

Abstract
The Toll signaling pathway, an essential innate immune response in invertebrates, is mediated via the serine protease cascade. Once activated, the serine proteases are irreversibly inactivated by serine protease inhibitors (serpins). Recently, we identified three serpin-serine protease pairs that are directly involved in the regulation of Toll signaling cascade in a large beetle, Tenebrio molitor. Of these, the serpin SPN48 was cleaved by its target serine protease, Spätzle-processing enzyme, at a noncanonical P1 residue of the serpin's reactive center loop. To address this unique cleavage, we report the crystal structure of SPN48, revealing that SPN48 exhibits a native conformation of human antithrombin, where the reactive center loop is partially inserted into the center of the largest β-sheet of SPN48. The crystal structure also shows that SPN48 has a putative heparin-binding site that is distinct from those of the mammalian serpins. Ensuing biochemical studies demonstrate that heparin accelerates the inhibition of Spätzle-processing enzyme by a proximity effect in targeting the SPN48. Our finding provides the molecular mechanism of how serpins tightly regulate innate immune responses in invertebrates.
Keywords: Crystal Structure, Innate Immunity, Serine Protease, Serpin, Signal Transduction, Toll Receptors
Introduction
Serine protease inhibitors (serpins) play essential roles in the down-regulation of extracellular signals exerted by serine protease proteolytic cascades (1, 2). In mammals, blood coagulation is specifically activated by a serine protease cascade and is tightly controlled by serpins (2). The reactive center loop (RCL)3 of serpins acts as bait to capture the target serine protease and is then cleaved at the P1 residue, forming an acyl-intermediate complex with the target serine protease (3). In the acyl-intermediate complex, serpins undergo dramatic conformational changes, and the RCL is inserted into the central strand of the largest β-sheet (sheet A), resulting in distorting and inactivating the catalytic machinery of the protease (3–5).
Thrombin, the key enzyme responsible for converting fibrinogen to fibrin in the mammalian blood clotting cascade, is mainly regulated by the target serpin human antithrombin (1, 6). Heparin can bind to both antithrombin and its target serine proteases, thrombin, factor Xa, and factor IXa, activating antithrombin through allosteric and/or bridging mechanisms (7, 8). In Drosophila, the Toll signaling pathway is triggered by invading pathogens and is amplified in hemolymph (insect blood) via the proteolytic cascade of the insect serine proteases largely consisting of a clip domain and a serine protease domain (9, 10). This pathway induces antimicrobial peptide gene expression through activation of the NF-κB transcription factor, and antimicrobial peptide then kills the pathogens (9).
Four Drosophila serpins related to innate immunity have been analyzed using genetic approaches (11, 12). Necrotic is a serpin that inhibits the clip domain serine protease Persephone (13), and necrotic mutant flies constitutively express the Toll signaling pathway (11). To date, only the crystal structures of serpin 1K and 1B from the tobacco hornworm, Manduca sexta are available among insect serpins (14, 15), but their target proteases remain unclear.
Our research group has solved structures and determined the biological functions of serine proteases involved in insect innate immunity (16–19). Recently, we identified three pairs of serine proteases and serpins involved in the Toll signaling pathway in the large beetle, Tenebrio molitor (19, 20). Of these, we demonstrated that the Spätzle-processing enzyme (SPE) can make a specific serine protease-serpin complex with a 48-kDa serpin (SPN48). Because the RCL of serpin is first recognized by the target serine protease, the RCL should be a good substrate for the target protease (3). In most serpins, the P1 residue of serpins is important for indication of specificity of the target protease (21). However, the RCL region of SPN48 does not contain an arginine or lysine residue, which is expected to be present as a target residue of trypsin-like SPE. When the cleaved site of SPN48 by activated SPE was determined biochemically, SPN48 was cleaved after the noncanonical P1 glutamic acid (Glu-353) of the RCL region (20).
To gain insight into how this SPN48 achieves specific reactivity to the target protease SPE in the innate immune response, we performed biochemical and structural studies on SPN48 with regard to the target serine protease SPE.
EXPERIMENTAL PROCEDURES
Overexpression, Purification, and Site-directed Mutagenesis
Overexpression and purification of recombinant SPN48 (residues 17–389) using a bacterial expression system were reported previously (22). To construct SPN48 mutants, DNA fragments encoding mutant SPN48 proteins were created using the overlap extension PCR method. The resulting PCR DNA was digested with NdeI and HindIII and ligated into the same sites in a modified vector from pPROEX-HTA (Invitrogen) in which the NcoI site was replaced with NdeI. To construct wild-type (residues 336–361) and mutant RCL-linker (E353K), DNA fragments encoding SPN48 RCL (residues 336–361) were amplified by PCR. The resulting PCR DNA was digested with BamHI and EcoRI and ligated into the same sites of the pGEXTEV-MliC vector, which expresses the N-terminal GST protein, C-terminal Pseudomonas aeruginosa MliC protein, and the tobacco etch virus protease cleavage site between the two proteins (23). The resulting vector contains the RCL instead of the tobacco etch virus protease cleavage site between the GST tag and P. aeruginosa MliC. The Linker-RCLs were purified using glutathione-agarose resins. To construct SPE, the clip domain (residues 19–84) and the serine protease domain (residues 85–385) were ligated into EcoRI and XhoI sites of pGEX-TEV vector. The sequences of the used primers in the DNA construction are listed in supplemental Table S1.
