

Abstract
Generation of the NF-κB p50 transcription factor is mediated by the proteasome. We found previously that p50 is generated during translation of the NFKB1 gene and that this cotranslational processing allows the production of both p50 and p105 from a single mRNA. We now demonstrate that the Rel homology domain in p50 undergoes cotranslational dimerization and that this interaction is required for efficient production of p50. We further show that this coupling of dimerization and proteasome processing during translation uniquely generates p50–p105 heterodimers. Accordingly, after the primary cotranslational event, additional posttranslational steps regulate p50 homodimer formation and the intracellular ratio of p50 and p105. This cellular strategy places p50 under the control of the p105 inhibitor early in its biogenesis, thereby regulating the pool of p50 homodimers within the cell.
Keywords: cotranslational dimerization/cotranslational processing/NF-κB1/proteasome
Introduction
Proteasome-mediated degradation is not only essential for the normal turnover of many cellular proteins, but is also central to the regulation of many biological processes (Hochstrasser, 1995; Baumeister et al., 1998; Ciechanover, 1998). For example, the proteasome plays a central role in the activation of the mammalian transcription factor NF-κB. In resting cells, NF-κB is sequestered in the cytosol by its interaction with an inhibitor termed IκB (Baeuerle and Baltimore, 1988, 1989). In the specific case of IκBα, a variety of inducing signals converges on two specific IκB kinases: IKKα and β, which phosphorylate serines 32 and 36 of IκBα (Stancovski and Baltimore, 1997). Subsequent ubiquitylation of this phosphorylated IκBα results in its rapid degradation by the proteasome (Chen et al., 1995; Scherer et al., 1995; Spencer et al., 1999) and the release of NF-κB. This transcription factor complex then translocates into the nucleus where it induces κB sequence-specific gene expression.
The 26S proteasome plays a critical role in the generation of the p50 subunit of the NF-κB complex, as demonstrated by both biochemical and genetic experiments (Fan and Maniatis, 1991; Palombella et al., 1994; Lin et al., 1998; Sears et al., 1998). Both p50 and the larger p105 protein are products of the NFKB1 gene (Ghosh et al., 1990; Kieran et al., 1990). The p50 polypeptide coincides with the N-terminal portion of p105 and spans the ∼300- residue Rel homology domain (RHD). The C-terminal portion of the p105 protein contains multiple ankyrin repeats, a hallmark of the IκB family of cytoplasmic Rel inhibitors. In this regard, p105 has been shown to function as an IκB (Rice et al., 1992). Our previous studies suggest that p50 is generated principally during translation of the NFKB1 mRNA and that p50 and p105 production may be balanced by the transient folding state of the p105 nascent polypeptide (Lin et al., 1998). Homeostasis of p105 and p50 appears to be physiologically important since transgenic mice expressing p50 but not p105 exhibit severe inflammation (Ishikawa et al., 1998). In contrast, NFKB1 knockout mice lacking expression of both p50 and p105 do not display such inflammatory changes and only manifest minor defects in B cell function (Sha et al., 1995). These in vivo results indicate that p105 likely plays an important role in regulating p50 function and that this property of p105 is not compensated for by the other IκBs. The cotranslational processing of the NFKB1 gene product leads to a natural balance of these two proteins, and thus ensures their distinct biological functions within the cell. The intracellular ratio of these two proteins can also be regulated by signal-induced kinases that trigger the complete degradation of p105, a process that may facilitate p50 homodimer formation (Belich et al., 1999; Heissmeyer et al., 1999).
NF-κB p105, a related Rel protein, p100 (Betts and Nabel, 1996; Heusch et al., 1999) and a Drosophila protein Ci (Ingham, 1998) are the only known examples of proteasome substrates that are not completely degraded to small oligopeptides. A 23 residue glycine-rich region (GRR) located downstream of the nuclear localization signal (NLS) in the NFKB1 gene products is essential for p50 generation (Lin and Ghosh, 1996) and likely both initiates the process and determines the site of cleavage (Lin and Ghosh, 1996; Heusch et al., 1999). However, it remains unclear how the translated N-terminal portion of the NFKB1 gene product corresponding to p50 is effectively cleaved yet escapes degradation by the proteasome. Additionally, the mechanism by which different NF-κB–Rel protein dimers form in vivo is poorly understood. Urban and Baeuerle (1990) observed that Rel protein dimers do not dissociate and reassociate freely and proposed that the NF-κB dimers assemble early in their biogenesis. The potential relationship between p50 production and dimerization has not been assessed.
NF-κB p105 possesses a unique domain organization that resembles the artificial H-Ras–DHFR fusion protein employed by Netzer and Hartl (1997) in studies that led to their sequential and cotranslational model of eukaryotic protein folding. The N- and C-terminal portions of p105, which serve independent functions, are separated by the GRR and thus may fold independently. In our previous work, we proposed that cotranslational folding of p50 might regulate the primary ratio of p50 and p105 (Lin et al., 1998). A study by Johnson et al. (1998), demonstrating that dimerization of the yeast mating factors MATα2 and MATa1 prevented the degradation of both factors by the proteasome, prompted us to examine the assembly status of p50. We now report that the RHD in the N-terminal portion of p105 dimerizes cotranslationally through its second subdomain (sd2) and that this dimerization is required for efficient p50 production. Our results further indicate that cotranslational dimerization and processing generate only p50–p105 heterodimers. After the primary cotranslational event, formation of p50 homodimers involves additional posttranslational steps. These results highlight a mechanism by which the p105 inhibitor gains very early control of the p50 transcription factor and provide a new perspective on Rel protein assembly.
Results
p50 exists naturally as a dimer
NF-κB p50 forms homodimers or heterodimers with p65 and other Rel proteins through sequences located in the second subdomain (sd2) of the RHD (Ghosh et al., 1995; Müller et al., 1995; Chen et al., 1998; Figure 1A). Specifically, two segments corresponding to residues 251–270 and 302–310 within sd2 are responsible for dimerization. Deletion of either of these segments prevents p50 from forming either homo- or heterodimers (data not shown). To confirm that p50 exists naturally as a dimer rather than a monomer, we translated mRNAs encoding full-length p50 (433 residues) and a mutant of p50 (Δ302–310) lacking one of these dimerization segments in vitro, and examined the products by gel filtration chromatography. Nearly all of the wild-type p50 eluted with a size consistent with dimers, while the p50 (Δ302–310) mutant protein eluted in later fractions consistent in size with monomers (Figure 1B and C).

Fig. 1. NF-κB p50 exists as a dimer. (A) Amino acid sequence of p50. The sequence of murine p50 and structural domains as described by Ghosh et al. (1995) are shown. Subdomains 1 and 2 (sd1 and sd2) of the Rel homology domain (RHD) are indicated by parentheses while the intervening loop is labeled as L3. The sd1 in this figure encompasses residues 39–240; however, in this work the N-terminal 1–240 residues are referred to as sd1. The sequences of segments involved in dimerization (residues 251–270 and 302–310) are shown in bold, and the NLS in bold italics. Residues participating in the dimer interface are indicated by diamonds underneath the amino acid code, and the number of residues are marked at the right of each line. (B) Elution profile of the Superdex 200 column. The curve is drawn according to the peak elution volumes for the indicated gel filtration standards detected by absorption at 280 nm. The estimated position of elution of the p50 dimer and monomer preparations is shown. (C) Gel filtration chromatography of [35S]methionine radiolabeled, in vitro-translated, gp10-tagged wild-type p50 or a dimerization mutant (Δ302–310) of p50. Fractions eluting between 20 and 31.5 ml were immunoprecipitated with anti-gp10 antibodies, analyzed by SDS–PAGE and visualized by fluorography. The elution volumes of p50 dimer and monomer peaks are indicated in lanes 9–12 and lanes 15–18.
