Ub chains on BAR (supplemental Fig. S2 and Fig. 2D), suggesting that BAR may also serve as an E3 ligase for catalyzing K63-linked poly-Ub chains. The ability of some E3 ligases to catalyze both K48- and K63-linked ubiquitin chains has been reported previously (38). In agreement with previous reports (39, 40), we detected an increase in the K63-linked poly-Ub signal after MG132 treatment (supplemental Fig. S2), which may be an indication of mixed K48/K63 chains whereby the K48 poly-Ub component mediates proteasomal degradation. BAR also probably mediates formation of non-K48/non-K63-linked poly-Ub chains, given the strong ubiquitination of BAR observed in cells expressing mutant ubiquitin (K48R). Recently, K11 has been identified as an abundant site of ubiquitin conjugation in vivo (16).
The finding that BAR is an ER-associated RING-type E3 ligase involved in the degradation of ER membrane proteins (e.g. BI-1) raised the possibility that BAR is a novel ERAD component that targets the degradation of a subset of misfolded proteins in the ER. To this end, we studied by immunoprecipitation experiments the interaction of BAR with MmUbc6 and MmUbc7, mammalian homologs of yeast ER-associated E2s, Ubc6 and Ubc7, respectively, which are implicated in ERAD (41). We found MmUbc7 associated with BAR in a RING-dependent manner (supplemental Fig. S13). However, when we further assessed the role of BAR in degradation of two bona fide ERAD substrates, TCR-α and CD3-δ (41, 42), overexpression of BAR did not affect the steady-state levels of either TCR-α or CD3-δ (supplemental Fig. S14). Thus, either BAR is not involved in ERAD or it plays a role distinct from other RING-type of E3 ligases that have been previously implicated in ERAD (43).
Analogous to BI-1, the BAR protein may have several functions in ER membranes besides the BI-1-directed E3 ligase activity documented here. In this regard, both BI-1 and BAR were originally discovered using a functional genomics method in which human cDNA libraries were screened in yeast for suppressors of cell death induced by ectopic expression of mammalian Bax (10, 11). Thus, BI-1 and BAR appear to share some unknown function by which they oppose the cytotoxic activity of Bax in yeast, despite the role of BAR as an antagonist of BI-1 with respect to regulation of protein degradation. Like BI-1, BAR reportedly associates with Bcl-2 and Bcl-XL (but not Bax) in ER membranes and inhibits apoptosis (10). In addition to at least one (possibly as many as three) transmembrane domain, the BAR protein also possesses a coiled-coil domain sharing sequence similarity with protein interaction domains found in the ER membrane protein Bap31, the Huntingtin-interacting protein (Hip), and the Hip protein interactor (Hippi) (21, 44). BAR also reportedly binds to these proteins (e.g. Bap31, Hip, and Hippi), which have all been reported to regulate apoptosis (21). Of these, Bap31 has been claimed to regulate ER Ca2+ homeostasis (45), reminiscent of BI-1 (33, 34). Thus, several intriguing connections intertwine BAR with BI-1 and other ER proteins implicated in regulation of cell life and death, serving as a foundation for future investigations of the mechanisms of these proteins in the context of ER stress.
Supplementary Material
Acknowledgments
We thank Tessa Siegfried and Melanie Hanaii for manuscript preparation, Dr. Ze'ev Ronai for critical review of the manuscript. We thank Drs. Claudio Hetz, Axel Methner, Ron Prywes, and Ze'ev Ronai for providing plasmids. We thank Drs. Ricardo Correa and Craig Tamble for technical support, Renata Sano for BI-1 inducible HeLa cells, and Temesgen Samuel for K48-only and K63-only ubiquitin constructs. We thank members of Reed lab for sharing reagents and helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant AG-15393 (to J. C. R.) and American Heart Association Western States Affiliate Postdoctoral Fellowship 0825021F (to J. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S14.
- ER
- endoplasmic reticulum
- PERK
- protein kinase RNA-like ER kinase
- ATF
- activating transcription factor
- UPR
- unfolded protein response
- TRAF2
- tumor necrosis factor receptor-associated factor 2
- ERAD
- ER-associated protein degradation
- BI-1
- Bax inhibitor-1
- BAR
- bifunctional apoptosis regulator
- poly-Ub
- polyubiquitin
- IRE1
- inositol-requiring protein-1.