Crystallization, Data Collection, and Structure Determination of SPN48
The crystallization of SPN48 and data collection were described previously (22). Initial phases were calculated from a molecular replacement program MOLREP (24) solution obtained using α1-antitrypsin (Protein Data Bank entry 3CWL) (25) using a search model. The resulting map revealed traceable electron density for the SPN48 molecule. The model was built in the program COOT (26). The structure was refined in the program CNS (27) and was further refined using PHENIX.REFINE (28). Another crystal grown in a reservoir solution (13% polyethylene glycol 8000 and 0.11 m MES (pH 6.0)) was obtained, from which a 1.9-Å resolution dataset with similar cell dimensions was collected. The final coordination of SPN48 was calculated by refining against this higher resolution dataset. Crystallographic data statistics are summarized in Table 1. Structure presentations were generated using PyMOL (29).
TABLE 1.
X-ray data collection and refinement statistics
| Data set | |
| Source | Beamline 4A at PLS |
| Wavelength | 1.0000 Å |
| Resolution limit | 50 to 1.9 Å (1.93 to 1.90 Å)a |
| Space group | P21 |
| Unit cell | a = 66.3, b = 43.0, c = 66.0 Å, α = γ = 90°, β = 105.1° |
| Redundancy | 6.7 (3.4)a |
| Rsym | 5.4% (27.6%)a |
| Completeness | 97.4% (89.9%)a |
| Refinement | |
| Resolution range | 30 to 1.9 Å |
| R-factor | 22.3% |
| Rfreeb | 27.8% |
| Average B value | 49.5 Å2 |
| r.m.s.d.c for bonds | 0.008 Å |
| r.m.s.d. for angles | 1.093° |
| Total no. of atoms | |
| No. of protein atoms | 2890 |
| No. of water atoms | 105 |
| No. of metal atoms | 0 |
| Ramachandran plot | |
| Most favored regions | 298 (90.6%) |
| Additional allowed regions | 28 (8.5%) |
| Generously disallowed region | 3 (0.9%) |
| Disallowed region | 0 (0.0%) |
a The numbers in parentheses are statistics for the highest resolution shell.
b Rfree was calculated with 10% of the data.
c r.m.s.d., root mean square deviation.
Amidase and Protease Activity Assays
Active SPE was obtained as described previously (20), and thrombin and trypsin were purchased from Sigma. The amidase activities of active SPE, thrombin, and trypsin were measured using a fluorescent substrate as described previously (19). The amidase activity of each sample was measured as the fluorescence intensity with excitation and emission wavelengths of 380 and 460 nm, respectively. To measure the protease activity of SPE and thrombin toward wild-type or the mutant RCL linker, 5 μg of the RCL linker protein was incubated with 0.15 μg of SPE or 25 units of thrombin at 30 °C for 2 h in buffer containing 20 mm Tris (pH 8.0) and 150 mm NaCl, and then resolved by SDS-PAGE.
Western Blot Analysis
Equal amounts of proteins were resolved by 15% SDS-PAGE and subsequently transferred onto a polyvinylidene difluoride (PVDF) membrane, which was pre-equilibrated in methanol. The membrane was blocked for 1 h with 3% skim milk in TBST (50 mm Tris, 150 mm NaCl, 0.05% Tween 20 (pH 8.0)) buffer, then incubated with anti-SPN48 antibody (20) (1:500) in 1% skim milk at 4 °C for about 12 h, and then washed three times with TBST. The membrane was then incubated with secondary antibody (anti-rabbit antibody) for 2 h at room temperature, and the SPN48 protein band was visualized by WEST-ZOL reagent (iNtRON, Korea) according to the manufacturer's instructions.
Heparin Binding Assay
Approximately 200 μg of each SPN48 protein was loaded onto a Hitrap-heparin column (1 ml; GE Healthcare) equilibrated with 50 mm Tris buffer (pH 7.5) containing 3 mm EDTA. The samples were eluted with a gradient of 0 to 1 m NaCl in 50 mm Tris buffer at a flow rate of 1.0 ml/min with detection at A280. The concentration of NaCl was monitored by a conductivity meter in the Acta FPLC system (GE Healthcare).
