

Abstract
Excessive proliferation and stabilization of the microtubule (MT) array in cardiac myocytes can accompany pathological cardiac hypertrophy, but the molecular control of these changes remains poorly characterized. In this study, we examined MT stabilization in two independent murine models of heart failure and revealed increases in the levels of post-translationally modified stable MTs, which were closely associated with STAT3 activation. To explore the molecular signaling events contributing to control of the cardiac MT network, we stimulated cardiac myocytes with an α-adrenergic agonist phenylephrine (PE), and observed increased tubulin content without changes in detyrosinated (glu-tubulin) stable MTs. In contrast, the hypertrophic interleukin-6 (IL6) family cytokines increased both the glu-tubulin content and glu-MT density. When we examined a role for ERK in regulating cardiac MTs, we showed that the MEK/ERK-inhibitor U0126 increased glu-MT density in either control cardiac myocytes or following exposure to hypertrophic agents. Conversely, expression of an activated MEK1 mutant reduced glu-tubulin levels. Thus, ERK signaling antagonizes stabilization of the cardiac MT array. In contrast, inhibiting either JAK2 with AG490, or STAT3 signaling with Stattic or siRNA knockdown, blocked cytokine-stimulated increases in glu-MT density. Furthermore, the expression of a constitutively active STAT3 mutant triggered increased glu-MT density in the absence of hypertrophic stimulation. Thus, STAT3 activation contributes substantially to cytokine-stimulated glu-MT changes. Taken together, our results highlight the opposing actions of STAT3 and ERK pathways in the regulation of MT changes associated with cardiac myocyte hypertrophy.
Keywords: Cardiac Hypertrophy, Cytokine, Cytoskeleton, ERK, MAP Kinases (MAPKs), Microtubules, Signal Transduction, STAT Transcription Factor
Introduction
In cardiac myocytes, the microtubule (MT)5 array comprises a major cytoskeletal element. MT organization changes between phases of assembly/disassembly with either fast (dynamic) or slow (stable) kinetics. MT stability can be regulated through direct association and regulation by numerous proteins such as MT-associated proteins (MAPs), plus-end tracking proteins (+TIPs) and tubulin-sequestering negative regulators of MT assembly (e.g. stathmins) (1–3). In turn, these direct modulators of MT stability are regulated by multisite phosphorylation by various kinases thus linking changes in MT dynamics with extracellular stimuli (2, 3). Furthermore, once assembled, tubulin polymers are modified by the addition of acetyl groups on Lys-40 (acetyl-tubulin) and the reversible removal of tyrosine from the α-tubulin C terminus (named glu-tubulin for the penultimate glutamate revealed) (4, 5). Post-translational modification of tubulin occurs as a direct function of the time a tubulin heterodimer spends in the polymerized state and therefore reports on MT age. Although not directly contributing to the stabilization of MTs per se, these modifications are markers of stabilized MTs that turnover slowly. In neurons, protein kinases such as the c-Jun N-terminal kinases and microtubule affinity-regulating kinases have been shown to be key mediators of the MT network (6, 7). In contrast, the signaling events that regulate MTs in cardiac myocytes are not as well defined.
In addition to well-established roles under physiological conditions, MT reorganization accompanies the development of cardiac disease (8). Increased MT numbers correlated with pathological cardiac hypertrophy induced by pressure overload (9, 10) and elevated MT density has been reported in various models of cardiac disease (11). Significant increases in MT density and number, as well as increases in MT stabilization have been reported during heart failure (12). A causal link between MT densification/stabilization and cardiac dysfunction was strengthened by observations that MT-targeting drugs that enhance MT disassembly could normalize defects in myocardial contractility in pressure overload hypertrophy (13). Importantly, MT changes alone were shown to be sufficient to precipitate a reduction in cardiac function (14). Thus, aberrant MT growth and stabilization can directly contribute to cardiac disease and promote dysfunction in the hypertrophic and failing heart.
Despite the important contribution of MTs to cardiac disease, the specific upstream signaling events that impact on MT regulatory proteins are not fully defined. Here we report the actions of extracellular signal-regulated kinase (ERK) and a member of the Signal Transducer and Activator of Transcription (STAT) family of transcription factors, STAT3, in regulating the cardiac myocyte MT array. The ERK family is activated by a highly conserved signaling cascade and phosphorylates several nuclear and cytoplasmic substrates. ERKs have been implicated in the regulation of hypertrophy (15) and, in transgenic mice, constitutive cardiac ERK activation is sufficient to trigger compensated hypertrophy (16). The activation of STAT3 has been observed in experimental models of cardiac hypertrophy (17–19) and can occur in parallel with ERK activation by cytokines such as those for the interleukin-6 (IL6) family (17, 20). A hallmark of active STAT3 is its tyrosine phosphorylation by Janus kinases (JAKs) followed by its nuclear translocation and up-regulation of specific gene targets. Importantly, MT regulatory functions have previously been ascribed to ERK and STAT3 signaling in non-cardiac settings (21–23). In our investigation of regulation of cardiac MTs, we reveal the opposing actions of ERK and STAT3 signaling in MT stabilization.
EXPERIMENTAL PROCEDURES
Reagents
The phospho-STAT3 and the phospho-ERK antibodies were purchased from BD Bioscience and Cell Signaling Technology, respectively. The GAPDH, ERK1/2, and STAT3 antibodies were purchased from Santa Cruz Biotechnology. Oncostatin-M (OSM), AG490, Stattic and U0126 were from Calbiochem. Cy2-/Cy3-conjugated secondary antibodies were obtained from Millipore and phalloidin-TRITC from Molecular Probes. Cell culture serum and reagents were from Invitrogen-Invitrogen. Leukemic inhibitory factor (LIF), phenylephrine (PE), nocodazole, anti-α-tubulin, anti-β-tubulin, and anti-acetylated tubulin were obtained from Sigma-Aldrich. The glu-tubulin antibody was purchased from Chemicon.
Murine Models of Heart Failure
The G203S human cardiac troponin I (TnI-203) transgenic, R403Q α-myosin heavy chain (MHC-403) knock-out/knock-in and double-mutant TnI-203/MHC-403 mouse models have been described previously (24). Dissection of heart tissue and preparation of protein extracts was performed as described previously (24). In addition, we examined changes in the hearts of 12-week-old male FVB/N mice undergoing pressure overload-induced hypertrophy following ascending aortic constriction for 2 weeks (25). Aortic-banded mice were subdivided into 2 groups: (i) compensated hypertrophy (ii) decompensated hypertrophy and heart failure. The delineation of these groups was based on differences in cardiac function as previously described in detail (25). In brief, the mice with decompensated hypertrophy and heart failure had been exposed to a greater load (aortic pressure gradient) that was associated with a significant reduction in systolic function compared with mice with compensated hypertrophy, as published previously (Ref. 25; functional and morphological data presented in supplemental Table S1). All experiments involving animals were approved by Institutional Animal Ethics Committees under direction from the Australian Bureau of Animal Welfare and conform with the United States NIH Guide for the Care and Use of Laboratory Animals.