REFERENCES
- 1. Ron D., Walter P. (2007) Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 2. Kim I., Xu W., Reed J. C. (2008) Nat. Rev. Drug. Discov. 7, 1013–1030 [DOI] [PubMed] [Google Scholar]
- 3. Lee K. P., Dey M., Neculai D., Cao C., Dever T. E., Sicheri F. (2008) Cell 132, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., Ron D. (2000) Science 287, 664–666 [DOI] [PubMed] [Google Scholar]
- 5. Xu C., Bailly-Maitre B., Reed J. C. (2005) J. Clin. Invest. 115, 2656–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin J. H., Li H., Yasumura D., Cohen H. R., Zhang C., Panning B., Shokat K. M., Lavail M. M., Walter P. (2007) Science 318, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lisbona F., Rojas-Rivera D., Thielen P., Zamorano S., Todd D., Martinon F., Glavic A., Kress C., Lin J. H., Walter P., Reed J. C., Glimcher L. H., Hetz C. (2009) Mol. Cell 33, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bailly-Maitre B., Fondevila C., Kaldas F., Droin N., Luciano F., Ricci J. E., Croxton R., Krajewska M., Zapata J. M., Kupiec-Weglinski J. W., Farmer D., Reed J. C. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 2809–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bailly-Maitre B., Belgardt B. F., Jordan S. D., Coornaert B., von Freyend M. J., Kleinridders A., Mauer J., Cuddy M., Kress C. L., Willmes D., Essig M., Hampel B., Protzer U., Reed J. C., Brüning J. C. (2010) J. Biol. Chem. 285, 6198–6207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang H., Xu Q., Krajewski S., Krajewska M., Xie Z., Fuess S., Kitada S., Pawlowski K., Godzik A., Reed J. C. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 2597–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu Q., Reed J. C. (1998) Mol. Cell 1, 337–346 [DOI] [PubMed] [Google Scholar]
- 12. Zapata J. M., Pawlowski K., Haas E., Ware C. F., Godzik A., Reed J. C. (2001) J. Biol. Chem. 276, 24242–24252 [DOI] [PubMed] [Google Scholar]
- 13. Deshaies R. J., Joazeiro C. A. (2009) Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 14. Joazeiro C. A., Wing S. S., Huang H., Leverson J. D., Hunter T., Liu Y. C. (1999) Science 286, 309–312 [DOI] [PubMed] [Google Scholar]
- 15. Pickart C. M., Fushman D. (2004) Curr. Opin. Chem. Biol. 8, 610–616 [DOI] [PubMed] [Google Scholar]
- 16. Xu P., Duong D. M., Seyfried N. T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J. (2009) Cell 137, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S. P. (2003) Nat. Biotechnol. 21, 921–926 [DOI] [PubMed] [Google Scholar]
- 18. Newton K., Matsumoto M. L., Wertz I. E., Kirkpatrick D. S., Lill J. R., Tan J., Dugger D., Gordon N., Sidhu S. S., Fellouse F. A., Komuves L., French D. M., Ferrando R. E., Lam C., Compaan D., Yu C., Bosanac I., Hymowitz S. G., Kelley R. F., Dixit V. M. (2008) Cell 134, 668–678 [DOI] [PubMed] [Google Scholar]
- 19. Deng L., Wang C., Spencer E., Yang L., Braun A., You J., Slaughter C., Pickart C., Chen Z. J. (2000) Cell 103, 351–361 [DOI] [PubMed] [Google Scholar]
- 20. Huang F., Kirkpatrick D., Jiang X., Gygi S., Sorkin A. (2006) Mol. Cell 21, 737–748 [DOI] [PubMed] [Google Scholar]
- 21. Roth W., Kermer P., Krajewska M., Welsh K., Davis S., Krajewski S., Reed J. C. (2003) Cell Death Differ. 10, 1178–1187 [DOI] [PubMed] [Google Scholar]
- 22. Wang Z., Cuddy M., Samuel T., Welsh K., Schimmer A., Hanaii F., Houghten R., Pinilla C., Reed J. C. (2004) J. Biol. Chem. 279, 48168–48176 [DOI] [PubMed] [Google Scholar]