In Vitro Binding Assay
GST-fused SPE clip domain (GST-clip) or GST-fused SPE serine protease domain (GST-SP) protein was bound to glutathione-coupled agarose resin. Equal amounts of hexahistidine-tagged SNP48 protein were added to both the GST-clip-bound resin and the GST-SP-bound resin in PBS (final 50 μl). The reaction mixtures were incubated for 1 h at 4 °C, washed with PBS, then centrifuged at 3000 rpm for 10 s, and further washed three times with RIPA buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, and 0.1% SDS).
RESULTS
SPN48 Is Highly Specific to SPE
To investigate the role of the noncanonical motif in the SPN48 RCL, the inhibitory activities of SPN48 on SPE, thrombin, and trypsin were measured using wild-type SPN48 and mutant SPN48 (E353K), which has the canonical sequence of SPE after substitution of the P1 glutamic acid residue (Glu-353) with a lysine residue. As shown in Fig. 1A, wild-type and mutant SPN48 (E353K) inhibited the amidase activity of SPE but not of thrombin and trypsin, and it is therefore distinct from other serpins, such as serpin 1K and Necrotic, which can inhibit chymotrypsin or trypsin (14, 30). Interestingly, the mutant SPN48 (E353K) showed a stronger inhibitory effect on SPE than the wild-type SPN48 (Fig. 1A), indicating that the noncanonical P1 Glu-353 residue is not optimal for the inhibition of SPE.
FIGURE 1.

Specific activity of SPN48. A, inhibitory effects of the wild-type and E353K SPN48 (400 ng each) on SPE, thrombin, and trypsin (100 ng each) were monitored by the amidase activity. The amidase activity of each protease was measured in the presence of wild-type SPN48 based on the fluorescence intensity liberated from the product. The peptide t-butyloxycarbonyl-Val-Pro-Arg-4-methylcoumaryl-7-amide was used as a substrate for SPE and thrombin, whereas the peptide t-butyloxycarbonyl-Phe-Ser-Arg-4-methylcoumaryl-7-amide was used for trypsin. Before measuring the amidase activities, the reaction mixtures were incubated for 4 h at 30 °C. Each experiment was performed in triplicate, and the standard deviations are shown as error bars. B, complex formation of wild-type and E353K SPN48 (400 ng each) with SPE (200 ng), thrombin (2 units), and trypsin (200 ng). The reaction mixtures were incubated for 30 min at 30 °C (for SPE) or 37 °C (for trypsin and thrombin) and then were subjected to SDS-PAGE and subsequent Western blotting using anti-SPN48 antibody. When SPN48 inactivates the target serine protease, a high molecular weight SDS-stable complex is formed with the target protease, as indicated. Alternatively, SPN48 is cleaved at the P1 residue without inactivating the target protease or is degraded, as indicated. C, comparison of wild-type SPN48 with the mutant SPN48 (E353K) in the specific inhibition of SPE in the presence of a nontargeting protease thrombin. Wild-type SPN48 (wt SPN48) forms a complex with SPE without being affected by thrombin, whereas thrombin severely interfered with the mutant SPN48 (E353K). The SPN48 was visualized by immunoblotting with anti-SPN48 antibody.
To further analyze the above result, Western blotting was performed with anti-SPN48 antibody. Wild-type SPN48 was resistant to proteolysis by thrombin and was degraded by trypsin without forming a complex, which is distinct from the complex formation of SPN48 and SPE. The mutant SPN48 (E353K) failed to form a complex with trypsin and thrombin even though the serpin was specifically cleaved probably at the mutated Lys residue (Fig. 1B). This result indicates that the noncanonical P1 residue of SPN48 may be important for the specificity in inhibition of SPE.
To test this idea, we co-incubated SPE and thrombin with wild-type or mutant SPN48, and we measured the inhibitory activity of each serpin. SPE was efficiently inhibited by wild-type SPN48 even in the presence of thrombin (Fig. 1C). In contrast, the mutant SPN48 (E353K) was cleaved at the mutated Lys residue by thrombin, and thus its inhibitory activity on SPE was abolished (Fig. 1C). Taken together, our finding suggests that the noncanonical P1 residue of SPN48 plays a crucial role in the prevention from the cleavage of SPN48 by the nontargeting serine protease, enhancing the specific reactivity of the serpin.
Structure Determination and Overall Structure of SPN48
To gain insight into the unique properties of SPN48, we determined the crystal structure of SPN48. The mature form of SPN48 was expressed in Escherichia coli and was successfully crystallized (22). The crystal structure of SPN48 was determined by the molecular replacement method using α1-antitrypsin as a search model (25). The final 1.9-Å resolution model contains residues 17–389 with 105 water molecules (Fig. 2A), whereas residues 348–356 are disordered in the crystal.
FIGURE 2.