Neonatal Rat Cardiac Myocyte Culture
Primary normal cardiac myocytes were isolated from 1–2-day-old Sprague-Dawley rats as previously described (26). Briefly, ventricular cells were isolated by collagenase digestion and then preplated to deplete non-myocyte cells. Cardiac myocytes were then plated on gelatin-coated 60-mm dishes in Dulbecco's modified Eagle's medium/Medium 199 (4:1 v/v) containing 10% (v/v) horse serum, 5% (v/v) fetal calf serum, and penicillin/streptomycin (100 units/ml). The serum was removed after 18 h, and cells starved for 24 h prior to experimental treatment. Cardiac myocytes were then plated on collagen-coated dishes or laminin-coated glass coverslips in serum-containing media. The serum was removed after 18 h, and cells starved for 24 h prior to experimental treatment and immunoblot/immunofluorescence analysis.
Protein Lysate Preparation and Immunoblotting
Protein lysates from cultured cardiac myocytes were lysed in RIPA buffer (50 mm Tris-HCl, pH 7.3, 150 mm NaCl, 0.1 mm EDTA, 1% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100, 0.2% (w/v) NaF, and 100 μm Na3VO4) supplemented with protease inhibitors. After lysis, cell debris was removed by centrifugation. Whole heart protein extracts were prepared by lysing heart tissue in solubilizing buffer (5 m urea, 2 m thiourea, 2% CHAPS, 2% sulfobetaine 3–10, 40 mm Tris, 65 mm dithiothreitol, and 2 mm tributyl phosphine) and insoluble material cleared by centrifugation. Protein concentrations were determined by Bio-Rad Bradford assay and lysates diluted with Laemmli sample buffer before SDS-PAGE and immunoblot analysis as previously described (21).
Immunofluorescence Confocal Microscopy and Image Analysis
MTs in cardiac myocytes were analyzed by immunofluorescence staining with antibodies to α-tubulin and post-translationally modified tubulin. Neonatal rat cardiac myocytes were plated on laminin-coated glass coverslips overnight in serum-containing medium. Following experimental treatments, cells were fixed in 4% (w/v) paraformaldehyde in PBS, permeabilized with 0.2% (v/v) Triton X-100 in PBS, and blocked with 10% (v/v) fetal calf serum in PBS. Cells were then stained with primary antibodies diluted (1:250) in 1% (w/v) bovine serum albumin (BSA) in PBS and detected with Cy2-/Cy3- conjugated secondary antibodies (1:500 in 0.1% (w/v) BSA in PBS). Following mounting, stained cells were examined by confocal microscopy (Leica TCS SP2 confocal microscope and laser unit).
Images were compiled and analyzed with Leica Confocal Software v2.61 (Leica, Germany) and Metamorph® analysis software (Molecular Devices). Microtubule densities (pixels/μm2) were determined by defining cell borders using phalloidin-stained actin filaments as a template. Phalloidin-TRITC staining of contractile filaments was also used to exclude non-myocytes, which typically exhibit faint, non-striated linear stress fibers with actin staining (27). The defined regions were then transferred to tubulin co-stained images and pixel intensity and cell area (μm2) measured (supplemental Fig. S1). A minimum of 100 cells was measured from 10 randomly selected fields. Microtubule densities (pixels/μm2) representative of three independent experiments (n = 3) were expressed as an average ± S.E.
STAT3 RNAi
STAT3 Stealth RNAi Duplexes were obtained from Invitrogen. The double stranded oligoribonucleotide sequences targeting STAT3 were target 1: 5′-AAGAGGUUAUGAAACACCAGAGUGG-3′ and 5′-CCACUCUGGUGUUUCAUAACCUCUU-3′, target 2: 5′-UUAGGCAUCUGACAGAUGUUGGAGA-3′ and 5′-UCUCCAACAUCUGUCAGAUGCCUAA-3′, and target 3: 5′-AUAUUGUCUAGCCAGACCCAGAACG-3′, and 5′-CGUUCUGGGUCUGGCUAGACAAUAU-3′. Stealth RNAi negative control Low GC Duplex (Invitrogen) was used as a control. RNAi were transfected into cardiac myocytes using LipofectamineTM 2000 according to the manufacturer's protocol.
Adenovirus Construction and Infection of Cultured Cardiac Myocytes
The AdMEK-EE virus was generously gifted by J. Molkentin (University of Cincinnatti). A constitutively active STAT3 (STAT3-C) mutant, in which the SH2 domain alanine 661 and asparagine 663 were substituted with cysteine residues (28), was generated by site-directed mutagenesis (QuikChange® Mutagenesis kit, Stratagene). The adenoviral construct expressing constitutively active STAT3-C (pAd-STAT3C) was generated using the AdEasy system following the manufacturer's instructions (Stratagene) and used to infect primary cardiac myocytes. STAT3-C was subcloned into the pAdtrack-CMV shuttle vector that expresses GFP from a cis-promoter. This was recombined with pAdEasy-1 in electro-competent BJ5183 cells.
AdMEK-EE, AdSTAT3C, and control adenoviruses were generated by transfection of recombined adenoviral constructs into Ad293 cells. Viral particles were expanded, isolated and purified on cesium chloride gradients followed by dialysis into 3% (w/v) sucrose in PBS. Titers of purified virus were then determined by colony formation assays before transduction of cardiac myocytes using a multiplicity of infection of 50:1. Positive viral transduction of cardiac myocytes was estimated to be 99% by the cis-promoter driven expression of GFP from the adenoviral construct.
Statistical Analysis
The statistical significance of observed differences in immunoblotting (α-tubulin, glu-tubulin, or ace-tubulin) or immunostaining (glu-tubulin) results was determined by unpaired t test. A p value <0.05 was considered statistically significant.
RESULTS
MT Stabilization Is Observed in Murine Models of Pathological Hypertrophy and Cardiomyopathy
We began by evaluating markers of MT stability in a mouse model of pressure overload hypertrophy. We had previously shown that depending on the severity of the pressure overload, mice exhibited either compensated cardiac hypertrophy (maintained systolic function) or had progressed to decompensated hypertrophy and failure with depressed contractile function (30% reduction in systolic function and dilation of ventricular chambers (supplemental Table S1) subsequent to aortic banding (25). The levels of α-tubulin were increased in pressure overloaded hearts when compared with control group (sham) (Fig. 1A) indicating an expansion of the MT network during pressure overload. We then demonstrated that the levels of acetyl- and glu-tubulin, that mark stable MTs, were also substantially elevated in aortic banded mice during both compensated (PO Comp) and decompensated stages (PO Decomp) (Fig. 1A). Densitometric analysis of acetyl- and glu-tubulin bands, normalized against changes in α-tubulin expression, indicated a significant 3-fold and 2-fold increase, respectively, in pressure overloaded animals compared with controls (Fig. 1, B and C). The levels of post-translationally modified tubulin were not statistically different between the compensated or decompensated hypertrophy groups. These results confirm stabilization of cardiac MTs during pressure overload.
FIGURE 1.

Elevated markers of cardiac MT stabilization, phosphorylated STAT3 and ERK during pressure overload hypertrophy. A, protein extracts from hearts of 12-week-old mice subjected to aortic banding for 2 weeks, resulting in compensated (PO comp; PO-pressure overload) or decompensated hypertrophy (PO decomp), were immunoblotted for phosphorylated STAT3 (pY-STAT3), phosphorylated ERK (pT/Y-ERK), STAT3, ERK, α-tubulin, acetyl-, and glu-tubulin. Sham-operated mice were used as the control group. B+C, acetyl-tubulin (B) and glu-tubulin (C) bands were quantitated and normalized against α-tubulin levels and values expressed as mean ± S.E. (**, p < 0.01, n = 3). D, pY-STAT3 bands were quantitated by densitometric analysis and normalize against total STAT3. Values represent mean ± S.E. (*, p < 0.05, n = 3).
