

Abstract
Whereas UDP-glucuronosyltransferase-2B7 is widely distributed in different tissues, it preferentially detoxifies genotoxic 4-OH-estradiol and 4-OH-estrone (4-OHE1) with barely detectable 17β-estradiol (E2) conversion following expression in COS-1 cells. Consistent with the UDP-glucuronosyltransferase requirement for regulated phosphorylation, we discovered that 2B7 requires Src-dependent tyrosine phosphorylation. Y236F-2B7 and Y438F-2B7 mutants were null and 90% inactive, respectively, when expressed in COS-1. We demonstrated that 2B7 incorporated immunoprecipitable [33P]orthophosphate and that 2B7His, previously expressed in SYF-(Src,Yes,Fyn)−/− cells, was Src-supported or phosphorylated under in vitro conditions. Unexpectedly, 2B7 expressed in SYF−/− and SYF+/− cells metabolized 4-OHE1 at 10- and 3-fold higher rates, respectively, than that expressed in COS-1, and similar analysis showed that E2 metabolism was 16- and 9-fold higher than in COS-1. Because anti-Tyr(P)-438–2B7 detected Tyr(P)-438–2B7 in each cell line, results indicated that unidentified tyrosine kinase(s) (TKs) phosphorylated 2B7 in SYF−/−. 2B7-transfected COS-1 treated with increasing concentrations of the Src-specific inhibitor PP2 down-regulated 4-OHE1 glucuronidation reaching 60% maximum while simultaneously increasing E2 metabolism linearly. This finding indicated that increasing PP2 inhibition of Src allows increasing E2 metabolism caused by 2B7 phosphorylation by unidentified TK(s). Importantly, 2B7 expressed in SYF−/− is more competent at metabolizing E2 in cellulo than 2B7 expressed in COS-1. To confirm Src-controlled 2B7 prevents toxicity, we showed that 2B7-transfected COS-1 efficiently protected against 4-OH-E1-mediated depurination. Finally, our results indicate that Src-dependent phosphorylation of 2B7 allows metabolism of 4-OHE1, but not E2, in COS-1, whereas non-Src-phosphorylated 2B7 metabolizes both chemicals. Importantly, we determined that 2B7 substrate selection is not fixed but varies depending upon the TK(s) that carry out its required phosphorylation.
Keywords: Drug Metabolism, Estrogen, Phosphotyrosine Signaling, Post-translational Modification, Tyrosine-protein Kinase (Tyrosine Kinase), Depurination, PKC, Phosphorylation, Src, UDP-glucuronosyltransferase
Introduction
Mammalian UDP-glucuronosyltransferase (UGT)4 isozymes constitute a chemical defense system that detoxifies an enormous number of lipophilic substrates. Each isozyme links an acceptor substrate to glucuronic acid to produce glucuronides with greatly increased water solubility and excretability. The endoplasmic reticulum-bound superfamily of UGT isozymes protects biological systems from toxic metabolites and ingested chemicals (1) encountered on a regular basis in dietary plants and environmental contaminants (2). To a detriment, UGTs clear many medications prematurely (3, 4). Isozymes are distributed primarily in hepatic tissues and differentially in a wide range of other tissues (2, 5). In the case of impaired UGTs−/−, lipophiles accumulate in tissues causing deleterious effects that include hepatotoxicities, developmental defects, mutagenesis, as well as initiation and promotion of cancer (6). Inheritance of defective bilirubin-conjugating isozyme (7) in Crigler-Najjar (UGT1A1−/−) children causes high serum bilirubin levels that deposit in the central nervous system, causing kernicterus.
Except for UGT1A10, studies reveal that UGTs poorly metabolize primary estrogens (8, 9), E2 and E1. To the contrary, cytochromes 1B1 and 1A1/1A2 primarily generate the catechol-estrogen (CE) metabolites (for review see Ref. 10), 4-OHE2/E1 and 2-OHE2/E1, respectively, that undergo extensive glucuronidation (11, 12). Highly genotoxic 4-OHE1 and 4-OHE2 are preferred 2B7 substrates, and minimally toxic 2-OHE2 and 2-OHE1 are also O-methylated (10, 13). Genotoxic 4-OHE2 and 4-OHE1 are associated with the initiation of breast cancer (14). Without detoxification, 4-OHE2 and 4-OHE1 compared with 2-OHE2 and 2-OHE1 undergo extensive redox cycling as semiquinone(s)-quinone(s) (14) producing superoxide anions (O2˙̄) that depurinate DNA-forming adducts that undergo excision, leaving behind abasic sites (14). Often an error-prone base excision repair process ensues with departed adenines rapidly replaced with fixed mutations, rendering the metabolites prime tumor initiators in breast (14, 15) and other tissues (14).
Moreover, breast tissue contains high concentrations of E2 and E1 (16) and moderately high levels of the monooxygenases that oxidize E1 and E2 to CEs as well as non-CE derivatives (for reviews see Refs. 10 and 14). The non-CE metabolite, 16α-OHE1, is positively associated with breast cancer patient survival (17). Generally, the least toxic 2-OH-CEs are synthesized in the highest amount (10, 14). When in vivo conditions generate imbalances in cytochrome P450 activities that increase 4-OH-CE levels in combination with low levels of antioxidant reactions and detoxifying enzymes, cellular environments favor carcinogenesis (14). 4-OH-CE-generated superoxide anions (14) are also suspected of promoting cancer invasiveness and metastases by activating matrix metalloproteinases that degrade the extracellular matrix, which is the barrier to tumor passage (18). Because chemical reactivity of 4-OH-CE metabolites can both initiate and promote cancer invasiveness, CE-detoxifying enzymes (8, 9, 11, 12) are critical to the normal health of mammary gland and other estrogen-responsive tissues.
Whereas 2B7 was considered a pivotal isozyme for detoxifying toxic estrogen metabolites, its distribution in both estrogen-responsive and other tissues (2, 5) suggested that the isozyme is critical to protection against mutagenizing estrogen metabolites without altering E2 levels. An immunocytochemical study (19) detailed 2B7 distribution in normal breast tissue, its elevation in in situ lesions within some carcinomas, and its dramatic reduction in invasive carcinomas that showed disorganized basement membranes. Separately, an immunohistochemical study showed 2B7 is located in normal breast ductal and endometrial epithelial cells (20). Contrariwise, family A UGT1A10, primarily distributed throughout gastrointestinal tissues (2, 5, 8) and bladder (5) without detection in mammary gland and liver (2, 5), avidly metabolizes primary estrogens, CEs, and phytoestrogens (8). Hence, 2B7 is likely the relevant UGT that detoxifies 4-OHE2 and 4-OHE1 in estrogen-responsive tissues and not 1A10 isozyme, which converts E1, E2, and their metabolites at robust rates (8).