Overall structure of SPN48. A, ribbon representation of the SPN48 structure. The RCL is shown in green. Helices are in cyan; helix D is in purple, and helix I is in blue. Sheet A is shown in magenta, and sheets B and C are in yellow and orange, respectively. B, superposition of SPN48 (green RCL and magenta elsewhere) and serpin 1K (gray RCL and lime elsewhere) using Cα atoms. Structural superposition was carried out using PyMOL (29). C, superposition of SPN48 (green RCL and magenta elsewhere) and antithrombin (cyan RCL and orange elsewhere) using Cα atoms. Structural superposition was carried out using PyMOL (29). D, stereo representation of the hinge region of SPN48 with the electron density map. 2FoFc electron density map is contoured at 1.0σ level.
The overall structure of SPN48 exhibits a typical serpin fold, revealing that SPN48 is most closely related to M. sexta serpin 1K and human antithrombin. SPN48 was superimposed with M. sexta serpin 1K with a root mean square deviation of 1.08 Å (between 271 Cα atoms; Fig. 2B) and with human antithrombin with root mean square deviation of 1.51 Å (between 274 Cα atoms; Fig. 2C). In particular, the overall RCL position of SPN48 is most similar to that of M. sexta serpin 1K, which is distinguished from that of other mammalian serpins (Fig. 2, B and C).
Native Conformation of SPN48, Similar to Antithrombin
The crystal structure of SPN48 revealed a subtle but important difference in the conformation of the N-terminal region of RCL (so-called hinge region) compared with those of other serpins, except for antithrombin. The N-terminal portion of RCL was partially inserted into sheet A forming the fourth strand in sheet A (Fig. 2A), as observed in the native conformation of antithrombin (31, 32). The native conformation of antithrombin is distinct from the canonical structure of active α1-antitrypsin and M. sexta serpin 1K, where the RCL is fully exposed without forming the fourth strand in sheet A. In the native conformation of antithrombin, the length of the RCL is reduced because of the partial insertion into sheet A, which may account for the poor inhibitory activity of antithrombin on factor Xa and IXa (33, 34) in the absence of heparin. However, antithrombin can efficiently inactivate thrombin in the native conformation (35), which suggests that the native conformation of antithrombin confines the inhibitory activity on thrombin. Likewise, the RCL conformation of SPN48 may contribute to the specific inhibitory activity of SPN48 on SPE as a tight regulator of the innate immune responses.
Highly Exposed RCL of SPN48 and Suboptimal Substrate for SPE
The sequence of RCL (P4–P1′) recognized by SPE was disordered in the crystal structure of SPN48, which may reflect the intrinsic flexibility of the RCL of SPN48. Because there was no conformational restraint in the protease-recognized sequence, we analyzed the proteolytic susceptibility of only RCL region in the absence of the main body of the serpin. The RCL regions (residues 336–361) from the wild-type and E353K mutant were inserted between two arbitrarily selected proteins as a linker (Linker-RCL; Fig. 3A) to mimic the peptide substrates but to exclude the role of the main body of the serpin.
FIGURE 3.

Intrinsic flexibility of the SPN48 RCL region. A, domain structure of linker-RCLs. The RCL region of SPN48 is inserted in the GST and P. aeruginosa MliC proteins. B, protease activity of SPE and thrombin on wild-type and mutant linker-RCLs. Wild-type or mutant linker-RCL was used as a substrate for thrombin or SPE for the specified time at 30 °C. Although the wild-type protein (wt Linker-RCL) remained uncleaved, the mutant protein (Linker-RCL (E353K)) was gradually degraded by both proteases. C, sequence alignment of SPN48 RCL and the SPE substrates T. molitor prophenol oxidase (Tm-pro-PO) and Spätzle around the cleavage sites, indicated by an arrow. Because T. molitor Spätzle has not been cloned, its homologue Tribolium castaneum Spatzle (Tc-Spatzle) was used instead.
As shown in Fig. 3B, wild-type Linker-RCL was not cleaved by SPE and thrombin, whereas the mutant Linker-RCL (E353K) was cleaved by SPE and thrombin. This observation indicates that the wild-type RCL cannot be recognized by the proteases without the main body of the serpin and that the mutant RCL with the canonical P1 residue is readily recognized by both the target protease SPE and a nontargeting protease thrombin. The sequence alignment of the SPN48 RCL with the cleavage sites from the SPE substrates Spätzle and pro-phenol oxidase shows that the RCL region has similarity at the P3 residue (Fig. 3C), suggesting that the P3 residue fits the active site of SPE even though the P1 residue is also important. This result indicates that the RCL is a suboptimal substrate of the target serine protease SPE that could not be recognized by the target protease without the main body of the serpin.