To investigate signaling mechanisms that could contribute to MT stabilization, we immunoblotted for phosphorylated STAT3 and ERK as an indicator of JAK/STAT and ERK MAPK pathway activation. In response to pressure overload, cardiac STAT3 phosphorylation was substantially increased in mice with compensated or decompensated hypertrophy (Fig. 1A) and correlated well with the elevated levels of post-translationally modified tubulin. STAT3 protein levels were also elevated in compensated or decompensated hypertrophy groups (Fig. 1A). This latter observation is consistent with a sustained time course of STAT3 activation acting in a positive feedback loop to increase its own transcription (29). When the levels of phosphorylated STAT3 were normalized against total STAT3, we found a significant increase in mice with pressure overload hypertrophy (Fig. 1D). A single sham operated animal (lane 3) exhibited a modest increase in phosphorylated STAT3, which correlated with a small increase in glu-tubulin levels. The reason for this is unclear as the animals in the sham group were morphologically and functionally indistinguishable and there was no overlap between sham and PO groups (data not shown). ERK was also found to be activated in pressure-overloaded mice (Fig. 1A). However, unlike phosphorylated STAT3, the levels of phosphorylated ERK between individual mice with pressure overload varied widely and did not closely match the increases in acetyl and glu-tubulin (Fig. 1A).
To evaluate the contribution of STAT3 without the complication of ERK activation, we next looked at MT stabilization in a double mutation (TnI-203/MHC-403) murine model of cardiomyopathy that recapitulates a rapid progression to heart failure observed in human patients (24). A striking feature of this model was an initial robust activation of cardiac STAT3, first observed in 10-day-old mice, prior to the onset of heart failure (24). Importantly, ERK activation was not found in this mouse model of heart failure (24). We first evaluated the levels of α-tubulin, glu-, and acetyl-tubulin in the TnI-203/MHC-403 mice alongside three control groups: nontransgenic littermates (NTG) and single-mutant mice (TnI-203 or MHC-403 alone) that did not rapidly develop cardiac failure. When hearts from 5-day-old mice were examined, immunoblotting for total α-tubulin, glu-tubulin, and acetyl-tubulin revealed no substantial differences between TnI-203/MHC-403 mice and the control groups (Fig. 2A). In contrast, glu-tubulin and acetyl-tubulin were markedly elevated in the failing hearts from 18 day old TnI-203/MHC-403 mice when compared with age-matched non-failing hearts from the single mutant and NTG control animals (Fig. 2B). Interestingly, in the failing hearts, total levels of α-tubulin levels were not markedly different from the levels in all control groups (Fig. 2, A and B).
FIGURE 2.

Increases in cardiac glu-tubulin levels in TnI-203/MHC-403 mice coincide with elevated STAT3 phosphorylation. Protein extracts from the hearts of 5-day-old (A) or 18-day-old (B) non-transgenic (NTG, n = 3), TnI-203 (n = 3), MHC-403 (n = 3), and double mutant TnI-203/MHC-403 mice (n = 4) were immunoblotted for α-tubulin as an indicator of the total MT network and post-translationally modified glu- and acetyl-tubulin as indicators of MT stabilization. Immunoblotting for GAPDH indicated equivalent protein loading in all samples. Protein extracts from the hearts of 5, 10, 14, and 18 day old NTG and TnI-203/MHC-403 mice were immunoblotted for glu-tubulin, α-tubulin (C), Tyr-705-phosphorylated STAT3 and STAT3 (E). Glu-tubulin (D) and pY705-STAT3 (F) bands were quantified by densitometric analysis, normalized for α-tubulin and total STAT3 levels, respectively, and values expressed as mean ± S.D. (n = 3).
A more detailed temporal analysis demonstrated glu-tubulin levels were significantly increased in 10, 14, and 18-day-old TnI-203/MHC-403 mice, compared with the NTG control group of the same age (Fig. 2, C and D). This coincided with significant increases in Tyr-705 phosphorylated STAT3 in 10-, 14-, and 18-day-old TnI-203/MHC-403 mice (Fig. 2, E and F). In addition, the levels of α-tubulin and STAT3 expression were not substantially different between NTG and TnI-203/MHC-403 groups at any of the time-points studied (Fig. 2, C and E). A closer kinetic analysis indicated a substantial increase of phosphorylated STAT3 in 10 day old TnI-203/MHC-403 mice (supplemental Fig. S2). The levels of phosphorylated STAT3 increased further in 14- and 18-day-old TnI-203/MHC-403 mice (supplemental Fig. S2). Similarly, an increase in glu-tubulin levels was evident in 10-day-old TnI-203/MHC-403 mice, with considerably elevated glu-tubulin levels observed in 14- and 18-day-old animals (supplemental Fig. S2). In comparison, the levels of phosphorylated STAT3 and glu-tubulin were not increased greatly by the age of 18 days in the NTG group (supplemental Fig. S2).
These results from two independent models of heart failure show a striking enhancement of markers of the stabilized MT network. In addition, the close correlation of these changes with enhanced STAT3 signaling raises the question of the signaling events and the contribution of STAT3 to the increases in the stable MT subset.
IL6 Family Cytokines Induce MT Stabilization in Cardiac Myocytes
In subsequent experiments to address the molecular events that trigger cardiac MT changes, we used primary cardiac myocytes stimulated with pro-hypertrophic agonists. We have previously shown that hypertrophy of these cells is accompanied by an enhanced MT network (27). In this study, we investigated tubulin changes in cardiac myoyctes following 24 h exposure to the α-adrenergic agonist phenylephrine (PE) or the IL6 family cytokines, LIF or OSM. The increases in the size of hypertrophic cardiac myocytes (μm2) were not substantially different between the agonists investigated (supplemental Fig. S3). We then confirmed by immunoblotting the increase of α-tubulin in cardiac myocytes following 24 h exposure to PE, LIF, or OSM (Fig. 3A). Comparable increases in β-tubulin were also observed (results not shown). To assess the stable MT population, we next examined glu-tubulin and acetyl-tubulin and showed elevated levels following 24-h exposure to LIF or OSM (Fig. 3A). After normalizing for α-tubulin changes, LIF- and OSM-stimulated a significant increase in glu-tubulin levels compared with control cells (Fig. 3B). However, acetyl-tubulin was not significantly different to control myocytes (Fig. 3C).
FIGURE 3.

Hypertrophic agonists stimulate increases in tubulin content and stability in cardiac myocytes. Primary cultures of neonatal rat cardiac myocytes were treated for 24 h with PE (100 μm), LIF (10 ng/ml), OSM (10 ng/ml), or 0.1% (w/v) BSA in PBS as a non-stimulated control (Con). A, protein lysates from treated cardiac myocytes were immunoblotted for α-tubulin and post-translationally modified glu- and acetyl-tubulin. Blotting for GAPDH demonstrated equivalent protein loading. Glu-tubulin (B) and acetyl-tubulin (C) bands from immunoblots (n = 5) were quantitated by densitometric analysis, normalized for α-tubulin, and expressed as average fold change + S.E. Significant changes in glu- and acetyl-tubulin compared with Con are denoted by †, p < 0.05 and *, p < 0.05, respectively. D, immunostaining for glu-tubulin (upper panels) and co-staining with TRITC-conjugated phalloidin (lower panels) revealed the stabilized MT network and the organization of contractile actin filaments, respectively. E, Con or LIF-stimulated cardiac myocytes were treated with nocodazole (NZ, 2 μm, 1 h) before immunostaining for α-tubulin. Scale bars, 50 μm.