Consistent with the recent discovery that UGTs require phosphorylation (3, 8, 21–24), 2B7 contains two tyrosine kinase (TK) sites, Tyr-236 and Tyr-438, that require phosphorylation (24). Despite our confirmation that 2B7 requires Src-supported phosphorylation in COS-1 cells, unexpectedly we demonstrate that phosphorylation of 2B7 in Src-free (SYF−/−) cells enhanced metabolism of 4-OHE1 10-fold over that in Src-containing COS-1 cells. Moreover, barely detectable E2 metabolism in COS-1 cells was increased 16-fold in SYF−/− cells. Although Src phosphorylation of 2B7 expressed in COS-1 cells is required, its overall rate of 4-OHE1 glucuronidation is greatly inferior to that in SYF−/− cells, with barely detectable turnover of E2. This finding indicates that UGT2B7 substrate selection is not fixed but is differentially controlled by TKs phosphorylation similar to our observations that differential PKCϵ phosphorylation of sites in UGT1A7 controlled its substrate selections (22).
EXPERIMENTAL PROCEDURES
Materials
COS-1, SYF−/− (Src, Yes, and Fyn), and SYF+/− cells were from ATCC (Manassas, VA); serum was from Intergen Company (Purchase, NY). BCA protein assay kit was from Pierce. PP2 and anti-v-Src were from Calbiochem (La Jolla, CA), and restriction enzymes were from New England Biolab. Lipofectamine 2000 was from Invitrogen. [33P]Orthophosphate and UDP-[14C]glucuronic acid were from PerkinElmer Life Sciences; the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide kit, UGTs acceptor substrates, and anti-β-actin were from Sigma. COS-1 and mouse SYF fibroblast cells were grown in DMEM with 4 and 10% fetal bovine serum, respectively. Antibodies against 2B7 were as follows: anti-UGT-1168 (25) detected protein backbone, and anti-Tyr(P)-438–2B7, generated by Syn-Pep (Dublin, CA), detected only Tyr(P)-438–2B7. Anti-Fgr was from Cell Signaling Technology. The reagents used in cell culture were solubilized in fresh Me2SO to allow 0.5% final concentration. The viability of cells treated with curcumin was monitored with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
Mutation of PKC and TK Phosphorylation Sites in UGT2B7
Because 2B7 contains three PKC and two tyrosine kinase phosphorylation sites, each site was changed independently and in various combinations as already described (24).
Transfection of COS-1 and SYF Cells with pSVL-based UGT2B7, 2B7 Mutant, or 2B7-His-tagged Construct
The 2B7 expression unit, pSVL-UGT2B7, was cloned (11). All of the pSVLUGT2B7 mutants (24) and the pSVLUGT2B7His construct (24) were generated as described. 80% confluent COS-1 cells or 50% confluent SYF cells were transfected with pSVL-based 2B7-cDNA expression unit using Lipofectamine 2000 (according to the manufacturer's protocol); COS-1 cells involving radiolabeling were initiated 72 h after transfection.
Glucuronidation Assay
Reactions contained the common donor substrate UDP-[14C]glucuronic acid (1.40 mm, 1.4 μCi/μmol), 200 μm unlabeled acceptor substrate, 4-OHE1 or E2 with 150 μg of cellular homogenate in 40 mm phosphate (pH 6.4) or triethanolamine (pH 7.6) buffer as previously described (2). Homogenates were treated with 0.5 mg of CHAPS/mg of protein before addition to a reaction, which incubated at 25 °C for 16 h or at 37 °C for 2 h. Following standardization of the exposure process, radioactivity contained in [14C]glucuronides resolved in TLC plates was stored in phosphor-based film over 1–2 h in preparation for transferring the same to the PerkinElmer Cyclone Storage Phosphor Imager for quantitation.
Production of Anti-Tyr(P)-438–2B7 and Anti-UGT-1168
Phosphorylated Tyr-438 peptide (CKRVINDPSY(P03)KENV) derived from 2B7 was used to generate rabbit antibody (SynPeP), which was preabsorbed against nonphosphorylated peptide and then positively purified over phosphorylated peptide-containing resin. Anti-UGT-1168 was generated against highly purified mouse Ugt2b5 as described (25).
Western Blot Analysis
COS-1 or SYF cells were washed, harvested in cold PBS, solubilized in loading buffer, resolved in a 4–15% SDS-PAGE system, and subjected to Western blot analysis as described (8, 22, 23). Western blots exposed to antibody directed against phospho-groups were blocked with 5% bovine serum albumin (8, 23); others were blocked with 5% milk as described (23). Anti-Tyr(P)-438–2B7, anti-UGT-1168, anti-v-Src, and anti-β-actin were used as needed with different blots.
Incorporation of [33P]Orthophosphate into UGT2B7 and Its Mutants
Sixty hours after transfection with UGT2B7 or its mutants, the cells were processed and treated with [33P]orthophosphate to establish incorporation of immunoprecipitable label into specific protein as previously demonstrated (22).
Preparation of Microsomes from UGT2B7His-transfected SYF−/− Cells
Microsomes isolated from cultured cells were prepared as described (2) with modifications. The cells were harvested in PBS containing 100 μm sodium orthovanadate, 5 mm NaF, and protease mixture inhibitor (Sigma; catalog number P8849) before homogenization in 0.25 m sucrose, 0.15 m KCl adjusted to pH 7.4 containing 100 μm sodium orthovanadate, 5 mm NaF, and protease mixture inhibitor using a Dounce system. Cellular material was further disrupted by spritzing through a 29-gauge needle before centrifugation at 10,000 × g for 10 min to remove mitochrondia. The resulting supernatant was centrifuged at 100,000 × g for 1 h, and the pellet was resuspended in 0.15 m KCl containing 10 mm EDTA to chelate ribosomes before microsomes were pelleted at 60,000 × g for 30 min and resuspended in 0.25 m sucrose, 0.15 M KCl.
In Vitro Phosphorylation of 2B7 by Src
To confirm Src phosphorylates 2B7 in vitro, microsomes isolated from nontransfected and 2B7His-transfected SYF−/− cells were solubilized (23, 24), affinity-purified, and subjected to [γ-33P]ATP-dependent phosphorylation with and without SrcTK. The 2B7His-transfected sample was divided into two equal aliquots, and [γ-33P]ATP was added to the two aliquots and to control sample; Src kinase, added to equal amounts of transfected and nontransfected sample, incubated 30 min before affinity purification. Parallel duplicate systems containing nonlabeled ATP were similarly prepared. All of the samples were electrophesed in a 4–15% SDS-PAGE system (23) and quantitated as described. The resolved gel was exposed to film for autoradiography, and parallel nonradiolabeled samples were Western blotted with anti-Tyr(P)-438 and anti-UGT-1168.
Glucuronidation of 17β-Estradiol in Cellulo by 2B7-transfected SYF−/− and COS-1
Thirty-four hours after 2B7 transfection, SYF−/− and COS-1 cells were subjected to conditioning in Dulbecco's modified Eagle's medium containing 4% charcoal-stripped fetal bovine serum (8) for 8 h and exposed to 50 μm 17β[14C]E2 (2.5 μCi/μm) in fresh Dulbecco's modified Eagle's medium/charcoal-stripped fetal bovine serum for 16 h. For analysis of in cellulo 17β[14C]E2 glucuronide formed during exposure to 17β[14C]E2, 500 μl of medium were utilized in triplicate and diluted 1:2 with cold ethanol before centrifugation to remove protein. Recovered supernatant was spun dry and resuspended in phosphate-buffered saline. One-half of each sample was treated with buffer or buffer containing 50 units of β-glucuronidase in 50 μm phosphate buffer (pH 6.8) for 4 h at 37°C. The samples were vacuum-dried, resuspended in 70% ethanol, and applied to TLC plates for elution (8) for quantitation as described under the glucuronidation assay.
