

Abstract
We have previously shown that β-amyloid (Aβ) treatment resulted in an age-dependent calpain activation leading to Tau cleavage into a neurotoxic 17-kDa fragment in a cellular model of Alzheimer disease. This detrimental cellular response was mediated by a developmentally regulated increase in membrane cholesterol levels. In this study, we assessed the molecular mechanisms by which cholesterol modulated Aβ-induced Tau cleavage in cultured hippocampal neurons. Our results indicated that these mechanisms did not involve the regulation of the binding of Aβ aggregates to the plasma membrane. On the other hand, experiments using N-methyl-d-aspartic acid receptor inhibitors suggested that these receptors played an essential role in cholesterol-mediated Aβ-dependent calpain activity and 17-kDa Tau production. Biochemical and immunocytochemical analyses demonstrated that decreasing membrane cholesterol levels in mature neurons resulted in a significant reduction of the NR1 subunit at the membrane as well as an increase in the number of large NR1, NR2A, and NR2B subunit clusters. Moreover, the majority of these larger N-methyl-d-aspartic acid receptor subunit immunoreactive spots was not juxtaposed to presynaptic sites in cholesterol-reduced neurons. These data suggested that changes at the synaptic level underlie the mechanism by which membrane cholesterol modulates developmental changes in the susceptibility of hippocampal neurons to Aβ-induced toxicity.
Keywords: Alzheimer Disease, Amyloid, Cholesterol, Membrane, Synapses, Tau
Introduction
Alzheimer disease (AD)2 is a devastating disorder growing in incidence among the elderly population. The most common form of the disease occurs sporadically; however, some cases are caused by familial genetic mutations (1). Although the cause of each type of AD differs, aging is the greatest factor for both familial and sporadic AD onset (reviewed in Refs. 2, 3). Thus, it is a common goal in the AD field to understand how aging increases neuronal susceptibility to this disease process. Individuals with either form of AD exhibit characteristic morphological changes in the brain, such as senile plaque formation and the assembly of intracellular neurofibrillary tangles (reviewed in Refs. 4–6). β-Amyloid (Aβ), the prominent molecular component of senile plaques, has long been suspected as the main initiator of the AD pathogenic cascade (reviewed in Refs. 7–10). One of the means by which Aβ exerts its toxic effects is by inducing post-translational modifications of the Tau protein, such as hyperphosphorylation (reviewed in Refs. 11–14). More recently, we have shown that Tau cleavage was an alternative mechanism by which Tau mediated Aβ toxicity (15). When mature neurons were cultured in the presence of Aβ, intracellular calcium (Ca2+) concentrations became significantly elevated (16). An increase in intracellular Ca2+ caused the activation of the calpain protease leading to Tau cleavage into the neurotoxic 17-kDa fragment (15, 16). However, Aβ did not initiate this detrimental cascade in young cultured neurons (17). Furthermore, our results indicated that membrane cholesterol levels were responsible, at least in part, for regulating the age-dependent susceptibility of cells to Aβ-dependent Tau cleavage. When membrane cholesterol levels were decreased in mature neurons, Aβ treatment did not initiate Ca2+ influx, calpain activation, and 17-kDa Tau production. On the other hand, Aβ triggered the activation of this cascade in young neurons only after cholesterol had been added to their membranes (17).
In this study, we assessed the mechanisms by which membrane cholesterol content alters the susceptibility of hippocampal neurons to Aβ-induced Ca2+ influx that leads to calpain activation and Tau cleavage into a 17-kDa fragment. Our results showed that reducing membrane cholesterol levels decreased NMDA receptor content at the plasma membrane and altered NMDA receptor localization in mature hippocampal neurons. In contrast, increasing membrane cholesterol levels caused an increase in synaptic number leading to an increase in Aβ-induced Ca2+ influx in young neurons. Taken together, these results suggested that age-dependent changes in membrane cholesterol might, at least in part, modulate the susceptibility of hippocampal neurons to Aβ-induced Ca2+ influx, calpain activation, and subsequent Tau toxicity in an NMDA receptor-dependent manner.