In contrast to cytokine stimulated effects, the levels of post-translationally modified tubulins were not enhanced by PE treatment despite increases in total α-tubulin levels (Fig. 3A). Rather, glu-tubulin and acetyl-tubulin levels adjusted for α-tubulin changes were significantly decreased compared with control cells (Fig. 3, B and C). Furthermore, glu-tubulin levels were not markedly elevated during short-term treatment (0–8 h) with PE, LIF or OSM (supplemental Fig. S4, A–C). These results suggest that elevated stable MT markers observed in IL-6 cytokine-treated myocytes may be the result of longer term molecular events that actively enhance MT stabilization in addition to a general increase in α-tubulin expression. Our observations also indicated that the levels of glu- and acetyl-tubulin are not correlated to α-tubulin levels in all cases.
Immunofluorescence staining of myocytes confirmed an increase in the glu-MT subset following 24 h treatment with LIF or OSM but not PE (Fig. 3D, upper panels). Co-staining for actin filaments confirmed the reorganization of the sarcomeres, a hallmark of cardiac myocyte hypertrophy (Fig. 3D, lower panels). Thus, increases in stable MTs were only in response to the IL6-family cytokines, further emphasizing stimulus-specific differences in cardiac MT organization.
We confirmed the stability of the cardiac MT network directly by exposing control or LIF-treated cardiac myocytes to a low dose of a MT-depolymerizing drug, nocodazole (NZ, 2 μm, 1 h). We showed a substantial number of MTs remained after NZ treatment of LIF-stimulated myocytes when compared with the low MT numbers observable in NZ-treated control myocytes (Fig. 3E). In addition, nocodazole-resistant MTs in LIF-treated myocytes were substantially longer when compared with short fragments in the controls (Fig. 3E). In contrast, a similar analysis in PE-stimulated myocytes treated with NZ revealed short MT fragments which were more abundant but comparable in length with MTs in NZ-treated control cells (supplemental Fig. S5). This indicated that MT proliferation is increased in response to PE but an increase in MT stability was not evident. Our studies therefore show increased MT stabilization during IL6 family cytokine stimulation of myocytes and raise the question of the signal transduction pathways that regulate these MT changes.
ERK Negatively Regulates MT Stabilization
LIF treatment of normal cardiac myocytes rapidly activates both STAT3 and ERK pathways (30). In contrast, PE exposure leads to rapid activation of the ERK pathway (15) but is not reported to activate STAT3 directly. We confirmed in normal cardiac myocytes that PE rapidly (15 min) increased ERK phosphorylation which was sustained over 8 h (supplemental Fig. S6A). In contrast, exposure to PE only modestly increased STAT3 phosphorylation (supplemental Fig. S6A). LIF or OSM treatment rapidly (15 min) and markedly increased STAT3 phosphorylation (supplemental Fig. S6, B and C). The phosphorylation of ERK was only modestly increased in response to LIF at 15 min (supplemental Fig. S6B) but was not increased by OSM treatment at the time points tested (supplemental Fig. S6C). Furthermore, we observed increased STAT3 phosphorylation following 24-h stimulation with LIF or OSM, but not PE (supplemental Fig. S6D). Phospho-ERK levels were not significantly increased by 24-h treatment with any agonist (supplemental Fig. S6D). LIF or OSM therefore stimulated potent STAT3 activation sustained during the course of cardiac myocyte hypertrophy, prompting us to explore contributions by STAT3 or ERK to MT stabilization.
We used the MEK-specific inhibitor, U0126, to evaluate a contribution by ERK signaling in promoting MT stabilization in cardiac myocytes. We confirmed that U0126 pretreatment (10 μm, 30 min) prevented rapid ERK phosphorylation (15 min) following PE or IL6-family cytokine stimulation whereas STAT3 Tyr-705 phosphorylation was unaffected (supplemental Fig. S7A). Following prolonged U0126 exposure (24 h), ERK activity remained inhibited below basal levels in all treatment groups (Fig. 4A). Although levels of glu-tubulin were unchanged following PE treatment in cardiac myocytes, glu-tubulin levels were unexpectedly enhanced in response to PE treatment in the presence of U0126 (Fig. 4A). Furthermore, U0126 treatment increased glu-tubulin levels in control cells or cells exposed to LIF or OSM (Fig. 4A). Similar observations were made when acetyl-tubulin levels were investigated (Fig. 4A).
FIGURE 4.
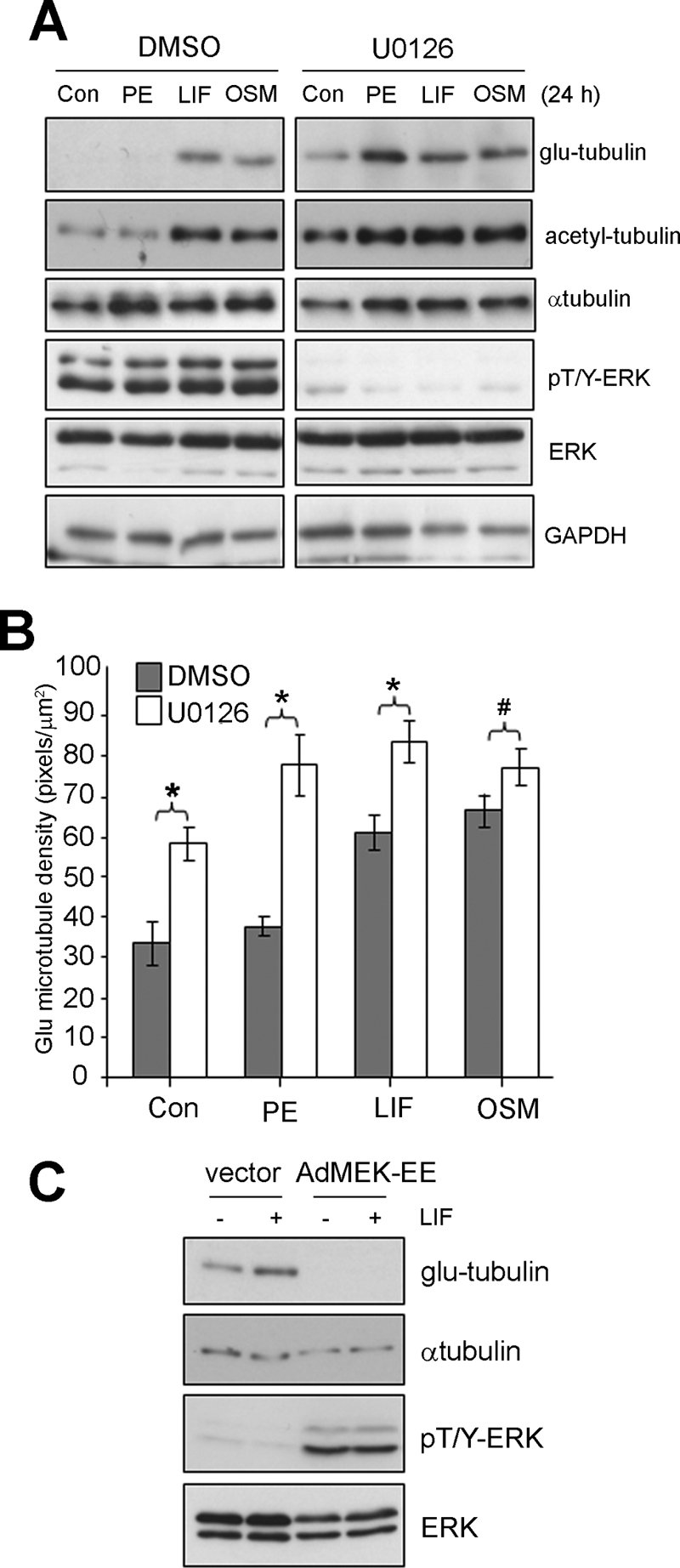
MEK/ERK signaling negatively regulates stable MTs in cardiac myocytes. Cultured cardiac myocytes were pretreated with U0126 (10 μm, 30 min) or a vehicle control (0.1% (v/v) DMSO), then stimulated (24 h) with agonists, PE (100 μm), LIF (10 ng/ml), OSM (10 ng/ml), or 0.1% (w/v) BSA in PBS as a control. A, Protein lysates were then prepared blotted with the indicated antibodies. B, Glu-MTs were immunostained to revealed the stabilized MT network and the density of glu-MTs quantitated by immunofluorescence image analysis and results expressed as an average ± S.E. (*, p < 0.01, #, p < 0.05, n = 3). C, cardiac myocytes were infected (50 moi) with adenovirus encoding MEK-EE (AdMEK-EE) or a shuttle vector only control (vector) before stimulating with LIF (10 ng/ml, 24 h) or equivalent volume of 0.1% BSA (w/v) in PBS. Protein lysates were then prepared and blotted with the indicated antibodies.