4-OHE1 Depurination of UGT2B7-transfected COS-1 Cells
To assess DNA damage in COS-1 cells following exposure to 4-OHE1 versus protection by 2B7 transfection, we measured damaged DNA by the comet assay (26). 4-OHE1-damaged DNAs are presented as short strands that migrate rapidly in an electrophoretic field and ultimately resemble a comet tail after staining. A visual representation of 4-OHE1-damaged DNA analyzed by the comet assay is shown in the appropriate figure below. To evaluate DNA damage accurately by the comet assay in this study, control vector- or 2B7-transfected COS-1 cells were grown for 48 h before replacing the medium with 4% charcoal-stripped FCS for 12 h. Finally, the medium was replaced with 4% charcoal-stripped FCS containing DMEM with 100 μm 4-OHE1 for 6 h. Cells washed with Ca2+/Mg2+-free PBS were placed on ice and harvested with the same ice-cold buffer by scraping with a rubber policeman. Harvested cells were subjected to the comet assay using a kit (Trevigen) with modification as described (26). Exceptions to the modifications (27, 28) included 12-h alkaline lysis at 4 °C, washing the surface of cell-containing gels with ice-cold water, followed by cold 70% ethanol wash. The gels were dried with a hair dryer in the cold room. Electrophoresis was carried out for 30 min as described (26) using 1 volt/centimeter for the cathode-anode distance.
Scoring Damaged DNA Using the Comet Assay
To assess the clinical relevance of 2B7, we determined its capacity to protect against 4-OHE1-dependent depurination following its expression in COS-1 cells. Immediately before imaging analysis, the slides were stained for 5 min at 4 °C with SYBR-Green diluted 1:10,000 according to the kit (27, 28). Observations were made at 20× magnification using a fluorescent microscope (Zeiss, Axiovert 200) with an Axiocam-HRC camera using Axiovision Rel 4.6 software. For purposes of visualizing the results of the comet assay, >70% saturation of the comet tail with damaged DNA is shown in the appropriate figure below. For accurate evaluation (28), we previously established 6 h as treatment time with 100 μm 4-OHE1 that generated <70% saturation of DNA in the comet tail. For each group, 200 processed cells were randomly and cumulatively counted on three slides representing three different experiments evaluated using Comet Score v1.5 software from TriTek Corporation (Sumerduck, VA). Damaged DNA is presented as the percentage of damaged DNA in the comet tail and as the extent of damage defined by tail moment, the most commonly reported parameter. Tail moment is the product of percentage DNA (fluorescence) in the comet tail and the distance between the means of the tail and head fluorescence distribution; mean is the profile center of gravity divided by 100. Tail moment is expressed in arbitrary units.
Statistical Analysis of Results Generated by the Comet Assay
Differences between DNA-damaged in treated control vector- and 2B7-transfected COS-1 cells were compared. Damaged DNA in the tail was evaluated as defined above, and undamaged DNA in the head is shown as fluorescent circles in the single-cell images in the appropriate figure below. The difference between the two treatment groups of cells was evaluated for significance using Student's t test. The results were highly significant at p = 0.001 with a 95% confidence interval.
RESULTS
UGT2B7 Requires Tyrosine Kinase Phosphorylation
Previously, we demonstrated that treatment of constitutive UGTs in human colon cell lines (2, 21) and recombinant UGT1A isozymes expressed in COS-1 cells (3, 23) is reversibly down-regulated by appropriate concentrations of dietary antioxidant, curcumin. Its inhibition of PKC isozymes led to the discovery that UGTs require phosphorylation. According to co-immunoprecipitation of signaling partners in complexes (22, 23) that are also distinguished by differential sedimentation profiles during centrifugation,5 each of three UGT1A isozymes studied requires phosphorylation by different PKC isozyme(s). To the contrary, the three PKC sites in 2B7 undergo phosphorylation (Fig. 1), but mutant PKC sites have little effect on activity (24). Because combined treatment with cycloheximide and curcumin was not different from curcumin alone, the evidence indicates that curcumin also down-regulates tyrosine kinase(s) as previously shown (29, 30). Hence, we have demonstrated that the agent inhibits phosphorylation of TK sites in 2B7, which completely down-regulated activity within 15 min with detectable recovery between 20 and 24 h (supplemental Fig. S1). As shown in Fig. 1A, site-directed mutagenesis of tyrosine residues, Tyr-236-2B7 and Tyr-438-2B7 (Fig. 1A, M4 and M5), caused null and 90% loss of activity, respectively, indicating that both residues require phosphorylation (Fig. 1B, left panel).
FIGURE 1.

Cartoon-defined distribution of kinase-specific sites in UGT2B7, specificity of UGT antibodies, and incorporation of [33P]orthophosphate into 2B7 and various 2B7 mutants expressed in COS-1 cells. A, specific kinase sites in 2B7 and their mutations are defined as shown. B, left panel, glucuronidation by 2B7 and its mutants at tyrosine-kinase sites in parallel with anti-Tyr(P)-438–2B7 specificities toward 2B7 phosphorylated tyrosine residues and anti-UGT-1168. Right panel, Western blot analysis of cellular homogenates of COS-1 cells transfected with UGT2B7 or different UGTs. Antibodies were generated (25) and used as described under “Experimental Procedures.” Activity by 2B7 and its TK site mutants were analyzed using 4-OHE1 in 2-h incubations at 37 °C and 150 μg of protein as described under “Experimental Procedures.” C, incorporation of [33P]orthophosphate into wild-type 2B7, 2B7 triple PKC sites mutant (M1), 2B7 double TK sites mutant (M2), and 2B7 combined M1/M2 penta-mutant (M3). The radiolabeling procedure is described under “Experimental Procedures.” Parallel unlabeled samples were Western blotted with anti-UGT-1168, and 4-OHE1 and E2 were glucuronidated in 2-h incubations at 37 °C as described for B.
Development and Characterization of Anti-Tyr(P)-438- UGT2B7
To monitor the status of Tyr(P)-438–2B7 residue, we carried out Western blot analysis with our custom developed anti-Tyr(P)-438-2B7 preparation that was purified as detailed (see “Experimental Procedures”). Anti-Tyr(P)-438-2B7 does not recognize family A UGTs: 1A1, 1A3, and 1A10, or other family B UGTs: 2B15 and 2B17 (Fig. 1B, right panel), which also contain two each TK phosphorylation sites.6 Because anti-Tyr(P)-438-2B7 appropriately detects Tyr(P)-438 in wild-type 2B7 and in Y236F-2B7 (M4), but not in Y438F-2B7 (Fig. 1B, left panel, M4 and M5), it is shown to be immunospecific toward 2B7. Finally, goat anti-UGT-1168, developed against highly purified mouse Ugt2b5 (25), recognizes the protein backbone of two of three family A isozymes (Fig. 1B, right panel) and of the three family B isozymes. Neither antibody recognized COS-1 control proteins.