Putative Heparin-binding Site of SPN48 Differs from That of Antithrombin
Because SPN48 was bound to heparin-coupled resin during the purification from the insect hemolymph (20), we confirmed the heparin binding ability of SPN48 (Fig. 4A). However, sequence alignment with human antithrombin revealed that the lysine residues at the classical heparin-binding site of antithrombin are not conserved in SPN48, suggesting that the heparin-binding site of SPN48 may differ from the classical heparin-binding site (Fig. 4B). In antithrombin, the classical heparin-binding site is linked to the helix D extension that is responsible for maintaining the native conformation; helix D moves and expels the hinge region of the RCL from sheet A upon heparin binding (8, 33).
FIGURE 4.

Heparin-binding site of SPN48. A, heparin binding capacity of wild-type and mutant SPN48. The heparin binding activity of wild-type and mutant SPN48 4KA (K293A/K294A/K297A/K298A) was assessed by heparin-affinity chromatography. The SPN48 protein peak was confirmed by Western blotting (data not shown). B, sequence alignment of SPN48 with antithrombin, focusing on the residues at the classical heparin-binding site in antithrombin. The lysine or arginine residues involved in heparin binding are indicated by a shaded box (33, 35). Of six residues, only two residues are conserved in SPN48. C, putative heparin binding region of SPN48. All lysine or arginine residues of SPN48 are displayed with secondary elements shown in gray. Helix I, which is located on a surface opposite Helix D, contains Lys-293, Lys-294, Lys-297, and Lys-298, indicated by a dotted circle.
Structural analysis of SPN48 revealed that several lysine residues are clustered on the surface of a lower part of SPN48, suggestive of a putative heparin-binding site (Fig. 4C). The lysine cluster of SPN48 is located at the opposite face (helix I) to the classical heparin-binding site, which is distinct from the classical heparin-binding region of antithrombin and plasminogen activator inhibitor, and heparin cofactor II (36). A mutant SPN48 protein harboring alanine substitutions at four lysine residues at helix I showed a reduced heparin binding ability (Fig. 4A), suggesting that helix I might serve as a heparin-binding site of SPN48.
Roles of Heparin and Calcium Ion in SPE Inhibition by SPN48
Given the heparin binding ability and functional similarity of SPN48 with antithrombin, we examined the involvement of heparin in the function of SPN48. Native full-length heparin accelerated the SPN48 inhibition of SPE (Fig. 5A), whereas a commercially available heparin pentasaccharide (fondaparinux), which has anti-coagulation activity in mammalian blood (37), did not alter the reactivity of SPN48. Another low molecular weight heparin (15-saccharide with molecular mass of ∼5000 Da) also did not affect the SPN48 activity like the heparin pentasaccharide. These results show that heparin enhances the reactivity of SPN48 with SPE depending on the length of the heparin chain. This dependence on the length of heparin chain was observed in the case of antithrombin and thrombin (38). Because only long chain heparin, which can combine the serpin and serine protease simultaneously, increased the reactivity of the serpin, our findings suggest that heparin plays only a bridging role. In these experiments, it can be excluded that heparins activate the enzyme by inducing the conformational change of SPE, because any heparin did not increase the proteolytic activity of SPE on Linker-RCL (E353K) (Fig. 5B). Consistently, the heparin-binding site of SPN48 is not linked to helix D inducing a conformational change in the RCL. Furthermore, we failed to observe any conformational change of SPN48 by the addition of heparins using fluorescence and circular dichroism spectra from the protein (data not shown).
FIGURE 5.

Heparin and calcium effects on SPE inhibition by SPN48. A, heparin effect on SPE inhibition by SPN48 depending on the length of heparin. Full-length heparin accelerated the inhibitory activity of SPN48, whereas pentasaccharide unit of heparin (fondaparinux) and low molecular weight heparin (LMWH, ∼15-saccharide unit) did not affect SPN48 activity. SPE (300 ng), SPN48 (600 ng), full-length heparin (200 ng), fondaprinux (200 ng), or low molecular weight heparin (200 ng) were used in this experiment in 50 mm Tris-HCl (pH 7.5) containing 100 mm NaCl. Amidase activity of SPE was measured after 2 h of incubation at 30 °C. B, heparin does not affect the proteolytic activity of SPE. Both full-length heparin (200 ng), pentasaccharide heparins (fondaparinux; 200 ng), and low molecular weight heparin (200 ng) did not cause a marked change in the enzymatic activity of SPE. The wild-type and the mutant (E353K) Linker-RCL (25 μg each) were used as substrates for SPE (50 ng) in 50 mm Tris-HCl (pH 7.5) containing 5 mm CaCl2. C, synergistic role of heparin and calcium ion on the inhibition of SPE by SPN48. The amidase activity of SPE was measured using the fluorescent peptide after 1 h of incubation at 30 °C. SPE (300 ng), SPN48 (600 ng), 5 mm CaCl2, and/or full-length heparin (200 ng) were used in 50 mm Tris-HCl (pH 7.5).