Dimeric p50 displays a different pattern of proteinase K digestion than monomeric p50
Since protease partial digestion profiles (PDPs) have been used successfully to monitor protein folding (Shuman et al., 1990; Frydman et al., 1994), we digested in vitro translated dimeric and monomeric forms of p50 with proteinase K (PK). The PK-treated p50 dimer-derived fragments were readily immunoprecipitated with anti-NLS antibodies, but were unreactive with antibodies to the T7 gp10 epitope tag located at the N-terminus (Figure 2A). These findings suggest that the N-terminus of p50 is cleaved by PK, while the NLS is protected when p50 is in a dimeric form. To exclude the possibility that these results derived from cleavage of the N-terminal epitope tag, we examined the PK PDP of untagged p50. The PK PDPs of both tagged and untagged p50 were indistinguishable (Figure 2B), indicating that PK cleavage occurred within the N-terminal sequences of p50, not at the joint of the epitope tag and p50. PK initially cleaved the p50 homodimer into a fragment of ∼37 kDa, which was degraded further to a 32 kDa fragment (Figure 2A). This 32 kDa fragment was resistant to PK digestion at 4°C for >2 h (data not shown). In contrast, PK treatment of monomeric forms of p50 yielded two smaller fragments of 27 and 24 kDa (Figure 2C), neither of which was immunoprecipitated by anti-NLS antibodies (Figure 2D). PK treatment was also performed using a shortened form of p50 (residues 1–364). Both the anti-NLS reactive 37 and 32 kDa fragments were generated from this smaller protein (Figure 2E), suggesting that a PK cleavage occurs downstream of residue 364. Together, these results indicate that differences in PK PDP and immunoreactivity with the anti-NLS antibody provide a biochemical approach to monitor dimerization of p50.

Fig. 2. PK PDP of p50 dimers and monomers. (A) PK PDP of p50 dimers. p50 dimers translated in vitro were treated with PK (10 μg/ml) at 4°C for the times indicated. This treatment resulted in two fragments of ∼37 (p37) and 32 kDa (p32). Both fragments could be immunoprecipitated by anti-NLS antidodies (lanes 6–10) but not by antibodies to the N-terminal T7 gp10 epitope tag (lanes 1–5). (B) PK cleavage occurs within the N-terminal sequences of the p50 dimer, not at the joint of p50 and the epitope tag. p50 dimers either containing (lanes 1–4) or lacking (lanes 5–8) the T7 gp10 epitope tag generated identical PK PDPs, as measured by immunoprecipitation of anti-NLS antibodies. (C) p50 monomers and p50 dimers exhibit different PK PDPs. mRNAs encoding wild-type p50 and p50 dimerization mutants (Δ251–270 and Δ302–310) were translated in vitro and treated with PK (10 μg/ml) at 4°C for 10 min. Aliquots (20 μl) of the reaction mixtures were analyzed directly by SDS–PAGE. Wild-type p50 generated p37 and p32 (lane 1), while both dimerization mutants generated two smaller fragments of ∼27 (p27) and 24 kDa (p24) (lanes 2 and 3). (D) The p27 and p24 PK-resistant fragments generated from the dimerization mutants of p50 are not immunoprecipitated by anti-NLS antibodies. Wild-type p50 and the p50 dimerization mutants were treated with PK and immunoprecipitated with anti-NLS antibodies. Neither p27 nor p24 reacted with anti-NLS antibodies (lanes 5–12) while the p37 and p32 proteins derived were immunoprecipitated (lanes 1–4). (E) Sequences located downstream of the p50 NLS are not included in the dimeric PK-resistant folding core. Both wild-type p50 and a p50 mutant containing only the 364 residues of p50 extending to the NLS were subjected to PK treatment. p50 (364 residues) generated a PDP (lanes 5–8) identical to that obtained with wild-type p50 (lanes 1–4). For an unknown reason, untreated p50 (364 residues) migrated as a doublet (lane 5). (F) Heterodimeric p50 generates a PK PDP indistinguishable from that of p50 homodimers. mRNAs encoding 780 residues of the N-terminal portion of p105 (p-780) or 497 residues (without stop codon, p-497/XhoI) were translated in vitro and treated with PK (10 μg/ml) at 4°C for 10 min and immunoprecipitated with anti-NLS antibodies. Lanes 1, 3 and 5 show untreated controls, and lanes 2, 4 and 6 show the PK-treated samples.
Translation of mRNA encoding a sufficiently long NF-κB1 fragment results in production of both the full-length protein and p50 (Lin et al., 1998). These translation mixtures contain a large portion of p50–full-length protein heterodimers as demonstrated by coimmunoprecipitation of p50 with antibodies specific for an epitope tag at the C-terminus of the full-length protein (data not shown). Consistent with this observation, p50–p105 heterodimers are readily detected in vivo (Naumann et al., 1993; Watanabe et al., 1997). To obtain the PK PDP of this translation mixture, we translated a stop codon-truncated mRNA encoding residues 1–497 of p105 (p-497/XhoI). PK treatment of this translation mixture revealed the 37 and 32 kDa fragments indicative of p50 dimer formation (Figure 2F, lanes 3 and 4). Although the mixture presumably contains homo- and heterodimers, anti-NLS antibodies did not immunoprecipitate additional fragments after PK PDP. Apparently, the C-terminal portion of the p-497 protein in the p50–p-497 heterodimer complex is cleaved and the NLS in monomeric p-497, like monomeric p50, is not protected during PK digestion. A priori, p50–p105 heterodimers possess a common folding core, and hence a PK PDP similar to that of p50 homodimers. A similar PK digestion pattern was obtained employing a larger fragment (residues 1–780, p-780) (Figure 2F, lanes 1 and 2). These results indicate that p50, either as a homodimer or heterodimer with p105 or its derivatives, exhibits a PK PDP in which the NLS region is protected. In contrast, monomeric p50 or p105 exhibits a different PK PDP in which the NLS is not protected.
p50 dimerizes during translation of the NFKB1 mRNA
We have suggested that the RHD located in the N-terminal portion of the translating NFKB1 gene product may fold to a conformation sufficient to support such activities as dimerization. In this regard, reovirus σ1 cell attachment proteins have been shown to trimerize during translation (Gilmore et al., 1996). To test whether the RHD cotranslationally dimerizes, an mRNA encoding p-780 was translated in vitro at 25°C for 20–30 min. The translation was then terminated by adding cycloheximide (CHX), and the mixture was subjected to sucrose gradient sedimentation and fractionation. All fractions were divided into two parts: one part was treated with PK and then immunoprecipitated with antibodies to p50 NLS, while the second part was immunoprecipitated directly with antibodies to the p50 NLS. The majority of the immunoreactive proteins present in the denser ribosome fractions were dimers, as indicated by the PK PDP (Figure 3A, right panel). Because the C-terminus of p-780 was predicted to be occluded in the ribosome channel and therefore inaccessible for immunoprecipitation, anti-NLS antibodies were used (Figure 3A, left panel). The dimeric forms observed here are likely to be p50–p-780 heterodimers instead of p50 homodimers, since the latter, when cleaved from the nascent chains, would appear in the low-density fractions at the top of the gradient due to its low Svedberg sedimentation coefficient. RNase treatment of the translation mixture before sucrose gradient sedimentation led to the release of all of the p50–p-780 dimeric forms from the ribosome fractions into the low-density fractions of the gradient (Figure 3B), thereby confirming that the heterodimers in the high-density fractions were associated with mRNA.