Phosphorylation of UGT2B7 Using [33P]Orthophosphate
Finding that phosphorylation of TK sites (24), but not PKC sites (24), in 2B7 is critical for activity led us to compare [33P]orthophosphate incorporation into 2B7 wild-type with its mutants at predicted PKC and TK phosphorylation sites. Results show the highest level of immunodetectable label in wild-type 2B7 with 5 kinase sites with progressively less in M2, M1, and M3, representing 3, 2, and 0 phosphorylation sites, respectively (Fig. 1C). Because all Src-interacting proteins contain Src homology binding domains with phosphotyrosine sequences (31, 32), it should be pointed out that the radiolabel in the 2B7 penta-mutant is likely due to phosphorylated residues in the SH2 domain uncovered by software available on-line.
Whereas the triple PKC site mutant in 2B7 had little effect on estriol and 17-epiestriol turnover (24), the results in Fig. 1C indicate that the mutant metabolizes 4-OHE1 at 63% of the rate of wild-type 2B7 (Fig. 1C, bottom row) following normalization. Wild-type 2B7 and its triple mutant, M1, failed to show detectable turnover of E2 in a 2-h incubation (Fig. 1C, bottom row). Typically, recombinant UGT2B7 expressed in cell culture has shown low or no detectable turnover of E2 (9, 11).
Identification of the Tyrosine Kinase Responsible for UGT2B7 Phosphorylation in COS-1 Cells
To identify the TK responsible for phosphorylation of 2B7, we examined effects of different TK effector molecules on constitutive 2B7 activity in LS-180 cells using 2B7-specific substrate, hyodeoxycholic acid. Treatment of the cells with Src activator (33), vitamin D, led to an increase in 2B7 activity, which suggested Src affected activity. We also demonstrated that the Src-specific inhibitor (34) PP2 inhibited 2B7 activity ∼60% maximally under conditions of equal specific protein by anti-UGT-1168 analysis (data shown in Fig. 4).
FIGURE 4.
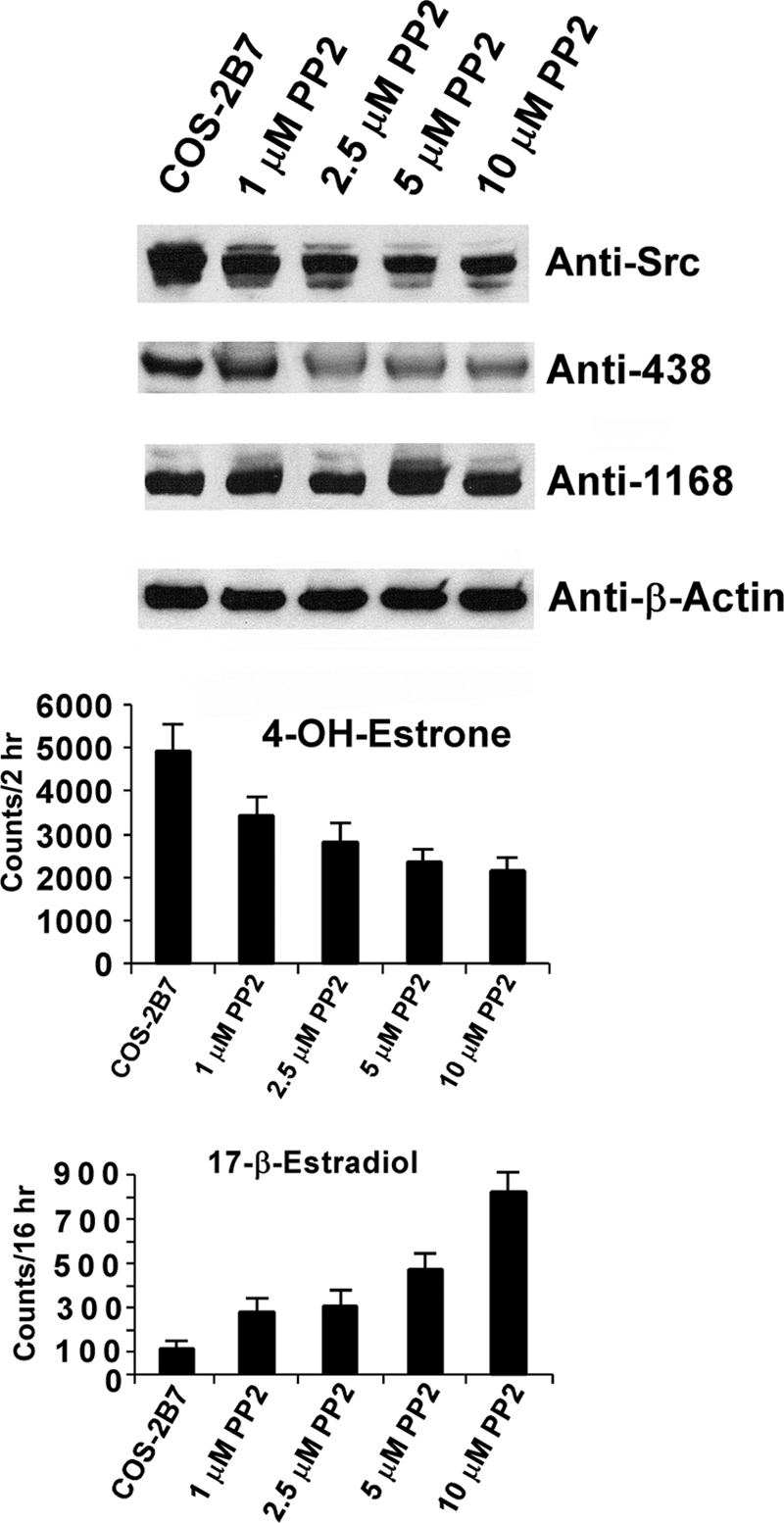
Effects of in cellulo treatment of 2B7-transfected COS-1 cells with increasing concentrations of PP2. Increasing concentrations of PP2 were added to 2B7-transfected COS-1 cells during the final 4-h of a 72-h culture period. The cells were harvested; homogenates were used for 4-OHE1 for 2 h at 37 °C and E2 glucuronidation assays at 25 °C for 16 h and for resolution in 4–15% gradient gels that were processed for Western blot analysis using antibodies shown as described under “Experimental Procedures.”