We confirmed these effects with glu-tubulin immunofluorescence staining in cardiac myocytes and estimation of glu-MT density. After U0126 for 24 h, we observed significant increases in glu-MT density in the absence of hypertrophic agonist stimulation (Fig. 4B). U0126 pretreatment further elevated glu-MT levels during co-treatment with LIF and OSM (Fig. 4B). In addition, U0126 pretreatment elevated glu-MT density in PE-treated myocytes even though PE treatment alone did not significantly increase glu-MT density (Fig. 4B). The effects of U0126 exposure on glu-MTs following PE treatment are shown in representative images (supplemental Fig. S8).
We next sought to determine the effect of specific activation of the ERK pathway, through adenoviral-mediated expression of an activated MEK1 mutant (AdMEK-EE) (16), on glu-tubulin levels. Whereas cardiac myocytes infected with a control adenovirus (vector) responded to LIF stimulation with increased levels of glu-tubulin (Fig. 4C), glu-tubulin was not detected in unstimulated cardiac myocytes expressing MEK-EE, nor following cytokine stimulation of MEK-EE expressing cells (Fig. 4C). We confirmed by immunoblot analysis that the levels of phosphorylated ERK were elevated in cardiac myocytes expressing MEK-EE but not in control infected cells (Fig. 4C). Immunoblotting for α-tubulin and ERK1/2 revealed expression levels of total proteins. Taken together, our results indicate that the ERK pathway negatively regulates the stable MT subset in cardiac myocytes.
Inhibition of STAT3 Signaling Prevents Cytokine-stimulated MT Stabilization
We next evaluated the contribution by STAT3 signaling to MT reorganization through pharmacological inhibition of JAK, a direct upstream STAT3 activator. We showed that AG490 pretreatment inhibited rapid (15 min) STAT3 activation following exposure to LIF or OSM (supplemental Fig. S7B). AG490 pretreatment also inhibited the sustained (24 h) STAT3 Tyr-705 phosphorylation stimulated by LIF or OSM (Fig. 5A). Because JAK signaling activates Shp2/Grb2/Ras pathway to activate ERK as well as STAT pathways (31), we found that AG490 treatment could also impact ERK activation stimulated by LIF in cardiac myocytes but did not impact on G-protein-coupled receptor mediated activation of ERK stimulated by PE (supplemental Fig. S7B).
FIGURE 5.

STAT3 inhibition attenuates IL-6 cytokine stimulated stable MT increases in cardiac myocytes. Cultured cardiac myocytes were pretreated AG490 (50 μm, 30 min) (A+B), Stattic (2 μm, 30 min) (C+D), or a vehicle control (0.1% (v/v) DMSO), then stimulated (24 h) with agonists, PE (100 μm), LIF (10 ng/ml), OSM (10 ng/ml), or 0.1% (w/v) BSA in PBS as a control. A+C, protein lysates were then prepared blotted with the indicated antibodies. B+D, Glu-MTs were immunostained to revealed the stabilized MT network and the density of glu-MTs quantitated by immunofluorescence image analysis and results expressed as an average ± S.E. (*, p < 0.01, n = 3).
When we looked at post-translationally modified tubulin, we found that AG490 treatment was sufficient to inhibit LIF- or OSM-stimulated increases in glu-tubulin and acetyl-tubulin (Fig. 5A), suggesting a role for the JAK/STAT pathway as a critical positive regulator in promoting stable MT increases. Furthermore, AG490 did not impact on α-tubulin levels in hypertrophic agonist stimulated myocytes (Fig. 5A) indicating that the reduction in post-translationally modified tubulin was not due to reduced tubulin content. Immunofluorescence staining of the glu-MTs demonstrated that, in the presence of AG490, glu-MT density was not significantly increased by LIF or OSM when compared with the control cells (Fig. 5B). AG490 pretreatment also did not significantly change glu-MT levels in unstimulated or PE-treated cells. LIF- or OSM-stimulated changes in the glu-MT network in the presence or absence of AG490 are shown in representative images (supplemental Fig. S9). These results reveal a role for JAK signaling in promoting IL-6 cytokine-induced increases in post-translationally modified stable MTs.
As AG490 is a JAK-specific inhibitor and attenuates STAT3 activation indirectly, we next utilized a new generation chemical inhibitor, Stattic that selectively disrupts active STAT3 dimers (32). We confirmed that Stattic pretreatment (2 μm, 30 min) prevented rapid STAT3 activation by LIF or OSM (supplemental Fig. S7C). Stattic pretreatment of cardiac myocytes prevented the up-regulation of STAT3 phosphorylation, glu-tubulin, and acetyl-tubulin levels in response to LIF or OSM treatment (Fig. 5C). As with AG490, α-tubulin levels were unaltered by Stattic (Fig. 5C). Immunofluorescence measurements indicated that glu-MT density in hypertrophic LIF-treated myocytes pretreated with Stattic was not statistically different to the density in unstimulated myocytes (Fig. 5D). Similarly, Stattic pretreatment prevented increases in glu-MT density in OSM-treated myocytes (Fig. 5D). Representative images show that treatment with Stattic inhibited LIF- or OSM-stimulated increased stabilization of cardiac MT array (supplemental Fig. S10).