Calcium ion is known to promote heparin bridging of antithrombin and factor IXa (39). We next examined the calcium ion effect in the SPN48 inhibition of SPE. As shown in Fig. 5C, calcium ion increased SPN48 reactivity with SPE only in the presence of heparin as observed in antithrombin and factor IXa (39). Like heparin, calcium ion did not show any conclusive evidence for the conformational change of SPN48 (data not shown). Thus, calcium ion might be involved only in the heparin binding and not in the conformational change of SPN48.
Intrinsic Affinities of SPE to SPN48
The Clip domains of some serine proteases of insects have been implicated in the involvement of the specific activation of the clip domain-mediated proteolytic cascades, such as Toll signaling and melanin-synthesis pathways (16, 17). To gain a clue of the role of the clip domain of SPE in the inhibition by SPN48, the clip domain and the serine protease domain of SPE were separately expressed as GST fusion proteins, and GST pulldown experiments were then performed. The result showed that both the clip domain and the serine protease domain exhibited an affinity to SPN48 (Fig. 6). Because the serine protease domain was in a zymogen form, the affinities from both domains to SPN48 should result from noncovalent bonds. Our finding suggests that these noncovalent interactions might be involved in cognate recognition of SPN48 with the target serine protease.
FIGURE 6.
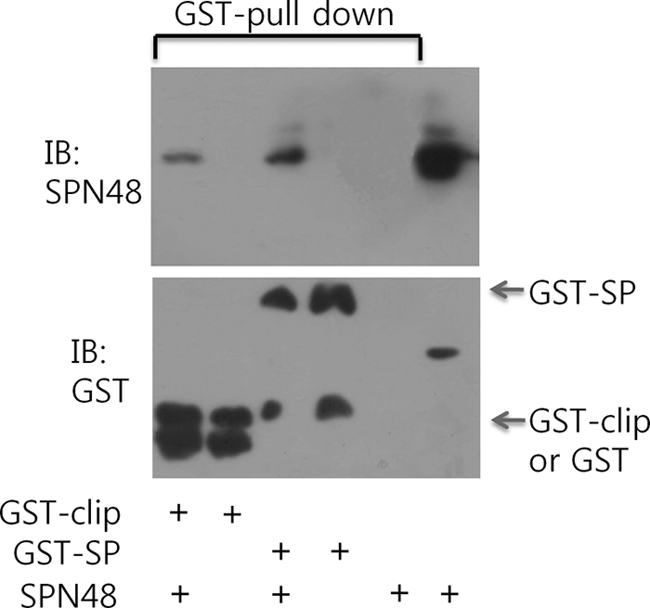
Interaction between SPE and SPN48. Bound SPN48 was pulled down by GST-fused SPE clip domain (GST-clip) or GST-fused SPE serine protease domain (GST-SP), which was immobilized in glutathione-coupled resin. The bound SPN48 was immunoblotted (top panel), and the GST-fused proteins were immunoblotted (IB) (bottom panel).
DISCUSSION
Elaborate regulation of the activation and inhibition of serine protease-mediated innate immune response is needed to prevent damage to the host (40). Serpins are responsible for the inhibition of serine proteases both in blood coagulation and innate immune responses. In this study, we determined the crystal structure of the insect serpin SPN48, which exhibits an unusual specificity for the target protease SPE. It should be noted that there is no lattice contact on the RCL region of SPN48. In most crystal structures of serpins, the RCL conformation was constrained by the lattice interaction. Thus, the crystal structure of SPN48 might better reflect the conformation of serpin in solution.
Furthermore, subsequent biochemical analyses suggested a putative heparin-binding site and demonstrated its role in enhancing the inhibitory activity of SPN48 in collaboration with calcium ion mainly through the bridging mechanism. Some serpins have noncanonical P1 residues, for example, kallistatin, a serpin against tissue kallikrein (41, 42), and protein Z-dependent protease inhibitor, a serpin against membrane bound factor Xa (43). However, the role of the noncanonical P1 residue remains unclear. Biochemical studies of SPN48 offered further insight into a novel strategy of serpins for selecting their target serine protease. The noncanonical P1 Glu-353 residue of SPN48 showed a reduced reactivity on SPE, compared with the mutant SPE with a canonical P1 residue. However, the wild-type SPN48 with the noncanonical P1 residue showed the inhibitory activity only on the target serine protease SPE, whereas the mutant SPE (E353K) showed a broad specificity. Our findings suggest that the noncanonical P1 residue prevents unwanted cleavage by a nontargeting serine protease that has substrate specificity similar to the targeting serine protease. In the insect hemolymph, many kinds of serine proteases are present, some of which exhibit a similar specificity to SPE. Because SPN48 is constitutively present in hemolymph, SPN48 would be nonspecifically cleaved and broken down by the SPE-like proteases, which might be harmful to the host. Thus, the noncanonical P1 residue of SPN48 contributed to the unusual specificity of SPN48.