EXPERIMENTAL PROCEDURES
Hippocampal Culture Preparation
Hippocampal neurons were isolated from embryonic day 18 Sprague-Dawley rat embryos as described previously (18). Briefly, hippocampi were dissected, and the meninges were removed. The cells were incubated for 15 min in 0.25% trypsin at 37 °C, followed by trituration with a fire-polished Pasteur pipette. The cells were plated in minimum essential medium with 10% horse serum (MEM10) on poly-l-lysine-coated dishes at high density (800,000 cells/60-mm dish). After 4 h, the medium was changed to glia-conditioned minimum essential medium containing 0.1% ovalbumin, 0.1 mm sodium pyruvate, and N2 supplements (N2 medium) (19). Neurons were also plated at low density (150,000 cells/60-mm dish) onto poly-l-lysine-coated coverslips in MEM10 for immunocytochemical analyses. After 4 h, the coverslips were transferred to dishes containing an astroglial monolayer and maintained in N2 medium. Neurons were kept in culture for 7 or 21 days to obtain young and mature cultures, respectively. Quantitative analysis showed no significant difference in the percentage of glial cells present in 21 days after plating cultures when compared with 7-day cultures (10 ± 1.2% versus 7.5 ± 1.1%, respectively; Student's t test, not significant; p > 0.05, n = 3).
Aβ Aggregation
Synthetic Aβ(1–40) (American Peptide, Sunnyvale, CA) was dissolved in N2 medium to a concentration of 0.5 mg/ml and incubated for 3 days at 37 °C to preaggregate (20). Neurons were cultured in the presence of the preaggregated peptide at final concentrations of 10 or 20 μm for 6–24 h (15).
Membrane Cholesterol Modification
Membrane cholesterol levels were modified as described previously (17). In short, 21 days in culture (mature) neuron membrane cholesterol was decreased to levels comparable with 7 days in culture (young) neurons using 2 μm methyl-β-cyclodextrin (MBCD; Sigma) for 30 min (17). To increase young neuron membrane cholesterol levels to that of mature cells, young neurons were treated with 30 μm cholesterol from a complex of MBCD and cholesterol (MBCD/cholesterol; Sigma) together with 2 μg/ml free cholesterol for 1 h (17). These agents were removed from the media for the duration of subsequent treatments.
NMDA Receptor Inhibition
NMDA receptor activity was blocked using memantine (Sigma), a universal NMDA receptor antagonist, or ifenprodil (Sigma), an NR2B-specific NMDA receptor antagonist. These agents were added to the culture medium of untreated and cholesterol-modified hippocampal neurons 1 h prior to the addition of Aβ at a final concentration of 10 and 5 μm, respectively. The antagonists remained in the culture medium for the duration of the Aβ incubation.
Subcellular Fractionation
Young and mature hippocampal neurons underwent subcellular fractionation to obtain cytosol and membrane fractions as described previously (16, 21, 22). Briefly, cells were scraped in 5 mm EDTA in phosphate-buffered saline (PBS) and pelleted by centrifugation for 10 min at 2300 × g at 4 °C. Pellets (P1) were resuspended in 100 μl of fractionation buffer (0.25 m sucrose, 1 mm magnesium chloride (MgCl2), 2 mm EGTA, 25 mm HEPES, pH 7.4) and lysed by three cycles of flash-freezing in liquid nitrogen. Lysates were then centrifuged at 100,000 × g for 30 min in a Beckman Airfuge (Beckman Coulter, Fullerton, CA), and the supernatants (cytosol fraction) were removed. The membrane-containing pellet (P2) was resuspended in 100 μl of fractionation buffer and an equivalent volume of Laemmli buffer (23). Total protein content was assessed by the modified Lowry technique to load equal protein for Western blotting (see below) (24, 25).