Fig. 3. Sucrose gradient sedimentation of p-780. (A) Analysis of CHX-terminated translation of p-780 by sucrose gradient sedimentation. p-780 was translated in vitro at 25°C for 20–30 min followed by adding CHX. The translation mixture was then sedimented on a 20–45% sucrose gradient (11 ml) (by centrifugation) at 40 000 r.p.m. for 3.75 h. Fractions (1 ml) were then collected from the gradient and divided into two parts. One part was immunoprecipitated directly with anti-NLS antibodies (left panel), and the other part was subjected to PK treatment (10 μg/ml) at 4°C for 30 min before immunoprecipitation (right panel). Due to the ratio of PK versus the substrate, most of the digestions yield only the more stable fragment p32. The fractions containing ribosomes are determined by absorption at 260 nm, and the top and bottom of the gradient are indicated (lanes 6–11). (B) Analysis of the RNase-treated p-780 translation mixture by sucrose gradient sedimentation. p-780 was translated as described in (A) and then treated with RNase (5 μg/ml) at 37°C for 15 min before sucrose gradient sedimentation. The resulting fractions were analyzed as in (A).
To confirm further the heterodimeric nature of NFKB1 gene products associated with the ribosome, we translated mRNAs encoding either residues 1–646 or 1–780. The stop codons were deleted from both mRNAs to promote tethering to the ribosome. As shown in Figure 4, when CHX was added to stabilize nascent chain–polysome complexes, a significant proportion of p50 was dimerized with the full-length nascent polypeptides that cosedimented with the polysome pellets (lanes 1, 2, 5 and 6). However, when puromycin was added, p50 and prematurely terminated partners of various length were released from the ribosomes and appeared in the supernatant (Figure 4, lanes 3, 4, 7 and 8). Since mRNAs remain intact under these conditions, this finding excludes the possibility that p50 cosediments with the polysome pellet by binding to mRNA.
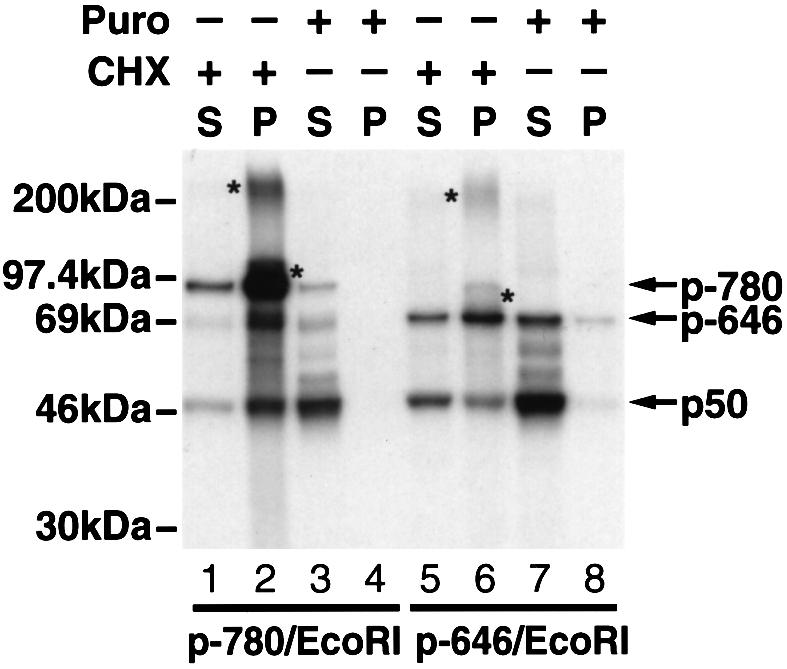
Fig. 4. Heterodimeric nature of the NFKB1 gene products associated with the ribosomes. mRNA encoding T7 gp10 epitope-tagged p105 N-terminal fragments of 646 or 780 residues (p-646/EcoRI and p-780/EcoRI, respectively) lacking stop codons were translated in vitro at 30°C for 20 min. The translation mixtures were then divided into two parts, one was treated with CHX (final concentration of 0.25 mg/ml) and the other was treated with puromycin (final concentration of 1 mM). Both samples were incubated at 30°C for an additional 20 min. After diluting with 100 μl of sucrose gradient buffer and centrifuging at 14 000 g for 10 min to remove insoluble material, the samples were loaded on to 350 μl of 25% sucrose and centrifuged at 100 000 r.p.m. for 40 min in a TLA.100.2 rotor. The pellets were washed twice with 0.5 ml of sucrose gradient buffer and suspended in 0.5 ml of the same buffer. Both pellets and supernatants were then immunoprecipitated with anti-T7 gp10 antibodies and analyzed by SDS–PAGE. Lanes 1, 2, 5 and 6 correspond to translation mixtures treated with CHX; and lanes 3, 4, 7 and 8 represent samples treated with puromycin. The supernatant (S) and pellet (P) fractions are also indicated. Asterisks appearing on the right side of bands indicate peptidyl-tRNA species, while those appearing on the left side of bands indicate possible ubiquitylation conjugates.
p50 dimerizes on the same polysome
To investigate whether p50 dimers assembled on the same or different polysomes, mRNAs encoding an N-terminally epitope-tagged wild-type p50 and an untagged shortened p50 (residues 1–364) were translated together. Anti-gp10 antibodies only immunoprecipitated tagged wild-type p50. Despite being effectively expressed and capable of dimerization, the 364-residue version of p50 lacking the epitope tag was not coimmunoprecipitated (Figure 5A, lanes 1 and 2). Similarly, anti-gp10 antibodies immunoprecipitated the tagged 364-residue version of p50 without coimmunoprecipitating untagged wild-type p50 when these two species of mRNA were translated together (Figure 5A, lanes 5 and 6). These findings suggest that dimer assembly is confined to p50 nascent chains translating on the same mRNA. Once assembled, the p50 dimer appears rather stable and fails to undergo disassociation and reassociation, since posttranslational mixing of these p50 species did not result in detectable heterodimer formation (Figure 5A, lanes 3 and 7).