In Vitro Phosphorylation of UGT2B7 by Src Tyrosine Kinase
To gain definitive evidence that Src supports or phosphorylates 2B7, we expressed the 2B7His construct or its vector in SYF−/− cells and isolated microsomes for solubilization as described under “Experimental Procedures.” To solubilized 2B7His in microsomal preparations from 2B7His-transfected SYF−/− cells, we added Src-TK and [γ-33P]ATP versus [γ-33P]ATP alone for 30-min incubations as described under “Experimental Procedures.” Following affinity purification of [γ-33P]2B7His, we detected 6-fold greater labeling following Src-TK and [γ-33P]ATP treatment versus [γ-33P]ATP alone (Fig. 2A). Also, His affinity-purified Src-supported [γ-33P]2B7His showed 17-fold greater incorporation of labeled phosphate at the 55–58-kDa position in the 4–15% SDS-PAGE system (Fig. 2B), considering 458 versus 26 cpm (background corrected) with no visible band for 2B7His treated with [γ-33P]ATP alone (lane 2). In parallel samples, 2B7His treated with Src and unlabeled ATP generated markedly higher Tyr(P)-438-2B7 content than ATP treatment alone (Fig. 2A, second panel, lanes 3 and 2); reactions in lanes 3 and 2 contained equal microsomal 2B7 by anti-UGT-1168 analysis (third panel). The finding that Src and ATP incubation led to more than 17-fold greater labeling than ATP alone indicates that Src avidly supports 2B7 phosphorylation or phosphorylates the protein as already described (24).
FIGURE 2.

In vitro Src phosphorylation of UGT2B7His expressed in SYF−/− mouse fibroblast cells. A, microsomes isolated from vector- or 2B7His-transfected SYF−/− cells after 60 h of incubation were solubilized and subjected to phosphorylation using 100 μm [γ-33P] ATP (3000 Ci/mm) without or with Src kinase in a 30-min incubation at 30 °C. All of the reactions were stopped by adding 10 mm EDTA (final concentration). Duplicate reactions were carried out with unlabeled ATP. 2B7His affinity-purified samples were resolved in duplicate 4–15% SDS-PAGE with labeled or unlabeled reaction. Gel I with [γ-33P]ATP was fixed, dried, and exposed to x-ray film (lane 1); gel II was transferred and processed for Western blot analysis using antibodies shown (lanes 2 and 3). B, 2B7His-specific protein was affinity-purified and counted by liquid scintillation.
Differential Substrate Selections by UGT2B7 Expressed in SYF−/−, SYF+/−, and COS-1 Cells
Despite finding that 2B7 expressed in COS-1 cells is dependent upon Src-TK for required phosphorylation, we unexpectedly uncovered 10-fold greater 4-OHE1 conversion by 2B7 following its expression in Src-free cells (SYF−/−) than that expressed in COS-1 (Fig. 3, second panel). For comparison, 2B7 expressed in SYF+/− cells allowed only a 3-fold increase in 4-OHE1 conversion above that expressed in COS-1 cells. The fact that expression of 2B7 in cells that lacked both or one Src allele led to 10- and 3-fold higher 4-OHE1 conversion, respectively, than 2B7 expressed in COS-1 demonstrated unexpectedly that Src had markedly reduced 4-OHE1 turnover by 2B7, despite its required phosphorylation or support for phosphorylating 2B7.
FIGURE 3.

In vitro glucuronidation of 4-OHE1 or 17β-estradiol by 2B7 expressed in SYF−/−, SYF+/−, and COS-1 cells. Cells transfected with vector or 2B7 that had been incubated for 72 h were harvested, and homogenates were used for 4-OHE1 or E2 glucuronidation at 25 °C for 16 h. Aliquots of corresponding homogenates were resolved in 4–15% gradient gels, transblotted, and processed for Western blot analysis using the antibody preparations shown. Note: anti-SrcTK detects regular Src between 56 and 60 kDa in cultured cells.
Considering the fact that 2B7 expressed in COS-1 (11) and HEK293 cells (9) generated nondetectable and barely detectable E2 conversion, respectively, the highly favorable 4-OHE1 conversion by 2B7 expressed in SYF−/− cells suggested that this isozyme preparation possibly increased E2 turnover. Conversion of E2 by 2B7 expressed in SYF−/− cells increased 16-fold, and that expressed in SYF+/− increased 9-fold over that in COS-1 following normalization. Because anti-Tyr(P)-438-2B7 and anti-UGT-1168 Western blot analysis showed that 2B7 was phosphorylated in the three cell lines despite far less 2B7 protein in SYF−/− cells (Fig. 3, first panel), the results indicate that unidentified tyrosine kinase(s) in SYF−/− cells can also phosphorylate or support phosphorylation of 2B7 that is far more productive than that by Src, as shown after normalization. Western blot analysis (Fig. 3, first panel) confirmed that SYF−/− and SYF+/− lacked Src or contained less active Src (top band), respectively, than COS-1. Hence, the data indicate that the presence of Src reduced both 4-OHE1 and E2 turnover, in parallel, in COS-1 such that 2B7 barely converted the primary estrogen. It is noted that E2 was also metabolized by constitutive activity in SYF cells that decreased with the addition of a Src allele. Most importantly, different substrate selections by 2B7 following its phosphorylation by different TKs indicate that the isozyme substrate pattern is not fixed but varies according to factors that determine the particular TK(s) carrying out its phosphorylation. Finding that 2B7 depends upon Src phosphorylation in COS-1 cells, which actually reduces its potential maximum activity, is a conundrum that controls substrate selection.
Effects of PP2 inhibition of 2B7 on 4-OHE1 and 17β-Estradiol Conversions
Because the absence of Src in SYF−/− cells allowed unidentified TK enzymes to phosphorylate 2B7 in a manner that enabled it to support E2 turnover, the question arose as to whether inhibition of Src in COS-1 permits other TK(s) to phosphorylate 2B7, operationally, so as to metabolize E2. Hence, we examined the effect of the Src-specific inhibitor (34) PP2 on E2 turnover by 2B7 expressed in COS-1. Without PP2 treatment, 2B7 metabolized 4-OHE1 during a 2-h incubation at 37 °C, whereas E2 requires 16-h incubation at 25 °C. Treatment with increasing concentrations of PP2 led to a maximum of 60% inhibition of 4-OHE1 metabolism, whereas E2 turnover was simultaneously increasing linearly with concentration (Fig. 4). Western blot analysis did not detect changes in the 2B7 level, whereas its phosphorylation status and the level of Src decreased. Hence, it remains to be seen whether the level of Tyr(P)-438-2B7 controls differential substrate selections for 4-OHE1 and E2. Nevertheless, counts describing preferential turnover of 4-OHE1 over E2 remain high at a ratio of 2500 to 850 or 2.9-fold (Fig. 4, 10 μm PP2). The results strongly suggest that at least two different TK systems can phosphorylate 2B7 and that Src has dominance over the unidentified TK(s) uncovered in the SYF−/− cells.