In addition to its inhibition of STAT3 phosphorylation, Stattic pretreatment increased the levels of phosphorylated ERK irrespective of hypertrophic agonist stimulation (supplemental Fig. S7C). To evaluate the specific contribution of STAT3 to hypertrophic MT changes without the complication of ERK activation, we next down-regulated STAT3 levels specifically using small interfering RNA (siRNA) targeting STAT3. We first demonstrated that transient transfection of three distinct STAT3-targeting siRNAs (STAT3 siRNA t1, t2, t3) specifically decreased STAT3 levels when compared with a control siRNA or mock-transfected myocytes without siRNA (Fig. 6A). Importantly, the levels of total and phosphorylated ERK, as well as levels of GAPDH were not altered by these STAT3 siRNA treatments (Fig. 6A). In addition, we showed that LIF and OSM-stimulated STAT3 phosphorylation was attenuated by STAT3 siRNA while phosphorylated ERK levels were unaltered (supplemental Fig. S7D). We next investigated post-translationally modified tubulin and found that STAT3 siRNA prevented LIF or OSM-stimulated increases in glu- or acetyl-tubulin (Fig. 6B). In contrast, a control siRNA did not prevent cytokine-stimulated increases in stable-MT markers (Fig. 6B). Furthermore, α-tubulin levels and GAPDH were not substantially altered by STAT3 siRNA expression (Fig. 6B). Our results indicate that the specific down-regulation of STAT3 was sufficient to inhibit IL-6 type cytokine-stimulated MT changes in hypertrophic cardiac myocytes.
FIGURE 6.

STAT3 knockdown attenuates increases in post-translationally modified tubulin stimulated by IL-6-type cytokines. A, cultured cardiac myocytes were transiently transfected with STAT3-targeting siRNAs (STAT3 siRNA t1, t2, or t3), a non-targeting negative control siRNA (Con siRNA) or mock-transfected without oligoribonucleotides. At 48 h post-transfection, protein lysates were prepared and blotted for STAT3, ERK expression, phosphorylated ERK, and GAPDH as a loading control. B, cardiac myocytes were transfected with STAT3-targeting siRNA or non-targeting control siRNA before stimulation with LIF (10 ng/ml, 24 h) or OSM (10 ng/ml, 24 h). Glu-, acetyl-tubulin, and α-tubulin levels were then determined by immunoblotting. GAPH was also blotted as a control for protein loading.
To delineate the effect of STAT3-mediated MT changes from STAT3 regulation of hypertrophy in general, we looked at the effect of Stattic on glu-tubulin in LIF-treated hypertrophied myocytes. Again, we showed that LIF stimulation (24 h) increased glu-tubulin levels (Fig. 7A). When STAT3 was then subsequently inhibited in LIF-stimulated hypertrophied myocytes, a rapid decrease in glu-tubulin levels was observed as early as 60 min treatment with Stattic (Fig. 7A). Blotting for phosphorylated STAT3 confirmed that Stattic treatment reduced STAT3 Y705 phosphorylation while total STAT3 levels were not perturbed (Fig. 7A). The effect of Stattic treatment on LIF-hypertrophied myocytes was similarly shown to reduce the numbers of glu-MTs through immunofluorescence studies (Fig. 7B). Stattic treatment did not alter the hypertrophic morphology of contractile sarcomeric units (Fig. 7B). These findings suggest that STAT3-mediated stabilization of MTs is distinct from the effect of cytokines on cardiac myocyte hypertrophy and not merely a consequence of cell enlargement. Together, our studies reveal a requirement for STAT3 signaling for IL6-family cytokine-induced MT stabilization in cardiac myocytes.
FIGURE 7.

STAT3 inhibition in hypertrophic cardiac myocytes reverses MT stabilization. A, hypertrophy in cultured cardiac myocytes were induced by LIF (10 ng/ml) treatment for 24 h before treatment with Stattic (2 μm) for the indicated times. Lysates were then immunoblotted for glu-tubulin and bands quantitated by densitometric analysis. Values are expressed relative to glu-tubulin levels in untreated control myocytes. Phosphorylated STAT3 and STAT3 expression was also determined by immunoblotting. B, hypertrophic cardiac myocytes induced by LIF (10 ng/ml) treatment for 24 h was subsequently treated with Stattic (2 μm) or and equivalent volume of DMSO for 2 h before immunostaining with glu-tubulin antibody. Untreated cardiac myocytes were also stained for glu-tubulin as a control (Con). Phalloidin-TRITC staining revealed cardiac myocyte contractile filaments. Scale bar, 20 μm.
Constitutive STAT3 Activation Is Sufficient to Promote MT Stabilization
To evaluate whether STAT3 activation is sufficient to trigger increased MT stabilization, we utilized adenoviral-mediated expression of a constitutively active mutant of STAT3 (Ad-STAT3C). This revealed that constitutive STAT3 activity in the absence of hypertrophic agonist treatment was sufficient to induce an apparent increase in MTs in normal cardiac myocytes as determined by immunostaining for α-tubulin (Fig. 8, compare panels i and iv). Furthermore, STAT3C expression also induced an increase in glu-MTs in cardiac myocytes without the requirement for hypertrophic agonist treatment (Fig. 8, compare panels vii and x). Positive viral transduction of myocytes was verified to be ∼99% of the cell population by the inclusion of a cis-promoter driven expression of GFP in the adenoviral construct. (Fig. 8, panels ii, v, viii, and xii). Thus, sustained activation of STAT3 was sufficient to trigger MT stabilization in the absence of cytokine stimulation.
FIGURE 8.

Constitutively active STAT3 is sufficient to promote MT network densification and stabilization. A constitutively active STAT3 mutant (STAT3C) co-expressed with GFP or the GFP-expressing shuttle vector were transfected into cardiac myocytes by adenoviral transduction (50 moi). Immunofluorescence analysis of the MT network and stabilized MT network were determined by immunostaining for α-tubulin (i, iv) and glu-tubulin (vii, x), respectively. Viral transduction of myocytes was confirmed by visualizing GFP fluorescence (ii, v, viii, xi). Cell nuclei were stained with DAPI (iii, vi, ix, xii). Scale bar, 20 μm.
DISCUSSION
Excessive increases in MT density have been shown to impede the movement of contractile filaments and reduce contractile function, leading to detrimental effects in chronic pressure overload associated end-stage heart failure (8). Our findings in two independent murine models of heart failure confirmed an increase in stabilized MTs in hearts with hypertrophy and contractile dysfunction. Our observed increase in MT stabilization in the TnI-203/MHC-403 mice is the first report of MT changes in a mouse model of familial hypertrophic cardiomyopathy. In addition, increased stable-MT markers were observed in pressure overload mice that had yet to exhibit contractile deficits (Fig. 1, PO Comp), which supports the notion that maladaptive reorganization of the cardiac MT network may precede and contribute to the onset of heart failure. This is also supported by kinetic analysis in TnI-203/MHC-403 mice where glu-tubulin increases precede the onset of severe heart failure. Thus, our findings suggest that increased stabilization of cardiac MTs is closely associated with heart failure pathology of different origins.
The increases in post-translationally modified forms of tubulin associated with cardiac hypertrophy in the mouse models prompted our evaluation of the changes in MTs in cardiac myocytes as these contractile cells contribute a significant proportion of protein in the heart. While differences in neonatal versus adult cardiac myocytes are appreciated, isolated adult cells have been restricted by a lack of flexibility under prolonged culture and problematic responses to MT-targeting chemical treatments (33). In addition, extensive studies in neonatal cardiac myocytes have continued to provide new information on molecular mechanisms involved in adult-onset diseases (16, 34–36). Thus, cardiac myocytes isolated from neonatal animals remains a useful and robust model system amenable to the experimental manipulation required for detailed mechanistic studies. Importantly, our studies suggest that the increased levels of post-translationally modified MTs associated with cardiac disease are regulated by changes in the activity/expression of molecular regulators of MT stability in addition to any increases in total tubulin content.