Detergent-resistant Membrane (DRM) Fraction Isolation
DRMs were isolated from the membranes of young and mature hippocampal neurons (26). Briefly, the P2 fraction obtained during subcellular fractionation was resuspended in ice-cold DRM buffer containing Triton X-100 (5 mm EDTA, 10 mm HEPES, 150 mm sodium chloride (NaCl), 1 mm phenylmethanesulfonyl fluoride, 0.01 mg/ml aprotinin, 1% protease inhibitor mixture, 0.5% Triton X-100). The membranes were incubated on ice for 30 min, and the remaining DRM fraction was pelleted by centrifugation at 13,000 × g for 30 min at 4 °C. The detergent-soluble membrane fraction (DSM; supernatant) was removed, and an equivalent volume of Laemmli was added. The DRM pellet was resuspended in the same DRM buffer volume as the DSM fraction, and an equivalent amount of Laemmli was added. Whole membrane fraction controls were prepared with the same number of cells in which the P1 fraction was dissolved in equal volume DRM buffer to Laemmli buffer.
Amplex Red Cholesterol Determination
Membrane cholesterol content was quantified using the Amplex Red cholesterol assay (Molecular Probes, Eugene, OR) per the manufacturer's instructions. Samples were diluted in reaction buffer, and an equivalent volume of Amplex Red working solution (300 μm Amplex Red, 2 units/ml cholesterol oxidase, 2 units/ml cholesterol esterase, and 2 units/ml horseradish peroxidase) was added. The samples were incubated for 30 min at 37 °C, after which the sample absorbance was measured at 568 nm using a Tecan Infinite M200 microplate reader and i-Control software (Tecan, Research Triangle Park, NC). Cholesterol levels were calculated using standard cholesterol solutions. The obtained values were normalized to protein content as measured by the modified Lowry technique (24, 25).
Immunocytochemistry
Young and mature hippocampal neurons were cultured on coverslips and treated with or without cholesterol-modifying agents, after which they were fixed in 4% paraformaldehyde in PBS containing 0.12 mm sucrose for 15 min and permeabilized in 0.3% Triton X-100 in PBS for 4 min. For some experiments, cells were fixed in ice-cold methanol for 10 min. Coverslips were then incubated in 10% bovine serum albumin in PBS at room temperature for 1 h before labeling with the primary antibody. The primary antibodies used were anti-tubulin (clone DM1A; 1:1000; Sigma), anti-synaptophysin (1:500, Santa Cruz Biotechnology, Santa Cruz, CA), and antibodies against the NMDA receptor subunits as follows: anti-NR1 (1:50; Santa Cruz Biotechnology), anti-NR2A (1:50; Santa Cruz Biotechnology), and anti-NR2B (1:50; BD Biosciences). Biotin-conjugated anti-goat (1:50; Invitrogen) or anti-mouse (1:50; Chemicon, Temecula, CA) secondary antibodies followed by fluorescein-avidin (1:50; Vector Laboratories, Burlingame, CA) incubation were used to detect NMDA receptor subunits. For all other primary antibodies, we used AlexaFluor secondary antibodies (1:200; Molecular Probes). Images were taken using a Photometrics Cool Snap HQ2 camera coupled to a fluorescent microscope (Nikon Diaphot, Melville, NY). Images were analyzed using MetaMorph Image Analysis software (Universal Imaging Corporation, Fryer Co. Inc., Huntley, IL).