In Cellulo Conversion of 17β-Estradiol by 2B7-transfected SYF−/−
To determine whether 2B7-transfected SYF−/− cells were also competent to metabolize E2, we compared its conversion in both 2B7-transfected SYF−/− and COS-1 cells. Normalized results for 2B7-transfected cells show SYF−/− generated 17-fold greater counts, 2680 versus 151, than COS-1 after 16 h of in cellulo metabolism (Fig. 5A, top panel). Sensitivity of the product to β-glucuronidase treatment confirmed that 2B7 generated E2 glucuronide product in both cells (Fig. 5A, bottom panel). Simultaneously, harvested cells were homogenized and assayed in vitro using nonlabeled E2 in the regular glucuronidation assay with co-substrate [14C]UDP-glucuonic acid. After normalization by Western blot analysis using anti-1168, anti-438, and anti-β-actin, the results show that 2B7 expressed in SYF−/− generated 3-fold more E2 glucuronide than 2B7 expressed in COS-1 cells in 16 h. Hence, it is evident that SYF−/− cells are more competent to metabolize E2 compared with COS-1 cells. Because of variability in SYF−/− cells growth and, hence, transfection efficiency, we observed a range between 3- and 20-fold higher efficacy of those cells to support 2B7 turnover of E2 than its expression in COS-1.
FIGURE 5.

Glucuronidation of 17β[14C]estradiol in cellulo by 2B7-transfected COS-1 or SYF−/− cells. A, after culturing both nontransfected and 2B7-transfected COS-1 and -SYF−/− cells for 34 h and conditioning for 8 h, the cells were exposed to 17β[14C]E2 for 16 h as described under “Experimental Procedures.” Triplicate 500 μl of culture medium was processed as described under “Experimental Procedures.” After dividing each sample into two parts, one part was processed as described under “Experimental Procedures,” and one was subjected to β-glucuronidase treatment to confirm glucuronide product before processing as described under “Experimental Procedures.” Finally, the samples were resolved by TLC for counting via phosphorimaging and data collection. B, simultaneously, in vitro E2 glucuronidation was carried out with homogenates of nontransfected and 2B7-transfected COS-1 and SYF−/− cells using the regular glucuronidation assay with common donor-substrate [14C]UDP-glucuronic acid in 16-h incubations at 25 °C as described under “Experimental Procedures.”
Lack of Fgr Kinase in SYF−/− Cells
To determine whether the companion Src family Fgr kinase is a candidate for the unknown TK(s) that phoshorylate 2B7 expressed in SYF−/− cells, we compared Western blot analysis of SYF−/− and COS-1 cells expressing 2B7 using anti-Fgr. Although Fgr kinase has been reintroduced into SYF+/− cells, it is not present in SYF−/− cells (supplemental Fig. S2). Hence, Fgr is not involved in 2B7 phosphorylation in SYF−/− cells.
Demonstration 2B7 Transfection Protects COS-1 Cells against 4-OHE1 Depurination
Whereas we have demonstrated that Src-supported phosphorylation of 2B7 in COS-1 cells detoxifies 4-OHE1 between 10 and 30% the rate 2B7 expressed in SYF−/−cells, it was of interest to determine whether that level of activity provides measurable protection against 4-OHE1 toxicity. Hence, we adapted the comet assay to score depurination following 4-OHE1 treatment as described under “Experimental Procedures” (26–28). To capture images to demonstrate DNA damage, we first treated nontransfected and 2B7-transfected COS-1 cells with sufficient 4-OHE1, to cause nearly all DNA to redistribute from the head into the comet tail due to damage revealed by SYBR green staining (Fig. 6A, left bottom panel) versus no major DNA redistribution following 4-OHE1 treatment of 2B7-transfected cells (Fig. 6A, right bottom panel). Also, results showed that Me2SO (vehicle) treatment of either nontransfected (Fig. 6A, top left panel) or 2B7-transfected cells (Fig. 6A, top right panel) caused no major change in DNA shift from the head.
FIGURE 6.

Effect of 2B7 transfection on 4-OHE1-damaged DNA in comet tails. A, depiction of 2B7 protection against 4-OHE1-based depurination. B, quantitation of protection against 4-OHE1-dependent depurination of COS-1 cells. The cells were treated with vehicle or 100 μm 4-OHE1 for 6 h, processed, and stained before evaluation of damaged DNA using Comet Score v1.5 software as described under “Experimental Procedures.” The results for 200 cells/group were randomly and cumulatively evaluated from three experiments, using two different parameters as shown: percentage of damaged DNA in tail and tail moment (arbitrary units). Student's t test generated a p value of 0.001.
For the purpose of scoring depurinated DNA redistribution to the comet tail, treatment of nontransfected COS-1 cells with 100 μm 4-OHE1 for 6 h underwent linear accumulation of DNA fragments in tails that averaged 50–60% protection, whereas similar treatment of 2B7-transfected cells and Me2SO treatment of nontransfected or 2B7-transfected cells scored by the percentage of damaged DNA in comet tail (Fig. 6B, left panel) or by tail moment (Fig. 6B, right panel) showed 92% protection against DNA redistribution of damaged DNA into comet tails. Computer analysis was carried out with software (see “Experimental Procedures”). The results demonstrate that 2B7 glucuronidation of 4-OHE1 expressed in COS-1 had a significant protective effect against depurination of DNA.
DISCUSSION
Previously, we demonstrated that UGT2B7 transfected into COS-1 cells preferentially metabolized genotoxic 4-OHE2 and 4-OHE1 at rates 10–100-fold higher than minimally toxic 2-OHE2 and 2-OHE1 (11, 12, 14). More recently, we showed that overexpression of Src kinase in co-transfection studies with the 2B7-cDNA construct in COS-1 cells yielded a 1.5-fold maximum increase in 2B7 activity (24) with a constant 2B7 protein level but increased levels of anti-Tyr(P)-438-2B7 and anti-regular Src signals. Presently, we have confirmed that 2B7 requires tyrosine phosphorylation at residues Tyr-236 and Tyr-438 as suggested by site-directed mutagenesis (24). Moreover, we monitored the phosphorylation status of Tyr-438 with our custom-developed and highly specific anti-Tyr(P)-438-2B7 that recognizes only Tyr(P)-438-2B7. Similar to results seen for three different UGT1A isozymes shown to require regulated phosphorylation at PKC sites (21–23), 2B7 activity is reversibly down-regulated by curcumin treatment following expression in COS-1 (supplemental Fig. S1), indicating that it also requires regulated phosphorylation but at tyrosine residues. Although 2B7 is not only down-regulated by general TK inhibitors (24), it is down-regulated by Src-specific PP2 inhibitor (34), which suggests that the isozyme requires Src-dependent phosphorylation (Fig. 4, top panel). Consistent with Src kinase-supported phosphorylation of 2B7 in COS-1 cells, the two proteins also showed co-localization that was partially disrupted by PP2 pretreatment (24).
To establish that Src supports 2B7 phosphorylation, we demonstrated that Src kinase activity was 6-fold more effective at generating [γ-33P]2B7His using [γ-33P]ATP compared with [γ-33P]ATP alone (Fig. 2B). Migration of the in vitro Src-generated product to a 55–56-kDa position in the SDS gradient gel verified that the tyrosine kinase supports 2B7 phosphorylation; furthermore, 9-fold greater label at that position generated by Src compared with ATP alone is further evidence that Src supports 2B7 activity. Moreover, 2B7His contained 10–12-fold greater anti-Tyr(P)-438-2B7 signal with unlabeled ATP and Src than ATP alone, which also verified its effects on 2B7His (Fig. 2A, second panel).