Fig. 5. NF-κB p50 dimerizes on the same polysome. (A) p50 dimerizes on the same polysome. mRNAs encoding T7 gp10-tagged p50 (433 residues) and untagged p50 (364 residues) were either cotranslated (Co-, lanes 1 and 2) or separately translated and posttranslationally mixed (Post-, lanes 3 and 4). The translation mixtures were divided into two parts; one part was immunoprecipitated with anti-T7 gp10 antibodies (lanes 1 and 3), and the other part was immunoprecipitated with anti-NLS (lanes 2 and 4). Similarly, mRNA encoding T7 gp10-tagged p50 (364 residues) and untagged p50 (433 residues) were either cotranslated (lanes 5 and 6) or translated separately and then combined posttranslationally (lanes 7 and 8), followed by immunoprecipitation with anti-T7 gp10 antibodies (lanes 5 and 7) or anti-NLS antibodies (lanes 6 and 8). (B) p50 and p65 form heterodimers when cotranslated. mRNAs encoding T7 gp10-tagged p50 and untagged p65 were cotranslated (Co-, lanes 1 and 2) or translated separately and then combined (Post-, lanes 3 and 4). The translation mixtures were divided into two parts; one part was immunoprecipitated with antibodies specific for the N-terminus of p65 [αp65 (A), lanes 1 and 3], and the other part was immunoprecipitated with antibodies to p65 and the T7 gp10 epitope tag present on p50 (lanes 2 and 4).
To address the possibility that the formation of p50 dimers on the same polysome was due to a high local concentration of the same species of p50, we cotranslated mRNAs encoding p50 and p65. As suggested by structural studies (Chen et al., 1998), p50 and p65 exhibit stronger affinity for each other than two p50 molecules. Antibodies to p65 coimmunoprecipitated a majority of p50 present in the mixture (Figure 5B, lanes 1 and 2), indicating that high local concentrations of p50 do not prevent its association with p65. Posttranslational mixing of p50 and p65 did not result in formation of p50–p65 heterodimers (Figure 5B, lanes 3 and 4), presumably reflecting the prior formation of stable p50 homodimers.
The in vivo evidence of cotranslational dimerization of p50 homodimers
To demonstrate cotranslational homodimerization of p50 in vivo, we designed a bicistronic expression vector that allows synthesis of two different forms of p50 from the same mRNA (Figure 6A). In this construct, a differently tagged, shorter form of p50 (364 residues) is expressed from an internal ribosome entry site (IRES), while the full-length p50 (433 residues) is expressed from the 5′ start codon. If the dimerization occurs posttranslationally and hence is completely random, antibodies recognizing one epitope-tagged version of p50 should coimmunoprecipitate significant quantities of the other p50 species. As shown in Figure 6B, antibodies specific to hemagglutinin (HA) epitope tag immunoprecipitate principally the full-length p50 (Figure 6B, lane 1). Similarly, antibodies to T7 gp10 tag immunoprecipitate the corresponding shorter form of p50 and only a small amount of the full-length p50 proteins (Figure 6B, lane 2). The level of expression of both p50 species is shown in Figure 6B, lane 3. Together, these results provide corroboratory in vivo evidence supporting cotranslational dimerization of NFKB1 gene products.

Fig. 6. In vivo evidence of cotranslational dimerization of p50 homodimers. (A) Diagram of the bicistronic construct that expresses two different epitope-tagged versions of p50 (also different in size). (B) Immunoprecipitation of p50 species expressed in CHO-CD14 cells by the bicistronic construct. The bicistronic expression vector was transfected into CHO-CD14 cells and labeled with [35S]methionine/cysteine for 1 h. The cell lysates were prepared and divided into three parts for immunoprecipitation. Lane 1, immunoprecipitation with anti-HA; lane 2, immunoprecipitation with anti-gp10; lane 3, immunoprecipitation with anti-HA and anti-gp10; lane 4, pIRES vector that expresses only the HA-tagged full-length p50; lane 5, pIRES vector that expresses only the gp10-tagged shorter form of p50; lane 6, pIRES vector immunoprecipitated with anti-HA and anti-gp10. Since the overall expression of the IRES-dependent gp10-p50 is less than that of HA-p50, twice the amount of lysates was used for immunoprecipitation in lanes 2 and 3.
Cotranslational dimerization of the RHD is required for efficient generation of p50
We next examined whether cotranslational dimerization plays a role in the generation of p50. NFKB1 mutants altered in the dimerization segments were transfected into CHO-CD14 cells, radiolabeled and immunoprecipitated with antibodies specific for the T7 gp10 tag present at their N-termini. In contrast to wild-type NFKB1, three different dimerization mutants, including two deletion mutations (Δ251–270 and Δ302–310) and the composite L267D/L269D point mutation (Sengchanthalangsy et al., 1999), generated markedly lower quantities of p50 (Figure 7A, lanes 2–4). The introduction of comparably sized deletions elsewhere within the sd1 or sd2 segments of the NFKB1 gene, which do not alter dimerization, did not alter p50 generation (Figure 7B, lanes 4 and 5). Furthermore, the L267D/L269D point mutant exhibited a similar PK-resistant pattern to the deletion mutants (data not shown), suggesting a strong correlation between the abilities to dimerize and to generate p50.

Fig. 7. Mutations affecting dimerization significantly reduce p50 production. (A) Mutations in sd2 affecting dimerization significantly reduce p50 production. The gp10-tagged wild-type p105 (WT) and the dimerization mutants (deletion and composite point mutation) were transfected into CHO-CD14 cells and labeled with [35S]methionine/cysteine for 1 h and immunoprecipitated with anti-gp10 antibodies. Lane 1, wild-type p105; lanes 2 and 3, dimerization mutants [p105 (Δ251–270) and p105 (Δ302–310), respectively]; lane 4, p105 containing two point mutations, Y267D and L269D; lane 5, vector control. (B) Deletions that do not affect dimerization do not affect p50 production. p105 mutants containing a deletion of a 33 amino acid region [residues 166–198, p105 (ΔNO)] in sd1, and deletion of the NLS (residues 358–366) located at the end of sd2 [p105 (ΔNLS), see Figure 1A] were expressed in CHO-CD14 cells, immunoprecipitated with antibodies to their gp10 tag and analyzed for p50 production as described in (A) (lanes 4 and 5). Lane 1, wild-type p105, lanes 2 and 3, dimerization mutants. (C) Pulse–chase radiolabeling studies of the p105 and p50 dimerization mutants. The wild-type p50 and p105 as well as p50 (Δ302–310) and p105 (Δ302–310) mutants were transfected into CHO-CD14 cells, pulse radiolabeled with [35S]methionine/cysteine for 1 h and chased for the time periods indicated. Lanes 1–5, wild-type p50; lanes 6–10, p50 (Δ302–310); lanes 11–15, wild-type p105 and lanes 16–20, p105 (Δ302–310).
To test whether dimerization of the RHD promotes greater stability of either p50 or p105 in the cell, pulse–chase radiolabeling studies were performed in CHO-CD14 cells transfected with either wild-type p50 and p105 or the p50(Δ302–310) and p105(Δ302–310) dimerization mutants. When compared with their wild-type counterparts, dimerization mutants of both p50 and p105 exhibited shortened half-lives (Figure 6C). However, the reduced production of p50 seen with both dimerization mutants was not due to instability of the p50 monomer, since p105, which also contains the same mutation and exhibits a similar half-life, was readily detected (Figure 7A and B). Based on these observations, we suggest that cotranslational dimerization between the RHDs is required for effective processing and likely prevents the N-terminal portion of p105 from undergoing proteolysis (see Discussion).