Electrophoresis and Immunoblotting
Whole cell, membrane, DRM, or DSM fraction lysates were loaded and run on SDS-polyacrylamide gels as described previously (23). The proteins were transferred onto Immobilon membranes (Millipore, Billerica, MA) and immunoblotted (20, 27). Primary antibodies used for immunodetection were as follows: anti-Aβ (clone 6E10; 1:1000; Covance, Gaithersburg, MD), phosphorylation-independent anti-Tau (clone Tau5; 1:1000; BioSource International, Foster City, CA), anti-spectrin (1:1000; Chemicon), anti-tubulin (clone DM1A; 1:200,000; Sigma), anti-NR1 (1:400; Santa Cruz Biotechnology), anti-NR2A (1:100; Santa Cruz Biotechnology), and anti-NR2B (1:200; BD Biosciences) antibodies. Incubation with secondary antibodies conjugated to horseradish peroxidase (1:1000; Promega, Madison, WI) preceded enhanced chemiluminescence for protein detection (28). A ChemiDoc XRS system and Quantity One Software (Bio-Rad) were used to image and analyze immunoreactive bands. Whole cell lysates were loaded using tubulin as an internal control, whereas membrane fractions were loaded by total protein as assessed by the modified Lowry assay (24, 25). Equal loading volumes of DRM and DSM samples were used to compare protein content between the two fractions.
Fura-2 Ca2+ Imaging
Young and mature neurons cultured on coverslips were treated with Aβ, cholesterol-modifying drugs, memantine, ifenprodil, or a combination of these substances. The cells were loaded with 2 μm fura-2-AM (Invitrogen) for 15 min at 37 °C, washed, and incubated for an additional 15 min at 37 °C to allow de-esterification of the AM ester (29, 30). The cells were mounted in a Series 20 chamber (Warner Instruments, Hamden, CT) in HEPES buffer (5 mm KCl, 140 mm NaCl, 2 mm calcium chloride, 1 mm MgCl2, 10 mm glucose, and 10 mm HEPES, pH 7.4). An inverted microscope (Nikon Diaphot) connected to a Photometrics Cool Snap HQ2 camera and MetaMorph/Metafluor Image Analysis software (Universal Imaging Corp.) were used for imaging. Base-line Ca2+ concentrations were established by acquiring 60-ms exposures every 10 s for 15 min (17, 31–33). Fura bound or unbound to Ca2+ was quantified by establishing a ratio between its fluorescence at 510 nm post-excitation at 340 and 380 nm, respectively. The calculated 340:380 ratio was used to quantify intracellular Ca2+ levels by comparing the ratio to that of standard Ca2+ solutions.
NMDA Receptor Puncta Analysis and Synapse Counts
NMDA receptor subunit antibodies were utilized for the detection of these proteins in young and mature hippocampal neurons in culture (see above). The resulting immunofluorescent spots per cell were counted, and their area was determined using MetaMorph Image Analysis software (Universal Imaging Corp.). The number of synapses per neuron was determined by double labeling young cultured hippocampal neurons with anti-synaptophysin and anti-tubulin antibodies as described above. The number of synapses per cell was calculated by counting the synaptophysin immunoreactive puncta using MetaMorph Image Analysis software and dividing these values by the total number of cells as observed with tubulin labeling.
Statistical Analyses
All experiments performed in this study were conducted in five independent cultures. The compiled data were analyzed across the experimental conditions using either a Student's t test (when comparing only two experimental conditions) or one-way analysis of variance followed by Fisher's LSD post hoc test (when comparing more than two experimental conditions). The values in the graphs represent the means ± S.E. The statistical significance is indicated in the graphs.