The fact that 2B7 phosphorylated in Src−/− cells was 8-fold more active toward 4-OHE1 and 16-fold more active toward E2 than when expressed in Src-containing COS-1 cells represented the first indication that 2B7 phosphorylation could be distinguished operationally by different TKs altering its active site so as to change substrate selection (Fig. 3). Hence, the capacity of 2B7 to glucuronidate E2 following PP2 inhibition of Src in COS-1 was evidently due to 2B7 phosphorylation by unidentified TK(s), enabling it to glucuronidate E2 as seen in SYF−/− cells. Our finding demonstrates that 2B7 undergoes Src-dependent regulated phosphorylation allowing “alternative active site” utilization with enhanced discrimination between substrates. This allows 4-OHE1 conversion with E2 exclusion. Such action is consistent with 1A7-regulated phosphorylation and substrate selection (see below) (22, 23). Here it appears that the 2B7 “maximum rate” of 4-OHE1 glucuronidation following its phosphorylation by unidentified TKs in SYF−/− is significantly reduced by Src phosphorylation, which on a relative scale prevents E2 glucuronidation, an essential physiological benefit regarding estrogen-responsive tissues.
Enzymatically, there are 19 human UGT isozymes, and each covalently links a lipophilic chemical to common donor-substrate glucuronic acid (1), generating inactive excretable metabolites and environmental contaminants. The mechanism utilized by a finite number of UGT isozymes to metabolize an indeterminate number of toxic, as well as nontoxic acceptor substrates, is unknown. Whereas preliminary evidence predicts that 13 UGTs undergo regulated phosphorylation (21–24), detailed evidence demonstrates an association of regulated phosphorylation and substrate expansion for 1A7 (Ref. 22 and see below), and here we demonstrate an association of regulated phosphorylation and substrate selection for 2B7. Hence cumulative results support the concept that phosphate signaling is possibly the underpinning of ongoing refolding and dynamic conformational changes of active site(s) for 2B7 and 1A7 carried out by an appropriate assembly of select proteins including UGT(s), kinase(s), phosphatase(s), and adapter/scaffold protein(s) (23). Such an adaptable mechanism of refolding at the active site(s) could provide an enormous substrate potential that is susceptible to molecular probes that affect diseases and cellular homeostasis. To date, each UGT is associated with a different kinase(s) (22, 23, 35), which is likely based on the over-arching role of each isozyme.
Earlier COS-1 cells expressing UGT1A7-S432G or UGT1A7 treated with antagonist PKCϵ-specific peptide that blocked phosphorylation of Ser-432 (22) (Figs. 4 and 5) revealed that dephosphorylation at Ser-432 in 1A7 enabled the isozyme to gain new substrates simultaneous with a catalytic pH shift from 8.0 to 6.5 (22). Hence, ongoing refolding of the active site(s) in the isozymes mediated by phosphate signaling is a plausible explanation of our data.
The model dietary agent and UGT substrate curcumin reversibly down-regulates both PKC- and TK-regulated UGT isozymes. Hence, phosphate signaling enables cells to invoke appropriate kinase(s) and confers different substrate specificity(s) to respond appropriately to new chemical pressures similar to the myriad of other cellular pressures and phosphate signaling that maintain cellular homeostasis. Moreover, evidence indicates that substrate selections can be manipulated by appropriate agents. This was shown by PP2 inhibition of Src kinase that enhanced 2B7 metabolism of E2 while reducing 4-OHE1 in this study and by PKCϵ-specific inhibition of 1A7 by PKCϵ-specific peptides that increased E2 glucuronidation (22), demonstrating that regulated phosphorylation is a target for manipulating UGT substrate activities. Hence, our findings suggest the possibility of advancements in drug development that could lead to specific and targeted treatments for chemically mediated diseases including breast cancer initiation (14).
Emerging evidence continues to demonstrate unexpected UGT flexibility controlled by regulated phosphorylation. Earlier we showed that the UGT system in tissues of the gastrointestinal tracts of mice could be temporarily down-regulated (3) by ingesting the antioxidant curcumin to allow enhanced uptake and efficacy of immunosuppressant mycophenolic acid, which otherwise undergoes extensive glucuronidation by gastrointestinally distributed UGTs. Although Src-regulated phosphorylation of 2B7 reduces 4-OHE1 glucuronidation evidently to prevent E2 metabolism, PKCϵ-regulated phosphorylation of 1A7 appears to expand its substrate selection (22). Hence, accumulating evidence indicates that UGT substrate selections are not fixed and that selections can be manipulated by treatment with appropriate agents that modulate select kinase(s) activities. Detection of UGT substrate changes noted here were made possible, no doubt, by en masse shifts away from basal ongoing signaling equilibria to a single equilibrium by chemical treatments, kinase-specific disruptions, or mutant UGT isozymes as cited.
Concerning a possible mechanism to explain the variation in substrate selection following differential phosphorylation of Tyr-438-2B7 by Src kinase versus unidentified TK(s), it is noted that PP2 treatment caused concurrent decreases in both detectable Tyr(P)-438 in 2B7 and its turnover of 4-OHE1 simultaneously with its increase in E2 turnover (Fig. 4). Hence, the possibility exists that the high level of detectable phosphorylation of 2B7 by Src kinase restricts access of 4-OHE1 to the catalytic cavity(s), whereas less phosphorylation of 2B7 by unidenitified TK(s) allows a more accessible 2B7 catalytic cavity(s) and entry of both 4OHE1 and E2 molecules. The apparent inverse relationships between 2B7 phosphorylation status and turnover of two different substrates suggest that two different catalytic cavities exist that can interchange. Highly charged but variably formed salt bridges involving phosphate moieties could possibly participate in establishing such changing 2B7 active site(s). If changing active site(s) are involved, then analysis of a single enzyme preparation to describe catalytic activity(s) may not describe all of the possibilities. Hence, it may be necessary to use a Src-phosphorylated 2B7 preparation and an “unidentified TK” phosphorylated preparation expressed in SYF−/− cells for eventual x-ray analysis.
In the case of mammary gland-distributed 2B7, it should be pointed out that Src is critical to its development based on rudimentary mammary gland structures in a Src knock-out mouse (36). Whereas Src is known to promote differentiation, controlling 2B7 specific substrate turnover via signaling is consistent with that function. Because 2B7 is among the most widely distributed UGTs (5), it is likely important to many other cellular detoxification reactions.
Supplementary Material
Acknowledgments
We acknowledge the assistance of Dr. Kushal Chakraborty with the in vitro phosphorylation UGT2B7-His and the comet assay.