A striking finding of our study has been the opposing actions of ERK and STAT3 in regulating the MT array in neonatal cardiac myocytes. Our attention was initially drawn to these two signal transduction pathways as each has been implicated in the development of cardiac hypertrophy. In considering a role for the ERK MAPK pathway, constitutive ERK activation in transgenic mice has been previously shown to stimulate hypertrophy that is compensated and maintains contractile function (16). However, a contribution by ERK signaling to microtubule changes during cardiac hypertrophy had not been previously reported. Our findings that MT stabilization is elevated following ERK inhibition and attenuated by constitutive ERK activation indicates that MEK/ERK signaling may negatively modulate levels of stable MTs and supports the notion that multiple mechanisms are involved in MT reorganization associated with cardiac hypertrophy. As MT reorganization is specifically linked to cardiac hypertrophy associated with contractile dysfunction (13, 37), our findings suggest that stable MTs could be promoted in the absence or down-regulation of ERK activity and is consistent with ERK function in promoting cardiac hypertrophy without contractile deficits. One complication arises with the recent study reporting modest elevation of glu-tubulin levels in PE-treated neonatal cardiac myocytes, although this study did not investigate effects of other hypertrophic stimuli such as the IL6 family of cytokines or the specific role for the ERK MAPKs (35).
In contrast to our findings with ERK, our results also indicate that STAT3 signaling is involved in cytokine-induced increases in MT stability and is sufficient to promote an increase in glu-tubulin in normal cardiac myocytes. From conditional gene knock-out animal studies, the function of cardiac STAT3 signaling appears to be cardioprotective in situations of acute cardiac insult (38). The protective actions of STAT3 have been largely attributed to its regulation of specific target genes involved in vascularization, antioxidant status, and the suppression of apoptosis (39, 40). However, studies have not adequately addressed the function of STAT3 activation during chronic cardiac disease.
We have previously demonstrated activation of STAT3 in explanted failing human hearts (41) and recently revealed sustained STAT3 activation in a double mutation mouse model of familial hypertrophic cardiomyopathy (24). In this study, we further confirmed robust STAT3 Tyr-705 phosphorylation in two distinct murine models of heart failure. Importantly, STAT3 activation preceded the onset of contractile deficits in both models. This highlights the potential role for STAT3-mediated events in regulating the development of heart failure. In contrast, transient activation of STAT3 during pressure overload hypertrophy have been previously reported (18–20). The differences in the kinetics of STAT3 activation, compared with the current study, may reflect species specific effects, differences in surgical methods or the severity of the aortic banding to induce pressure overload. For example, in a previous report of transient STAT3 activation following aortic constriction in rabbits, no reduction in contractile function (i.e. a decrease in fractional shortening) was observed at 4 weeks post-constriction (18). Furthermore, although maximum STAT3 activation is rapid and transient in response to step loading, submaximal levels of STAT3 phosphorylation, that remain elevated over controls, are observed at the longest time points (typically 1–2 weeks) investigated in several studies (19, 20). Indeed, elevated STAT3 phosphorylation has been reported as long as 9 weeks after aortic constriction in a murine model of pressure overload hypertrophy (42). Thus, persistent activation of STAT3 has been observed in several animal models of pressure overload hypertrophy.
Our results indicate that persistent STAT3 activation is linked to MT changes associated with cardiac hypertrophy and suggest that sustained STAT3 activity plays a key role in the context of heart failure. Indeed, a previous report highlighted that persistent STAT3-mediated gene expression changes induced by angiotensin II contributed to diastolic dysfunction and cardiac myocyte loss during post-infarction remodeling of the myocardium (43). More recently, a compelling study demonstrated that dysregulated gp130-STAT3 signaling, characterized by a sustained STAT3 activation kinetic, was detrimental during cardiac remodeling following myocardial infarction in mice (44). Thus, STAT3 activation in the heart may not be universally protective. However, STAT3 regulates MT dynamics to control normal cellular processes in non-myocytes (21, 23). A similar role for STAT3 in controlling MT-driven processes in cardiac myocytes that are beneficial is also likely. Further investigation to delineate STAT3-regulated MT functions linked specifically with contractile dysfunction would be prudent.
Another question remains as to how STAT3 activation increases stabilization of MTs. We and others have demonstrated that, in non-cardiac cells, STAT3 can negatively regulate the activity of a key cytoplasmic MT-destabilizing factor, stathmin, through direct protein-protein interaction to impact on MT dynamics (21, 23). Thus, it would be interesting to investigate STAT3 binding of stathmin in hypertrophied or failing hearts in future studies. However, transcriptional regulation of factors that contribute to MT assembly by sustained STAT3 activity is also likely. Clearly, the precise mechanisms through which STAT3 modulates the hypertrophic cardiac MT array warrants further characterization. In summary, our results have revealed the contrasting functions of ERK and STAT3 signaling in regulating MT stability in cardiac myocytes. Our findings support further study to determine the contribution of these mechanisms in regulating MT changes linked to severe decompensated hypertrophy and subsequent cardiac failure.
Supplementary Material
This work was supported, in whole or in part, by NHFA Grants-in-aid (G06P2517, G09M4435; to D. C. H. N.) and NHMRC Project Grants (353592, to C. S. and 353592, to M. A. B.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S10 and Table S1.
- MT
- microtubule
- LIF
- leukemic inhibitory factor
- MAP
- microtubule-associated protein
- MHC
- myosin heavy chain
- NTG
- non-transgenic
- OSM
- oncostatin-M
- PE
- phenylephrine
- PO
- pressure overload
- TnI
- troponin I.