RESULTS
Amount of Membrane-bound Aβ Was Independent of Cholesterol Levels in Cultured Hippocampal Neurons
Our previous findings indicated that an age-dependent increase in membrane cholesterol modulated developmental changes in Aβ-induced Tau cleavage by regulating Ca2+ influx and calpain activation (17). However, the mechanisms underlying these effects are not completely known. Several studies have identified cholesterol as a facilitator of membrane-Aβ interactions, an essential step leading to its toxic effects (34–40). Thus, we assessed whether age-dependent changes in membrane cholesterol correlated with changes in the amount of membrane-bound Aβ in cultured hippocampal neurons. Membrane fractions from Aβ-treated young and mature neurons were isolated and immunoblotted using an antibody against this peptide. Low molecular weight Aβ immunoreactive bands corresponding to small oligomeric species were detected in the membrane fractions of both young and mature hippocampal neurons. However, quantitative analysis of Aβ band density indicated that membranes from mature neurons cultured in the presence of this peptide contained significantly more Aβ than young ones (Fig. 1, A and D). To assess whether membrane cholesterol levels determine the amount of Aβ associated with neuronal membranes, we pharmacologically reduced its levels in mature neurons to equal that of young cells and increased young neuron membrane cholesterol levels to equal that of mature ones using MBCD and a cholesterol complex, respectively, as described previously (17, 41). Treatment of mature neurons with 2 mm MBCD sufficiently decreased their membrane cholesterol content to the level of young neurons (52.6 ± 5.0 versus 58.8 ± 2.7 ng of cholesterol per μg of membrane protein, respectively. Student's t test, not significant; p > 0.05, n = 5) (see also Ref. 17). On the other hand, 30 μm complexed cholesterol increased young neuron membrane cholesterol to levels comparable with that of mature cells (81.3 ± 3.5 versus 85.4 ± 3.0 ng per μg of membrane protein, respectively. Student's t test, not significant; p > 0.05, n = 5) (see also 17). After membrane cholesterol levels were modified, neurons were incubated with Aβ and membrane fractions prepared 6 h later (36). Quantitative Western blot analysis showed no significant changes in membrane-bound Aβ content in MBCD-treated mature neurons nor in cholesterol-treated young ones cultured in the presence of Aβ when compared with neurons treated with Aβ alone (Fig. 1, B, C, E, and F). These results indicated that the developmental changes in the amount of Aβ associated with neuronal membranes were independent of their cholesterol content.
FIGURE 1.

Age-related changes in Aβ-membrane binding were independent of membrane cholesterol content. A–C, membrane samples were prepared from untreated young and mature cultured neurons (A), mature neurons (B) treated with Aβ (20 μm) for 6 h after preincubation with (+) or without (−) MBCD (2 mm), or young neurons (C) treated with Aβ (20 μm) for 6 h after preincubation with (+) or without (−) cholesterol (30 μm). These lysates were loaded by total protein and immunoblotted using an antibody against Aβ (clone 6E10). Although more Aβ was bound to mature versus young membranes, changing membrane cholesterol content did not alter Aβ immunoreactivity for either age. D–F, quantitative analysis of membrane-bound Aβ from cells treated as described above. Values represent the mean ± S.E. from five independent experiments per condition. *, differed from 7 days in culture cells, p < 0.05. C, control; CH, cholesterol.
Membrane Cholesterol-regulated Aβ-induced Tau Cleavage in an NMDA Receptor-dependent Manner
We assessed next whether membrane cholesterol mediated 17-kDa Tau production by affecting NMDA receptors at the cell surface because these receptors were identified as the primary players in Aβ-induced Ca2+ influx leading to calpain activation in mature hippocampal cultures (16, 42). For these experiments, we harvested whole cell lysates from mature neurons that had been previously treated in the absence or presence of Aβ, the universal NMDA receptor antagonist memantine, or both. Blots of these samples were labeled with a phosphorylation-independent Tau antibody (Tau5), which detects both full-length Tau (∼48 and ∼66 kDa) as well as the 17-kDa Tau fragment (15, 17). As we have shown previously, Aβ treatment caused a reduction in full-length Tau immunoreactivity and a concomitant increase in 17-kDa Tau band density leading to an increase in the overall 17-kDa/full-length Tau ratio as compared with untreated controls (Fig. 2, A and B) (17). In contrast, no reduction of full-length Tau was detected, and the production of 17-kDa Tau was almost completely abrogated when memantine was added to the culture medium of mature hippocampal neurons 1 h before the addition of Aβ. Therefore, the 17-kDa/full-length ratio in these neurons was significantly lower than in neurons treated with Aβ alone and comparable with those of controls and cells treated with memantine alone (Fig. 2, A and B; see also supplemental Fig. 1).