This work was supported, in whole or in part, by the National Institutes of Health NICHD Intramural Research Program.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
N. K. Basu, M. Basu, and I. S. Owens, manuscript in preparation.
S. Chakraborty, N. K. Basu, M. Basu, and I. S. Owens, manuscript in preparation.
- UGT
- UDP-glucuronosyltransferase
- 4-OHE2
- 4-OH-estradiol
- 4-OHE1
- 4-OH-estrone
- E2
- 17β-estradiol
- TK
- tyrosine kinase
- CE
- catechol-estrogen
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
REFERENCES
- 1. Dutton G. (1980) in Glucuronidation of Drugs and Other Compounds (Dutton G. J. ed) pp. 3–28, CRC Press, Boca Raton, FL [Google Scholar]
- 2. Basu N. K., Ciotti M., Hwang M. S., Kole L., Mitra P. S., Cho J. W., Owens I. S. (2004) J. Biol. Chem. 279, 1429–1441 [DOI] [PubMed] [Google Scholar]
- 3. Basu N. K., Kole L., Basu M., McDonagh A. F., Owens I. S. (2007) Biochem. Biophys. Res. Commun. 360, 7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burger D. M., Huisman A., Van Ewijk N., Neisingh H., Van Uden P., Rongen G. A., Koopmans P., Bertz R. J. (2008) Clin. Pharmacol. Ther. 84, 698–703 [DOI] [PubMed] [Google Scholar]
- 5. Nakamura A., Nakajima M., Yamanaka H., Fujiwara R., Yokoi T. (2008) Drug Metab. Dispos. 36, 1461–1464 [DOI] [PubMed] [Google Scholar]
- 6. Wells P. G., Mackenzie P. I., Chowdhury J. R., Guillemette C., Gregory P. A., Ishii Y., Hansen A. J., Kessler F. K., Kim P. M., Chowdhury N. R., Ritter J. K. (2004) Drug Metab. Dispos. 32, 281–290 [DOI] [PubMed] [Google Scholar]
- 7. Ritter J. K., Yeatman M. T., Ferreira P., Owens I. S. (1992) J. Clin. Invest. 90, 150–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Basu N. K., Kubota S., Meselhy M. R., Ciotti M., Chowdhury B., Hartori M., Owens I. S. (2004) J. Biol. Chem. 279, 28320–28329 [DOI] [PubMed] [Google Scholar]
- 9. Lépine J., Bernard O., Plante M., Têtu B., Pelletier G., Labrie F., Bélanger A., Guillemette C. (2004) J. Clin. Endocrinol. Metab. 89, 5222–5232 [DOI] [PubMed] [Google Scholar]
- 10. Zhu B. T., Conney A. H. (1998) Carcinogenesis 19, 1–27 [DOI] [PubMed] [Google Scholar]
- 11. Ritter J. K., Sheen Y. Y., Owens I. S. (1990) J. Biol. Chem. 265, 7900–7906 [PubMed] [Google Scholar]
- 12. Ritter J. K., Chen F., Sheen Y. Y., Lubet R. A., Owens I. S. (1992) Biochemistry 31, 3409–3414 [DOI] [PubMed] [Google Scholar]
- 13. Fotsis T., Zhang Y., Pepper M. S., Adlercreutz H., Montesano R., Nawroth P. P., Schweigerer L. (1994) Nature 368, 237–239 [DOI] [PubMed] [Google Scholar]
- 14. Cavalieri E., Chakravarti D., Guttenplan J., Hart E., Ingle J., Jankowiak R., Muti P., Rogan E., Russo J., Santen R., Sutter T. (2006) Biochim. Biophys. Acta. 1766, 63–78 [DOI] [PubMed] [Google Scholar]
- 15. Fernandez S. V., Russo I. H., Russo J. (2006) Int. J. Cancer 118, 1862–1868 [DOI] [PubMed] [Google Scholar]
- 16. Pasqualini J. R., Chetrite G., Blacker C., Feinstein M. C., Delalonde L., Talbi M., Maloche C. (1996) J. Clin. Endocrinol. Metab. 81, 1460–1464 [DOI] [PubMed] [Google Scholar]
- 17. Castagnetta L. A., Granata O. M., Traina A., Ravazzolo B., Amoroso M., Miele M., Bellavia V., Agostara B., Carruba G. (2002) Clin. Cancer Res. 8, 3146–3155 [PubMed] [Google Scholar]
- 18. Paquette B., Bisson M., Baptiste C., Therriault H., Lemay R., Cantin A. M. (2005) Int. J. Cancer 113, 706–711 [DOI] [PubMed] [Google Scholar]
- 19. Gestl S. A., Green M. D., Shearer D. A., Frauenhoffer E., Tephly T. R., Weisz J. (2002) Am. J. Pathol. 160, 1467–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thibaudeau J., Lépine J., Tojcic J., Duguay Y., Pelletier G., Plante M., Brisson J., Têtu B., Jacob S., Perusse L., Bélanger A., Guillemette C. (2006) Cancer Res. 66, 125–133 [DOI] [PubMed] [Google Scholar]
- 21. Basu N. K., Kole L., Owens I. S. (2003) Biochem. Biophys. Res. Commun. 303, 98–104 [DOI] [PubMed] [Google Scholar]
- 22. Basu N. K., Kovarova M., Garza A., Kubota S., Saha T., Mitra P. S., Banerjee R., Rivera J., Owens I. S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 6285–6290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Basu N. K., Kole L., Basu M., Chakraborty K., Mitra P. S., Owens I. S. (2008) J. Biol. Chem. 283, 23048–23061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mitra P. S., Basu N. K., Owens I. S. (2009) Biochem. Biophys. Res. Commun. 382, 651–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mackenzie P. I., Hjelmeland L. M., Owens I. S. (1984) Arch. Biochem. Biophys. 231, 487–497 [DOI] [PubMed] [Google Scholar]
- 26. Olive P. L., Banáth J. P. (2006) Nat. Protocols 1, 23–29 [DOI] [PubMed] [Google Scholar]
- 27. Klaude M., Eriksson S., Nygren J., Ahnström G. (1996) Mutat. Res. 363, 89–96 [DOI] [PubMed] [Google Scholar]
- 28. Orlow I., Park B. J., Mujumdar U., Patel H., Siu-Lau P., Clas B. A., Downey R., Flores R., Bains M., Rizk N., Dominguez G., Jani J., Berwick M., Begg C. B., Kris M. G., Rusch V. W. (2008) J. Clin. Oncol. 26, 3560–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leu T. H., Su S. L., Chuang Y. C., Maa M. C. (2003) Biochem. Pharmacol. 66, 2323–2331 [DOI] [PubMed] [Google Scholar]
- 30. Huang W. C., Chen J. J., Chen C. C. (2003) J. Biol. Chem. 278, 9944–9952 [DOI] [PubMed] [Google Scholar]
- 31. Newman D. K. (2009) J. Thromb. Haemost. 7, (Suppl. 1) 195–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rivera G. M., Vasilescu D., Papayannopoulos V., Lim W. A., Mayer B. J. (2009) Mol. Cell 36, 525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buitrago C., Vazquez G., De Boland A. R., Boland R. L. (2000) J. Cell Biochem. 79, 274–281 [DOI] [PubMed] [Google Scholar]
- 34. Kerman K., Chikae M., Yamamura S., Tamiya E. (2007) Anal. Chim. Acta. 588, 26–33 [DOI] [PubMed] [Google Scholar]
- 35. Volak L. P., Court M. H. (2010) Xenobiotica 40, 306–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim H., Laing M., Muller W. (2005) Oncogene 24, 5629–5636 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.