Discussion
p50 homodimers form during translation of the NFKB1 mRNA
In this study, we have further explored the biochemical basis for production of the NF-κB p50 subunit. Using PK PDP to monitor the assembly status, we find that the RHD dimerizes during translation. Deletion or point mutations that abrogate dimerization alter the PK PDP (Figure 2C) and inhibit the production of p50 production (Figure 6A). In contrast, other comparably sized deletions in the flanking region do not alter the PK PDP (Figure 2E) or inhibit the generation of p50 (Figure 7B). Based on these results, we conclude that cotranslational dimerization of the RHD is essential for effective biogenesis of p50.
In the sucrose gradient sedimentation studies, we observe only small quantities of p50 in the top fractions, suggesting a low production of p50 homodimers (Figure 3A). Instead, we observe that both p50 and p-780 are present in the ribosome fractions (Figure 3A). During this brief and disrupted translation, the majority of the proteins have not completed their synthesis and remain ribosome bound. Such polypeptides are particularly sensitive to proteolysis and are largely degraded. Accordingly, only the near-full-length p-780 survives due to its stable conformation. This conclusion is bolstered by data provided in Figure 4 where puromycin treatment releases a ladder of incompletely translated proteins. Detection of these proteins likely reflects their final folding following puromycin-mediated release from the ribosome and the acquisition of resistance to proteolytic degradation. Accordingly, a ladder of ‘p50–p105’ species is observed in the supernatant. These studies demonstrate the heterodimeric nature of the translating nascent chains (Figure 4, lanes 3 and 7) and confirm our previous conclusion that p50 generation occurs principally during translation.
Based on these results, we propose here a general model that combines cotranslational dimerization and processing (Figure 8A). In this model, a ‘quasi-dimer’ is formed between the nascent RHDs produced by the leading and trailing ribosome. The capture and cleavage of the first nascent chain by the proteasome generates p50 that is already bound to the nascent chain of the following ribosome. Release of p50 from both the ribosome and the proteasome, which could represent physical obstacles for maintenance of the ‘quasi-dimer’, likely stabilizes the dimer and additionally appears to facilitate the folding of trailing nascent chain promoting its escape from proteasome capture. These events lead to the selective formation of p50–p105 heterodimers. In the situation where a nascent chain is not captured and processed by the proteasome, translation of the C-terminal region of the corresponding p105 protein likely promotes disassembly of the ‘quasi-dimer’ owing to the interaction of the C-terminal IκB-like structure with the N-terminal RHD. A monomeric form of p105 is thus produced.

Fig. 8. Models for p50 homodimer formation (see text for more detailed description). (A) Formation of p50–p105 heterodimer. (B) Posttranslational processing model. (C) Chaperone-assisted dimerization model.
Although this working model must be validated by additional experimentation, it is consistent with the hypothesis that protein folding regulates the primary ratio of p50 and p105 (Lin et al., 1998). These experiments also provide the first evidence for cotranslational dimerization of a mammalian protein. This work may explain the previous finding that preformed NF-κB complexes undergo little or no reassortment (Urban and Baeuerle, 1990). We similarly find that mixing of two p50 species after their translation does not lead to their cross-dimerization (Figure 5A). Furthermore, results obtained from in vivo experiments (Figure 6B) involving the generation of an IRES-containing bicistronic mRNA are consistent with the in vitro studies. Since the expression directed by IRES is less than that obtained in the natural setting, if the dimerization is random, the less-expressed shortened p50 should dimerize predominantly with the full-length p50 that is expressed in greater quantity. The insignificant cross-dimerization between the two p50 species in vivo certainly argues against a random posttranslational dimerization mechanism. The assembly of p50-containing dimers may thus be restricted principally to the time of translation, involving peptide chains present on the same polysome. Once formed, the p50 homodimers are quite stable. If this is the case, how then do p50–p65 heterodimers form? In contrast to p50 homodimers, p65 homodimers are highly unstable and significant quantities of p65 remain in a monomeric state. Such monomers of p65 could complete with the unstable p50 ‘quasi-dimer’-promoting formation of the p50–p65 heterodimer. Because of the higher affinity between p50 and p65 (Chen et al., 1998), the majority of complexes from cotranslation of p50 and p65 are p50–p65 heterodimers (Figure 5B). However, the possibility of such cotranslational dimerization between p50 and p65 in vivo may be small. Brief radiolabeling of CHO-CD14 or HeLa cells followed by immunoprecipitation with anti-p65 antibodies revealed that the majority of endogenous complexes formed are p65–p105 heterodimers (data not shown). This result suggests that p105 may function as a de facto ‘p50 monomer’ in vivo by maintaining a conformation sufficient to support posttranslational dimerization with p65. The in vivo pathway of p50–p65 heterodimer formation remains an area of active study.
Additional posttranslational steps regulate the intracellular p50–p105 ratio after the primary cotranslational event
Assessment of the dimerization status of p50 reveals added complexity in the cotranslational biogenesis of p50 involving a linkage between assembly and production. Furthermore, our findings provide new insights into the controversy that has surrounded cotranslational versus posttranslational production of p50. As demonstrated in Figures 3 and 4, the primary ratio of p50 versus p105 is indeed generated during the translation of NFKB1 mRNA as reported previously (Lin et al., 1998). However, after the primary cotranslational event, the intracellular ratio of p50 and p105 may also be influenced either by cellular factors such as BCL-3, which mediates p50 homodimer formation (Naumann et al., 1993; Watanabe et al., 1997), or by phosphorylation of p105, which triggers degradation of this protein (MacKichan et al., 1996; Belich et al., 1999; Heissmeyer et al., 1999).
Since cotranslational dimerization and proteasome processing generate primarily p50–p105 heterodimers, additional posttranslational steps must be involved in p50 homodimer formation. Two possible models for p50 homodimer formation seem plausible including: posttranslational processing of the p105 partner (Figure 8B) and chaperone-assisted reassortment (Figure 8C). The first model requires selective degradation of the p105 C-terminal portion in the p50–p105 heterodimer. This possibility is supported by a series of in vitro studies (Palombella et al., 1994; Orian et al., 1995, 1999; Coux and Goldberg, 1998). In this regard, the 155 kDa Drosophila transcription factor Ci, which has no homology to the Rel proteins, is processed to a 75 kDa form by the proteasome (Ingham, 1998). This apparently posttranslational processing of Ci requires the protein complex with microtubules and three additional proteins, suggesting that the formation of this complex confers resistance to complete degradation by the proteasome. However, clear evidence for a similar processing of p105 to p50 in vivo has not yet been assembled.