- 23. Tcherpakov M., Broday L., Delaunay A., Kadoya T., Khurana A., Erdjument-Bromage H., Tempst P., Qiu X. B., DeMartino G. N., Ronai Z. (2008) Mol. Biol. Cell 19, 5019–5028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krajewski S., Tanaka S., Takayama S., Schibler M. J., Fenton W., Reed J. C. (1993) Cancer Res. 53, 4701–4714 [PubMed] [Google Scholar]
- 25. Chae H. J., Ke N., Kim H. R., Chen S., Godzik A., Dickman M., Reed J. C. (2003) Gene 323, 101–113 [DOI] [PubMed] [Google Scholar]
- 26. Lee G. H., Kim H. K., Chae S. W., Kim D. S., Ha K. C., Cuddy M., Kress C., Reed J. C., Kim H. R., Chae H. J. (2007) J. Biol. Chem. 282, 21618–21628 [DOI] [PubMed] [Google Scholar]
- 27. Watanabe N., Lam E. (2008) Plant Signal. Behav. 3, 564–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Isbat M., Zeba N., Kim S. R., Hong C. B. (2009) J. Plant. Physiol. 166, 1685–1693 [DOI] [PubMed] [Google Scholar]
- 29. Bolduc N., Ouellet M., Pitre F., Brisson L. F. (2003) Planta 216, 377–386 [DOI] [PubMed] [Google Scholar]
- 30. Kawai-Yamada M., Ohori Y., Uchimiya H. (2004) Plant Cell 16, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei M. C., Zong W. X., Cheng E. H., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., Korsmeyer S. J. (2001) Science 292, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hetz C., Bernasconi P., Fisher J., Lee A. H., Bassik M. C., Antonsson B., Brandt G. S., Iwakoshi N. N., Schinzel A., Glimcher L. H., Korsmeyer S. J. (2006) Science 312, 572–576 [DOI] [PubMed] [Google Scholar]
- 33. Kim H. R., Lee G. H., Ha K. C., Ahn T., Moon J. Y., Lee B. J., Cho S. G., Kim S., Seo Y. R., Shin Y. J., Chae S. W., Reed J. C., Chae H. J. (2008) J. Biol. Chem. 283, 15946–15955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu C., Xu W., Palmer A. E., Reed J. C. (2008) J. Biol. Chem. 283, 11477–11484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grzmil M., Thelen P., Hemmerlein B., Schweyer S., Voigt S., Mury D., Burfeind P. (2003) Am. J. Pathol. 163, 543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grzmil M., Kaulfuss S., Thelen P., Hemmerlein B., Schweyer S., Obenauer S., Kang T. W., Burfeind P. (2006) J. Pathol. 208, 340–349 [DOI] [PubMed] [Google Scholar]
- 37. Schmidt S. M., König T., Bringmann A., Held S., von Schwarzenberg K., Heine A., Holderried T. A., Stevanovic S., Grünebach F., Brossart P. (2009) Leukemia 23, 1818–1824 [DOI] [PubMed] [Google Scholar]
- 38. Xia Z. P., Chen Z. J. (2005) Sci. STKE 2005, pe7. [DOI] [PubMed] [Google Scholar]
- 39. Bennett E. J., Shaler T. A., Woodman B., Ryu K. Y., Zaitseva T. S., Becker C. H., Bates G. P., Schulman H., Kopito R. R. (2007) Nature 448, 704–708 [DOI] [PubMed] [Google Scholar]
- 40. Meierhofer D., Wang X., Huang L., Kaiser P. (2008) J. Proteome Res. 7, 4566–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tiwari S., Weissman A. M. (2001) J. Biol. Chem. 276, 16193–16200 [DOI] [PubMed] [Google Scholar]
- 42. Bonifacino J. S., Cosson P., Klausner R. D. (1990) Cell 63, 503–513 [DOI] [PubMed] [Google Scholar]
- 43. Kostova Z., Tsai Y. C., Weissman A. M. (2007) Semin. Cell Dev. Biol. 18, 770–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mukasa T., Santelli E., Reed J. C., Pascual J. (2007) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63, 297–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Breckenridge D. G., Stojanovic M., Marcellus R. C., Shore G. C. (2003) J. Cell Biol. 160, 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.