FIGURE 2.

Memantine significantly hindered Aβ-induced 17-kDa Tau generation, calpain activation, and Ca2+ influx in mature neurons. A, immunoblot analysis of 17-kDa Tau production and calpain activity, by means of spectrin (spec) degradation, in whole cell lysates prepared from mature hippocampal cultures incubated in the presence (+) or absence (−) of Aβ peptide (20 μm), memantine (10 μm), or both. The 17-kDa Tau fragment and calpain activation were nearly absent in Aβ-treated lysates that were preincubated with memantine as compared with those treated with Aβ alone. B, quantification of the 17-kDa/full-length Tau ratio as well as the 150:240-kDa spectrin ratio in whole cell lysates treated as described in A. Tubulin (tub) was used as an internal control, and the values were expressed as a percent of Aβ-treated neurons. C, using fura-2 imaging, intracellular Ca2+ levels were calculated in live mature neurons treated as mentioned in A after their base-line levels had been established. Ca2+ influx induced by Aβ treatment was abrogated in cells that were pretreated with memantine. Values represent the mean ± S.E. from five independent experiments per condition. *, differs from Aβ-treated cells (B) or untreated controls (C), p < 0.01. Mem, memantine.
We next examined whether the preventive effect of this NMDA receptor antagonist treatment on Aβ-dependent 17-kDa Tau production correlated with a decrease in Aβ-induced calpain activation as assessed by spectrin degradation. Spectrin cleavage from its full-length form (240 kDa) to a 150-kDa fragment is specific for calpain activity and is commonly used as a surrogate marker of this protease's activity (15, 43). Blotting membranes were probed with a spectrin antibody, and a ratio between calpain-cleaved and full-length spectrin was established. Comparable with what we have previously reported, a strong spectrin immunoreactive band at 240 kDa and very little of the 150-kDa spectrin cleavage product were detected in untreated mature neurons, whereas the opposite pattern was observed in Aβ-treated neurons leading to an increase in the 150:240-kDa spectrin ratio indicative of increased calpain activation (Fig. 2, A and B) (17). In contrast, preincubation with memantine significantly attenuated Aβ-induced calpain activation as shown by decreased 150-kDa spectrin immunoreactivity and maintained normal full-length spectrin levels resulting in a 150:240-kDa spectrin ratio similar to that of untreated controls or neurons treated only with memantine (Fig. 2, A and B).
To address whether the NMDA receptor antagonist-dependent decrease in Aβ-induced calpain activation leading to the 17-kDa Tau production was the result of decreased extracellular Ca2+ influx, we performed fura-2 imaging in live mature neurons treated as described above. Ca2+ concentrations in fura-2-loaded cells were determined using Metafluor Image Analysis software (see “Experimental Procedures”). Untreated neurons displayed basal Ca2+ levels (111 ± 14 nm), whereas intracellular Ca2+ in Aβ-treated cells was nearly four times greater than those of untreated neurons (Fig. 2C). On the other hand, no changes in intracellular Ca2+ levels were detected in mature neurons treated with memantine before the addition of Αβ when compared with untreated controls or neurons incubated with memantine alone (Fig. 2C). Similar changes in Tau cleavage, calpain activation, and intracellular Ca2+ concentrations were detected when hippocampal neurons were treated with ifenprodil, an NR2B antagonist, before the addition of Aβ (supplemental Fig. 2, A–C).