The second model involves the action of BCL-3, which has been implicated in p50 homodimer formation in the absence of proteolytic processing (Naumann et al., 1993; Watanabe et al, 1997; Heissmeyer et al., 1999). BCL-3 could act as a chaperone binding the p50–p105 complex and promoting its disassembly while preserving the proper conformation in p50 for dimerization with other disassembled p50 monomers. It would be interesting to explore the possibility of whether a similar cellular reassortment mechanism promotes the formation of p50–p65 heterodimers.
p105 modulates p50 homodimer in vivo
The formation of homo- or heterodimers is a common intracellular event involving many proteins; however, the rules that regulate such assemblies are not well understood (Lamb and McKnight, 1991; Tjian and Maniatis, 1994). Based on the observation that NF-κB dimers are tightly joined and undergo only low-level dissociation and reassociation, Urban and Baeuerle (1990) proposed that NF-κB dimerization occurs early in their biogenesis. Unlike p50–p65 heterodimers, p50 homodimers are not effectively regulated by IκBs. Therefore, a large pool of p50 homodimers in the cell could interfere adversely with normal cellular functions. In this regard, transgenic mice that produce p50, but not p105, accumulate high levels of p50 homodimers and exhibit significant inflammatory symptoms and increased susceptibility to bacterial infections (Ishikawa et al., 1998). In contrast, mice lacking both p50 and p105 exhibit only rather subtle immunological deficits. Our studies demonstrating cotranslational assembly of p50 and p105 highlight a cellular strategy whereby p105 control over p50 is established at a very early step in its biogenesis.
Cis elements required for efficient p50 production
In addition to the GRR, our studies now identify a second element, the NFKB1 dimerization domain, which is critical for p50 generation. Deletion of either dimerization segment present in the sd2 of the NFKB1 gene leads to a significant reduction in p50 production (Figure 6A and C). It is highly unlikely that p50 is generated from these mutant NFKB1 gene products, but not detected due to its rapid degradation. Our finding that the p105 dimerization mutant is easily detected and exhibits approximately the same intracellular half-life as the corresponding p50 dimerization mutant argues against rapid degradation of p50. Rather, we suspect that the nascent polypeptide chains of these mutants are recognized and bound normally for processing. However, in the absence of dimerization, the proteolysis initiated downstream progresses towards the N-terminus. Alternatively, the lack of dimerization may lead to a conformation within the sd2 region that is ineffective for the support of processing.
Johnson et al. (1998) found that heterodimerization of the yeast mating factors MATa1 and MATα2 masks an element that otherwise serves as the target for ubiquitylation and proteasome degradation. Dimerization in this setting thus serves as a cellular strategy to determine the yeast mating type via the ubiquitin–proteasome pathway. Thus far, we find no evidence based on deletion mapping for such a destabilizing element within the RHD that is masked by its dimerization. Although a more extensive mutagenesis is required to exclude this possibility completely, we currently favor the idea that the presence of a stable, well separated folding block in the RHD may be sufficient for efficient p50 production. It is also possible that dimerization activates subsequent proteasome processing. In this regard, ubiquitylation and proteasomal degradation of activating transcription factor 2 (ATF2) depends on its dimerization with c-Jun (Fuchs and Ronai, 1999). Such dimerization generates a conformation in ATF2 that effectively presents the protein to an E3, leading to its ubiquitylation. Whether similar ubiquitylation is involved in the cotranslational processing of p50 remains unknown. Both sd1 and sd2 contain structures that resemble the immunoglobulins (Igs) (Ghosh et al., 1995; Müller et al., 1995), raising the possibility that either domain could be recognized by an E3 broadly recognizing Ig-like proteins, or alternatively, by the PA700 regulatory component of the proteasome (Ma et al., 1994). Recently, Orian et al. (1999) located putative ubiquitylation targets downstream of the GRR in the NF-κB1 gene product and showed that these sites are important for posttranslational processing in vitro. However, the fact that sequences downstream of the GRR can be replaced with entirely foreign sequences without blocking processing (Lin and Ghosh, 1996; Lin et al., 1998) argues that additional elements directing the primary processing of p50 in vivo reside elsewhere, specifically upstream of the GRR in the RHD.
Regulation of the generation of p50 by the proteasome
Our previous results with NF-κB, and recent reports by others with the cystic fibrosis transmembrane conductance regulator and apolipoprotein B100, indicate that the proteasome can degrade proteins cotranslationally (Lin et al., 1998; Sato et al., 1998; Zhou et al., 1998). Thus, the proteasome may function during the earliest phases of protein biosynthesis, thereby utilizing proteolysis in a novel way to determine the composition of select protein complexes. In the case of NF-κB, the Ig-like folding of the RHD in the N-terminal portion of p105 may be recognized by the components of the ubiquitin–proteasome pathway and the long loosely folded GRR triggers the proteolysis. In contrast, the stably dimerized RHD may be protected from degradation by virtue of its conformation. In this manner, proteasome-mediated cleavage would generate the N-terminal polypeptide corresponding to p50 but further degradation would be blocked. Accordingly, mutations in the RHD that destabilize dimerization would render the entire protein susceptible to proteolytic attack and eliminate the production of p50. In this sense, an undimerized sd2 may function as a negative regulator that either promotes complete degradation or prevents processing. Currently, we cannot distinguish between these two possibilities.
The proteasome may be able to function cotranslationally because of its ability to interact with unfolded nascent polypeptides or unstructured regions that normally separate folded domains. Although the PA700 subcomplex of the 26S proteasome is best characterized for its interaction with polyubiquitin chains that mark proteins for degradation, recent work has demonstrated that PA700 can also interact directly with unfolded polypeptides that are not ubiquitylated (Strickland et al., 2000). Thus, regardless of whether p105 is ubiquitylated during translation, its structural features determine its susceptibility or resistance to proteolysis. Some subunits of PA700 are homologous to the translation initiation factor eIF3 (Glickman et al., 1998). Although the functional significance of this unexpected recent observation is unclear, it is intriguing to consider the possibility that the proteasome may participate in anabolic events in addition to its well recognized role in protein catabolism.
Materials and methods
Antibodies and enzymes
Antibodies specific for the p50 NLS and p65 were from Santa Cruz Biotechnology Inc.; antibodies to the T7 gp10 epitope tag were from Novagen. Proteinase K was from Sigma.
Construction of NF-κB1 mutants
All mutants were constructed using the PCR employing murine NFKB1 cDNA (Ghosh et al., 1990) as the template. The cDNAs were inserted into PEVRF mammalian expression vector or its modified form containing a T7 gp10 epitope tag, as described previously (Lin et al., 1998). For in vitro translation, cDNAs were inserted into the pET vector. All constructs used in these experiments were confirmed either by sequencing or restriction mapping. For bicistronic expression of p50, HA-tagged full-length p50 sequences were inserted into the multiple cloning sites of the Clontech vector pIRES2-EGFP, and the EGFP sequences were subsequently replaced by the sequences of the gp10-tagged shortened form of p50.
In vivo and in vitro protein expression
CHO-CD14 cells were transfected, radiolabeled with 10 mCi/ml of [35S]methionine/cysteine (Du Pont, NEN) immunoprecipitated and analyzed by SDS–PAGE as described previously (Lin et al., 1998). In vitro translations in rabbit reticulocyte lysates (Promega) and immunoprecipitations of the translated products were conducted as described previously (Lin et al., 1998).