It is interesting that SPE was not able to cleave the RCL of SPN48 without the main body of SPN48, although the full-length SPN48 can inhibit SPE. This observation suggests that a specific interaction between the main body of SPN48 and the SPE, i.e. an exosite interaction, might be required to enhance the enzymatic activity of SPE on the suboptimal substrate of the RCL. Supporting this idea, we observed that both the clip domain and the serine protease domain exhibited an affinity for SPN48. This observation indicates that the clip and/or serine protease domain of SPE might provide an exosite for the recognition of the cognate serpin. Rapid exosite-mediated inhibition is observed in the case of antithrombin with thrombin, factor Xa, or factor IXa (35, 44). It is noteworthy that the clip domain also plays a crucial role in the specific activation of the serine protease. We previously reported that the clip domain of PPAF-I, an SPE homologue (17), played an important role in the activation of pro-phenol oxidase activating factor-I (PPAF-I) by the upstream protease in a large beetle, Holotrichia diomphalia (16). Taken together, our findings suggest that specific recognition by SPN48 is achieved by contriving a noncanonical P1 residue and exosite-mediated cognate interaction with the target protease.
In conclusion, we have presented the three-dimensional structure of SPN48, which specifically inhibits SPE via a noncanonical cleavage of RCL. The heparin binding ability and clip domain of SPE were biochemically analyzed with bound calcium and the highly exposed activation loop (17). Based on the structure, heparin binds SPN48 with a different region than human antithrombin, and heparin binding dramatically enhances the inhibitory activity toward SPE. We have also proposed a plausible explanation of the molecular regulation of the innate immune response by the insect serpin. Finally, our finding provides new insight into how serpins are tightly regulated at a molecular level in various biological processes.
Supplementary Material
Acknowledgment
This study made use of beamline 4A at Pohang Accelerator Laboratory (Korea).
This work was supported by Bio-Scientific Research Grant PNU-2008-0596-000 from Pusan National University (to N. C. H.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
The atomic coordinates and structure factors (code 3OZQ) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- RCL
- reactive center loop
- SPE
- Spätzle-processing enzyme.
REFERENCES
- 1. Rau J. C., Beaulieu L. M., Huntington J. A., Church F. C. (2007) J. Thromb. Haemost. 5, Suppl. 1, 102–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gooptu B., Lomas D. A. (2009) Annu. Rev. Biochem. 78, 147–176 [DOI] [PubMed] [Google Scholar]
- 3. Johnson D. J., Langdown J., Li W., Luis S. A., Baglin T. P., Huntington J. A. (2006) J. Biol. Chem. 281, 35478–35486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huntington J. A., Read R. J., Carrell R. W. (2000) Nature 407, 923–926 [DOI] [PubMed] [Google Scholar]
- 5. Carrell R. W., Owen M. C. (1985) Nature 317, 730–732 [DOI] [PubMed] [Google Scholar]
- 6. Macfarlane R. G. (1964) Nature 202, 498–499 [DOI] [PubMed] [Google Scholar]
- 7. Walenga J. M., Petitou M., Samama M., Fareed J., Choay J. (1988) Thromb. Res. 52, 553–563 [DOI] [PubMed] [Google Scholar]
- 8. Whisstock J. C., Pike R. N., Jin L., Skinner R., Pei X. Y., Carrell R. W., Lesk A. M. (2000) J. Mol. Biol. 301, 1287–1305 [DOI] [PubMed] [Google Scholar]
- 9. Khush R. S., Leulier F., Lemaitre B. (2001) Trends Immunol. 22, 260–264 [DOI] [PubMed] [Google Scholar]
- 10. Lemaitre B., Hoffmann J. (2007) Annu. Rev. Immunol. 25, 697–743 [DOI] [PubMed] [Google Scholar]
- 11. Levashina E. A., Langley E., Green C., Gubb D., Ashburner M., Hoffmann J. A., Reichhart J. M. (1999) Science 285, 1917–1919 [DOI] [PubMed] [Google Scholar]
- 12. Ligoxygakis P., Pelte N., Ji C., Leclerc V., Duvic B., Belvin M., Jiang H., Hoffmann J. A., Reichhart J. M. (2002) EMBO J. 21, 6330–6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang L., Ligoxygakis P. (2006) Immunobiology 211, 251–261 [DOI] [PubMed] [Google Scholar]