REFERENCES
- 1. Akhmanova A., Hoogenraad C. C. (2005) Curr. Opin Cell Biol. 17, 47–54 [DOI] [PubMed] [Google Scholar]
- 2. Cassimeris L. (2002) Curr. Opin Cell Biol. 14, 18–24 [DOI] [PubMed] [Google Scholar]
- 3. Drewes G., Ebneth A., Mandelkow E. M. (1998) Trends Biochem. Sci. 23, 307–311 [DOI] [PubMed] [Google Scholar]
- 4. Webster D. R., Borisy G. G. (1989) J. Cell Sci. 92, 57–65 [DOI] [PubMed] [Google Scholar]
- 5. Webster D. R., Gundersen G. G., Bulinski J. C., Borisy G. G. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 9040–9044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang L., Jones Y., Ellisman M. H., Goldstein L. S., Karin M. (2003) Dev. Cell 4, 521–533 [DOI] [PubMed] [Google Scholar]
- 7. Drewes G., Ebneth A., Preuss U., Mandelkow E. M., Mandelkow E. (1997) Cell 89, 297–308 [DOI] [PubMed] [Google Scholar]
- 8. Cooper G., 4th (2006) Am. J. Physiol. Heart Circ. Physiol. 291, H1003–H1014 [DOI] [PubMed] [Google Scholar]
- 9. Tsutsui H., Ishihara K., Cooper G., 4th (1993) Science 260, 682–687 [DOI] [PubMed] [Google Scholar]
- 10. Watkins S. C., Samuel J. L., Marotte F., Bertier-Savalle B., Rappaport L. (1987) Circ. Res. 60, 327–336 [DOI] [PubMed] [Google Scholar]
- 11. Webster D. R. (2002) Cardiovasc. Toxicol. 2, 75–89 [DOI] [PubMed] [Google Scholar]
- 12. Belmadani S., Poüs C., Ventura-Clapier R., Fischmeister R., Méry P. F. (2002) Mol. Cell Biochem. 237, 39–46 [DOI] [PubMed] [Google Scholar]
- 13. Koide M., Hamawaki M., Narishige T., Sato H., Nemoto S., DeFreyte G., Zile M. R., Cooper G., IV, Carabello B. A. (2000) Circulation 102, 1045–1052 [DOI] [PubMed] [Google Scholar]
- 14. Cheng G., Zile M. R., Takahashi M., Baicu C. F., Bonnema D. D., Cabral F., Menick D. R., Cooper G., 4th (2008) Am. J. Physiol. Heart Circ. Physiol. 294, H2231–2241 [DOI] [PubMed] [Google Scholar]
- 15. Bogoyevitch M. A., Sugden P. H. (1996) Int. J. Biochem. Cell Biol. 28, 1–12 [DOI] [PubMed] [Google Scholar]
- 16. Bueno O. F., De Windt L. J., Tymitz K. M., Witt S. A., Kimball T. R., Klevitsky R., Hewett T. E., Jones S. P., Lefer D. J., Peng C. F., Kitsis R. N., Molkentin J. D. (2000) EMBO J. 19, 6341–6350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kunisada K., Negoro S., Tone E., Funamoto M., Osugi T., Yamada S., Okabe M., Kishimoto T., Yamauchi-Takihara K. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 315–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miyamoto T., Takeishi Y., Takahashi H., Shishido T., Arimoto T., Tomoike H., Kubota I. (2004) Basic Res. Cardiol. 99, 328–337 [DOI] [PubMed] [Google Scholar]
- 19. Willey C. D., Palanisamy A. P., Johnston R. K., Mani S. K., Shiraishi H., Tuxworth W. J., Zile M. R., Balasubramanian S., Kuppuswamy D. (2008) Int. J. Biol. Sci. 4, 184–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yasukawa H., Hoshijima M., Gu Y., Nakamura T., Pradervand S., Hanada T., Hanakawa Y., Yoshimura A., Ross J., Jr., Chien K. R. (2001) J. Clin. Invest. 108, 1459–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ng D. C., Lin B. H., Lim C. P., Huang G., Zhang T., Poli V., Cao X. (2006) J. Cell Biol. 172, 245–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vaillant A. R., Zanassi P., Walsh G. S., Aumont A., Alonso A., Miller F. D. (2002) Neuron 34, 985–998 [DOI] [PubMed] [Google Scholar]
- 23. Verma N. K., Dourlat J., Davies A. M., Long A., Liu W. Q., Garbay C., Kelleher D., Volkov Y. (2009) J. Biol. Chem. 284, 12349–12362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsoutsman T., Kelly M., Ng D. C., Tan J. E., Tu E., Lam L., Bogoyevitch M. A., Seidman C. E., Seidman J. G., Semsarian C. (2008) Circulation 117, 1820–1831 [DOI] [PubMed] [Google Scholar]
- 25. McMullen J. R., Sherwood M. C., Tarnavski O., Zhang L., Dorfman A. L., Shioi T., Izumo S. (2004) Circulation 109, 3050–3055 [DOI] [PubMed] [Google Scholar]
- 26. Grounds H. R., Ng D. C., Bogoyevitch M. A. (2005) J. Cell. Biochem. 95, 529–542 [DOI] [PubMed] [Google Scholar]
- 27. Ng D. C., Gebski B. L., Grounds M. D., Bogoyevitch M. A. (2008) Cell Motil Cytoskeleton 65, 40–58 [DOI] [PubMed] [Google Scholar]
- 28. Bromberg J. F., Wrzeszczynska M. H., Devgan G., Zhao Y., Pestell R. G., Albanese C., Darnell J. E., Jr. (1999) Cell 98, 295–303 [DOI] [PubMed] [Google Scholar]
- 29. Ichiba M., Nakajima K., Yamanaka Y., Kiuchi N., Hirano T. (1998) J. Biol. Chem. 273, 6132–6138 [DOI] [PubMed] [Google Scholar]
- 30. Ng D. C., Long C. S., Bogoyevitch M. A. (2001) J. Biol. Chem. 276, 29490–29498 [DOI] [PubMed] [Google Scholar]
- 31. Heinrich P. C., Behrmann I., Müller-Newen G., Schaper F., Graeve L. (1998) Biochem. J. 334, 297–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siddiquee K., Zhang S., Guida W. C., Blaskovich M. A., Greedy B., Lawrence H. R., Yip M. L., Jove R., McLaughlin M. M., Lawrence N. J., Sebti S. M., Turkson J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 7391–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rothen-Rutishauser B. M., Ehler E., Perriard E., Messerli J. M., Perriard J. C. (1998) J. Mol. Cell Cardiol. 30, 19–31 [DOI] [PubMed] [Google Scholar]
- 34. Adams J. W., Sakata Y., Davis M. G., Sah V. P., Wang Y., Liggett S. B., Chien K. R., Brown J. H., Dorn G. W., 2nd. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 10140–10145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fassett J. T., Xu X., Hu X., Zhu G., French J. P., Chen Y., Bache R. J. (2009) Am. J. Physiol. Heart Circ. Physiol. 297, H523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yue H., Li W., Desnoyer R., Karnik S. S. (2010) Cardiovasc Res. 85, 90–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsutsui H., Tagawa H., Kent R. L., McCollam P. L., Ishihara K., Nagatsu M., Cooper G., 4th (1994) Circulation 90, 533–555 [DOI] [PubMed] [Google Scholar]
- 38. Hilfiker-Kleiner D., Hilfiker A., Drexler H. (2005) Pharmacol. Ther. 107, 131–137 [DOI] [PubMed] [Google Scholar]
- 39. Negoro S., Kunisada K., Fujio Y., Funamoto M., Darville M. I., Eizirik D. L., Osugi T., Izumi M., Oshima Y., Nakaoka Y., Hirota H., Kishimoto T., Yamauchi-Takihara K. (2001) Circulation 104, 979–981 [DOI] [PubMed] [Google Scholar]
- 40. Negoro S., Kunisada K., Tone E., Funamoto M., Oh H., Kishimoto T., Yamauchi-Takihara K. (2000) Cardiovasc Res. 47, 797–805 [DOI] [PubMed] [Google Scholar]
- 41. Ng D. C., Court N. W., dos Remedios C. G., Bogoyevitch M. A. (2003) Cardiovasc Res. 57, 333–346 [DOI] [PubMed] [Google Scholar]
- 42. Gao X. M., Wong G., Wang B., Kiriazis H., Moore X. L., Su Y. D., Dart A., Du X. J. (2006) J. Hypertens. 24, 1663–1670 [DOI] [PubMed] [Google Scholar]
- 43. Mascareno E., Siddiqui M. A. (2000) Mol. Cell Biochem. 212, 171–175 [PubMed] [Google Scholar]
- 44. Hilfiker-Kleiner D., Shukla P., Klein G., Schaefer A., Stapel B., Hoch M., Müller W., Scherr M., Theilmeier G., Ernst M., Hilfiker A., Drexler H. (2010) Circulation 122, 145–155 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.