Gel filtration chromatography
mRNAs encoding either p50 or p50 (Δ302–310) were translated in vitro at 30°C for 1 h, and then centrifuged at 14 000 g for 10 min to remove insoluble material. The supernatant was subsequently loaded with gel filtration standards (Bio-Rad) on a 1 × 50 cm column (Bio-Rad) containing packed Superdex 200 resin (Pharmacia Biotech). Fractions (0.5 ml) were collected and the elution of the molecular weight standards was identified by absorption at 280 nm. The fractions were immunoprecipitated with antibodies specific for the T7 gp10 tag, and analyzed by SDS–PAGE followed by fluorography.
Partial digestion with PK
The PK partial digestions were performed according to Frydman et al. (1994). mRNAs (20 μl) were translated in vitro at 30°C for 1 h, followed by dilution of the translation mixture in 200 μl with PK buffer [20 mM Tris pH 7.4, 80 mM KOAC, 5 mM Mg(OAC)2]. PK partial digestion (with PK present at a final concentration of 10 μg/ml) was performed at 4°C for various periods of time. At each time point, the reaction was terminated by adding the protease inhibitor phenylmethylsulfonyl fluoride (PMSF, final concentration 2 mM). The reaction mixture was then diluted with 300 ml of ELB buffer (Lin et al., 1998), immunoprecipitated and analyzed.
Sucrose gradient sedimentation
Sucrose gradient sedimentation was performed as described by Nelson et al. (1992) and Frydman et al. (1994) with minor modifications. mRNA encoding the p-780 NFKB1 protein was translated in vitro at 25°C for 20–30 min and the translation was terminated by adding CHX at a final concentration of 10 μg/ml. The lysates were then centrifuged at 14 000 r.p.m. for 10 min to remove insoluble material, and the supernatant was diluted four times with sucrose gradient buffer [20 mM HEPES buffer pH 7.4, 5 mM Mg(OAC)2, 10 mM KCl, 1 mM EGTA] and loaded on an 11 ml 20–45% sucrose gradient. After 3.75 h sedimentation at 40 000 r.p.m. in an SW41Ti rotor, 1 ml fractions were collected and examined by absorption at 260 nm. The fractions were divided into two parts; one part was immunoprecipitated directly with the anti-NLS antibodies, while the other part was incubated with PK (10 μg/ml) at 4°C for 30 min before immunoprecipitation with the same antibodies. The immunoprecipitates were analyzed on SDS–PAGE and visualized by fluorography.
Acknowledgments
Acknowledgements
We thank Dr U.Hartl for suggesting the PK PDP experiments; Dr M.Hochstrasser for suggesting the bicistronic experiment, and for his critique and discussions; Dr E.Schaeffer for comments on the manuscript; Drs G.Ghosh and J.–K.Wang for the kind gifts of p50 Y267D/L269D mutant and pIRES2-EGFP vector, respectively; and Dr W.Hansen for advice on puromycin releasing experiments. We also thank R.Givens, N.Shea, S.Gonzales, J.Carroll, C.Goodfellow, S.Ordway and G.Howard for assistance in preparing the manuscript. This work is supported in part by Gladstone Institutes and the University of California, San Francisco Center for AIDS Research (P30A127763) and by the National Institutes of Health (RO1 DK46181 to G.N.D.). L.L. is a recipient of an Investigator Award from the Arthritis Foundation.
References
- Baeuerle P.A. and Baltimore,D. (1988) IκB: a specific inhibitor of the NF-κB transcription factor. Science, 242, 540–546. [DOI] [PubMed] [Google Scholar]
- Baeuerle P.A. and Baltimore,D. (1989) A 65-kD subunit of active NF-κB is required for inhibition of NF–κB by IκB. Genes Dev., 3, 1689–1698. [DOI] [PubMed] [Google Scholar]
- Baumeister W., Walz,J., Zühl,F. and Seemüller,E. (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell, 92, 367–380. [DOI] [PubMed] [Google Scholar]
- Belich M.P., Salmerón,A., Johnson,L.H. and Ley,S.C. (1999) TPL-2 kinase regulates the proteolysis of the NF-κB-inhibitory protein NF-κB 1 p105. Nature, 397, 363–368. [DOI] [PubMed] [Google Scholar]
- Betts J.C. and Nabel,G.J. (1996) Differential regulation of NF-κB2 (p100) processing and control by amino-terminal sequences. Mol. Cell. Biol., 16, 6363–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.E., Huang,D.B., Chen,Y.Q. and Ghosh,G. (1998) Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature, 391, 410–413. [DOI] [PubMed] [Google Scholar]
- Chen Z., Hagler,J., Palombella,V., Melandri,F., Scherer,D., Ballard,D. and Maniatis,T. (1995) Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin–proteasome pathway. Genes Dev., 9, 1586–1597. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. (1998) The ubiquitin–proteasome pathway: on protein death and cell life. EMBO J., 17, 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coux O. and Goldberg,A.L. (1998) Enzymes catalyzing ubiquitination and proteolytic processing of the p105 precursor of nuclear factor κB1. J. Biol. Chem., 273, 8820–8828. [DOI] [PubMed] [Google Scholar]
- Fan C.-M. and Maniatis,T. (1991) Generation of p50 subunit of NF-κB by processing of p105 through an ATP-dependent pathway. Nature, 354, 395–398. [DOI] [PubMed] [Google Scholar]
- Frydman J., Nimmesgern,E., Ohtsuka,K. and Hartl,F.U. (1994) Folding of nascent polypeptide chains in high molecular mass assembly with molecular chaperones. Nature, 370, 111–117. [DOI] [PubMed] [Google Scholar]
- Fuchs S.Y. and Ronai,Z. (1999) Ubiquitination and degradation of ATF2 are dimerization dependent. Mol. Cell. Biol., 19, 3289–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh G., van Duyne,G., Ghosh,S. and Sigler,P.B. (1995) Structure of NF-κB p50 homodimer bound to a κB site. Nature, 373, 303–310. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Gifford,A.M., Riviere,L.R., Tempst,P., Nolan,G.P. and Baltimore,D. (1990) Cloning of the p50 DNA binding subunit of NF-κB: homology to rel and dorsal. Cell, 62, 1019–1029. [DOI] [PubMed] [Google Scholar]
- Gilmore R., Coffey,M.C., Leone,G., McLure,K. and Lee,P.W.K. (1996) Co-translational trimerization of the reovirus cell attachment protein. EMBO J., 15, 2651–2658. [PMC free article] [PubMed] [Google Scholar]
- Glickman M.H., Rubin,D.M., Coux,O., Wefes,I., Pfeifer,G., Cjeka,Z., Baumeister,W., Fried,V.A. and Finley,D. (1998) A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell, 94, 615–623. [DOI] [PubMed] [Google Scholar]
- Heissmeyer V., Krappmann,D., Wulczyn,F.G. and Scheidereit,C. (1999) NF-κB p105 is a target of IκB kinases and controls signal induction of Bcl-3–p50 complexes. EMBO J., 18, 4766–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch M., Lin,L., Geleziunas,R. and Greene,W.C. (1999) The generation of nfkb2 p52: mechanism and efficiency. Oncogene, 18, 6201–6208. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. (1995) Ubiquitin, proteasomes and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol., 7, 215–223. [DOI] [PubMed] [Google Scholar]