- 14. Li J., Wang Z., Canagarajah B., Jiang H., Kanost M., Goldsmith E. J. (1999) Structure 7, 103–109 [DOI] [PubMed] [Google Scholar]
- 15. Ye S., Cech A. L., Belmares R., Bergstrom R. C., Tong Y., Corey D. R., Kanost M. R., Goldsmith E. J. (2001) Nat. Struct. Biol. 8, 979–983 [DOI] [PubMed] [Google Scholar]
- 16. Piao S., Song Y. L., Kim J. H., Park S. Y., Park J. W., Lee B. L., Oh B. H., Ha N. C. (2005) EMBO J. 24, 4404–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Piao S., Kim S., Kim J. H., Park J. W., Lee B. L., Ha N. C. (2007) J. Biol. Chem. 282, 10783–10791 [DOI] [PubMed] [Google Scholar]
- 18. Piao S., Kim D., Won Park J., Leul Lee B., Ha N. C. (2005) Biochim. Biophys. Acta 1752, 103–106 [DOI] [PubMed] [Google Scholar]
- 19. Kim C. H., Kim S. J., Kan H., Kwon H. M., Roh K. B., Jiang R., Yang Y., Park J. W., Lee H. H., Ha N. C., Kang H. J., Nonaka M., Söderhäll K., Lee B. L. (2008) J. Biol. Chem. 283, 7599–7607 [DOI] [PubMed] [Google Scholar]
- 20. Jiang R., Kim E. H., Gong J. H., Kwon H. M., Kim C. H., Ryu K. H., Park J. W., Kurokawa K., Zhang J., Gubb D., Lee B. L. (2009) J. Biol. Chem. 284, 35652–35658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chuang Y. J., Swanson R., Raja S. M., Bock S. C., Olson S. T. (2001) Biochemistry 40, 6670–6679 [DOI] [PubMed] [Google Scholar]
- 22. Park S. H., Piao S., Kwon H. M., Kim E. H., Lee B. L., Ha N. C. (2010) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 198–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yum S., Kim M. J., Xu Y., Jin X. L., Yoo H. Y., Park J. W., Gong J. H., Choe K. M., Lee B. L., Ha N. C. (2009) Biochem. Biophys. Res. Commun. 378, 244–248 [DOI] [PubMed] [Google Scholar]
- 24. Collaborative Computational Project No. 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 25. Pearce M. C., Morton C. J., Feil S. C., Hansen G., Adams J. J., Parker M. W., Bottomley S. P. (2008) Protein Sci. 17, 2127–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. DeLano W. (2002) The PyMOL User's Manual, DeLano Scientific LLC, Carlos, CA [Google Scholar]
- 30. Robertson A. S., Belorgey D., Gubb D., Dafforn T. R., Lomas D. A. (2006) J. Biol. Chem. 281, 26437–26443 [DOI] [PubMed] [Google Scholar]
- 31. Schreuder H. A., de Boer B., Dijkema R., Mulders J., Theunissen H. J., Grootenhuis P. D., Hol W. G. (1994) Nat. Struct. Biol. 1, 48–54 [DOI] [PubMed] [Google Scholar]
- 32. Carrell R. W., Stein P. E., Fermi G., Wardell M. R. (1994) Structure 2, 257–270 [DOI] [PubMed] [Google Scholar]
- 33. van Boeckel C. A., Grootenhuis P. D., Visser A. (1994) Nat. Struct. Biol. 1, 423–425 [DOI] [PubMed] [Google Scholar]
- 34. Johnson D. J., Langdown J., Huntington J. A. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 645–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li W., Johnson D. J., Esmon C. T., Huntington J. A. (2004) Nat. Struct. Mol. Biol. 11, 857–862 [DOI] [PubMed] [Google Scholar]
- 36. Pratt C. W., Church F. C. (1993) Blood Coagul. Fibrinolysis 4, 479–490 [DOI] [PubMed] [Google Scholar]
- 37. Walenga J. M., Jeske W. P., Samama M. M., Frapaise F. X., Bick R. L., Fareed J. (2002) Expert Opin. Investig. Drugs 11, 397–407 [DOI] [PubMed] [Google Scholar]
- 38. Olson S. T., Björk I., Sheffer R., Craig P. A., Shore J. D., Choay J. (1992) J. Biol. Chem. 267, 12528–12538 [PubMed] [Google Scholar]
- 39. Bedsted T., Swanson R., Chuang Y. J., Bock P. E., Björk I., Olson S. T. (2003) Biochemistry 42, 8143–8152 [DOI] [PubMed] [Google Scholar]
- 40. Ferrandon D., Imler J. L., Hetru C., Hoffmann J. A. (2007) Nat. Rev. Immunol. 7, 862–874 [DOI] [PubMed] [Google Scholar]
- 41. Chai K. X., Chen V. C., Ni A., Lindpaintner K., Rubattu S., Chao L., Chao J. (1997) Biochim. Biophys. Acta 1353, 277–286 [DOI] [PubMed] [Google Scholar]
- 42. Chen V. C., Chao L., Chao J. (2000) J. Biol. Chem. 275, 38457–38466 [DOI] [PubMed] [Google Scholar]
- 43. Gettins P. G. (2002) Chem. Rev. 102, 4751–4804 [DOI] [PubMed] [Google Scholar]
- 44. Izaguirre G., Olson S. T. (2006) J. Biol. Chem. 281, 13424–13432 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.