- Ingham P.W. (1998) Transducing hedgehog: the story so far. EMBO J., 17, 3505–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H., Claudio,E., Dambach,D., Raventos-Suarez,C. and Bravo,R. (1998) Chronic inflammation and susceptibility to bacterial infectious in mice lacking the polypeptide (p)105 precursor (NF-κB1) but expressing p50. J. Exp. Med., 187, 985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P.R., Swanson,R., Rakhilina,L. and Hochstrasser,M. (1998) Degradation signal masking by heterodimerization of MATα2 and MATa1 blocks their mutual destruction by the ubiquitin–proteasome pathway. Cell, 94, 217–227. [DOI] [PubMed] [Google Scholar]
- Kieran M. et al. (1990) The DNA binding subunit of NF-κB is identical to factor KBF1 and homologous to the rel oncogene product. Cell, 62, 1007–1018. [DOI] [PubMed] [Google Scholar]
- Lamb P. and McKnight,S.L. (1991) Diversity and specificity in transcriptional regulation: the benefits of heterotypic dimerization. Trends Biol. Sci., 16, 417–422. [DOI] [PubMed] [Google Scholar]
- Lin L. and Ghosh,S. (1996) A glycine-rich region in NF-κB p105 functions as a processing signal for the generation of the p50 subunit. Mol. Cell. Biol., 16, 2248–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L., DeMartino,G.N. and Greene,W.C. (1998) Cotranslational biogenesis of NF-κB p50 by the 26S proteasome. Cell, 92, 819–828. [DOI] [PubMed] [Google Scholar]
- Ma C.-P., Vu,J.H., Proske,R.J., Slaughter,C.A. and DeMartino,G.N. (1994) Identification, purification and characterization of a high molecular weight, ATP-dependent activator (PA700) of the 20S proteasome. J. Biol. Chem., 269, 3539–3574. [PubMed] [Google Scholar]
- MacKichan M.L., Logeat,F. and Israël,A. (1996) Phosphorylation of p105 PEST sequences via a redox-insensitive pathway up-regulates processing to p50 NF-κB. J. Biol. Chem., 271, 6084–6091. [DOI] [PubMed] [Google Scholar]
- Müller C.W., Rey,F.A., Sodeoka,M., Verdine,G.L. and Harrison,S.C. (1995) Structure of the NF-κB homodimer bound to DNA. Nature, 373, 311–317. [DOI] [PubMed] [Google Scholar]
- Naumann M., Wulczyn,F.G. and Scheidereit,C. (1993) The precursor p105 and the proto-oncogene product Bcl-3 are IκB molecules and control NF-κB nuclear translocation of NF-κB. EMBO J., 12, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R.J., Ziegelhoffer,T., Nicolet,C., Werner-Washburne,M. and Craig,E.A. (1992) The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell, 71, 97–105. [DOI] [PubMed] [Google Scholar]
- Netzer W.J. and Hartl,F.U. (1997) Recombination of protein domains facilitated by co-translational folding in eukaryotes. Nature, 388, 343–349. [DOI] [PubMed] [Google Scholar]
- Orian A., Whiteside,S., Israël,A., Stancovski,I., Schwartz,A.L. and Ciechanover,A. (1995) Ubiquitin-mediated processing of NF-κB transcriptional activator precursor p105. J. Biol. Chem., 270, 21707–21714. [DOI] [PubMed] [Google Scholar]
- Orian A., Schwartz,A.L., Israël,A., Whiteside,S., Kahana,C. and Ciechanover,A. (1999) Structural motifs involved in ubiquitin-mediated processing of the NF-κB precursor p105: roles of the glycine-rich region and a downstream ubiquitination domain. Mol. Cell. Biol., 19, 3664–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palombella V.J., Rando,O.J., Goldberg,A.L. and Maniatis,T. (1994) The ubiquitin–proteasome pathway is required for processing the NF-κB precursor protein and the activation of NF-κB. Cell, 78, 773–785. [DOI] [PubMed] [Google Scholar]
- Rice N.R., MacKichan,M.L. and Israël,A. (1992) The precursor of NF-κB has IκB-like functions. Cell, 71, 243–253. [DOI] [PubMed] [Google Scholar]
- Sato S., Ward,C.L. and Kopito,P.R. (1998) Cotranslational ubiquitination of cystic fibrosis transmembrane conductance regulator in vitro. J. Biol. Chem., 273, 7189–7192. [DOI] [PubMed] [Google Scholar]
- Scherer D.C., Brockman,J.A., Chen,Z., Maniatis,T. and Ballard,D.W. (1995) Signal-induced degradation of IκBα requires site-specific ubiquitination. Proc. Natl Acad. Sci. USA, 92, 11259–11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears C., Olesen,J., Rubin,D., Finley,D. and Maniatis,T. (1998) NF-κB p105 processing via the ubiquitin–proteasome pathway. J. Biol. Chem., 273, 1409–1419. [DOI] [PubMed] [Google Scholar]
- Sengchanthalangsy L.L., Shrimati,D., Huang,D.-B., Anderson,E., Braswell,E.H. and Ghosh,G. (1999) Characterization of the dimer interface of transcription factor NF-κB p50 homodimer. J. Mol. Biol., 289, 1029–1040. [DOI] [PubMed] [Google Scholar]
- Sha W.C., Liou,H.-C., Tuomanen,E. and Baltimore,D. (1995) Target disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell, 80, 321–330. [DOI] [PubMed] [Google Scholar]
- Shuman J.D., Vinson,C.R. and McKnight,S.L. (1990) Evidence of changes in protease sensitivity and subunit exchange rate on DNA binding by C/EBP. Science, 249, 771–774. [DOI] [PubMed] [Google Scholar]
- Spencer E., Jiang,J. and Chen,Z.J. (1999) Signal-induced ubiquitination of IκBα by the F-box protein Slimb/β-TrCP. Genes Dev., 13, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancovski I. and Baltimore,D. (1997) NF-κB activation: the IκB kinase revealed? Cell, 91, 299–302. [DOI] [PubMed] [Google Scholar]
- Strickland E., Hakala,K., Thomas,P.J. and DeMartino,G.N. (2000) Recognition of misfolding proteins by PA700, the regulatory subcomplex of the 26S proteasome. J. Biol. Chem., 275, 5565–5572. [DOI] [PubMed] [Google Scholar]
- Tjian R. and Maniatis,T. (1994) Transcriptional activation: a complex puzzle with few easy pieces. Cell, 77, 5–8. [DOI] [PubMed] [Google Scholar]
- Urban M.B. and Baeuerle,P.A. (1990) The 65-kD subunit of NF-κB is a receptor for IκB and a modulator of DNA-binding specificity. Genes Dev., 4, 1975–1984. [DOI] [PubMed] [Google Scholar]
- Watanabe N., Iwamura,T., Shinoda,T. and Fujita,T. (1997) Regulation of NF-κB1 proteins by candidate oncoprotein BCL-3: generation of NF-κB homodimers from the cytoplasmic pool of p50–p105 and nuclear translocation. EMBO J., 16, 3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Fisher,E.A. and Ginsberg,H.N. (1998) Regulated co-translational ubiquitination of apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein. J. Biol. Chem., 273, 24649–24653. [DOI] [PubMed] [Google Scholar]