We next determined to what extent NMDA receptors were also involved in Aβ-induced calpain activation and 17-kDa Tau production and in cholesterol-enriched young neurons. For these series of experiments, young neurons were treated with Aβ, cholesterol, memantine, or a combination of these agents, after which lysates were prepared and immunoblotted with Tau and spectrin antibodies. Tau cleavage into the 17-kDa fragment was not detected in control and Aβ-treated lysates, whereas a faint Tau immunoreactive band at 17 kDa was observed in cells treated with cholesterol alone. In agreement, only basal levels of active calpain and intracellular Ca2+ were detected in young cells under these experimental conditions (Fig. 3, A–C). Young neurons treated with cholesterol and Aβ revealed a decrease in full-length Tau immunoreactivity and an enhanced Tau-reactive band at 17 kDa resulting in an increased 17-kDa/full-length Tau ratio when compared with young neurons treated with cholesterol alone (Fig. 3, A and B) (17). The production of the 17-kDa Tau fragment was prevented when cholesterol-treated young neurons were incubated with memantine prior to Aβ treatment. Under these same experimental conditions, memantine also attenuated the Aβ-induced calpain activation as indicated by reduced spectrin cleavage from 240 to 150 kDa (Fig. 3, A and B). Finally, in young neurons pretreated with cholesterol, memantine also reduced intracellular Ca2+ levels caused by Aβ to levels comparable with untreated controls (124 ± 12 nm versus 117 ± 10 nm, respectively) (Fig. 3C).
FIGURE 3.

Memantine reduced Aβ-dependent 17-kDa Tau production, calpain activation, and Ca2+ influx in cholesterol-enriched neurons. A, whole cell lysates prepared from young hippocampal cultures incubated in the presence (+) or absence (−) of Aβ peptide (20 μm), cholesterol (30 μm), memantine (10 μm), or a combination of these agents were blotted with Tau and spectrin (spec) antibodies to detect 17-kDa Tau production and calpain activation, respectively. Memantine greatly reduced Aβ-dependent Tau cleavage and calpain activity in cholesterol-enriched cells. B, quantification of the 17-kDa/full-length Tau ratio as well as the 150:240-kDa spectrin ratio in whole cell lysates treated as described in A. Tubulin (tub) was used as an internal control, and the values were expressed as a percent of Aβ-treated neurons. C, fura-2 imaging was used to quantify intracellular Ca2+ levels in young neurons treated as mentioned in A after their base-line levels had been established. The significant increase in Ca2+ influx caused by Aβ treatment of cholesterol-enriched neurons was prevented with memantine pretreatment. Values represent the mean ± S.E. from five independent experiments per condition. *, differs from Aβ-treated cells (B) or untreated controls (C), p < 0.01. Mem, memantine; CH, cholesterol; N/D, not detectable.
Membrane Cholesterol Alterations Induced Synapse Formation Only in Young Neurons
The results described above suggested that NMDA receptors were involved in Aβ-induced Ca2+ influx and subsequent calpain activation and Tau cleavage mediated by age-dependent or pharmacologically induced changes in membrane cholesterol. Because cholesterol is a necessary component for synaptic formation and maintenance, it was possible that changes in its levels might affect the total number of synapses and, hence, functional membrane NMDA receptors (44–48). To address this possibility, we labeled control and cholesterol-altered mature and young neurons with an antibody specific for synaptophysin, an integral synaptic vesicle membrane protein commonly used as a synaptic marker (49–51). Synaptophysin immunoreactive puncta were easily detectable around cell bodies and along the neurite processes extended by both mature and young untreated hippocampal neurons (supplemental Fig. 3, A–H). However, the number of these immunoreactive spots was significantly higher in mature neurons when compared with young ones (113 ± 6 versus 71 ± 4 synapses per cell, respectively; p < 0.01) (supplemental Fig. 3, G, I and J). The number of synapses formed by mature neurons was not significantly changed when their cholesterol content was decreased by MBCD (118 ± 6 synapses per cell; see also supplemental Fig. 3J). In contrast, quantitative analysis showed a significant increase in the number of synaptophysin immunoreactive puncta in cholesterol-enriched young neurons when compared with their age-matched untreated controls (85 ± 2 synapses per cell; p < 0.05; see also supplemental Fig. 3I).
Reduction in Membrane Cholesterol Decreased NR1 Content and Altered the Localization of NMDA Receptor Subunits in Membrane Microdomains of Mature Neurons
We analyzed next whether altering cholesterol content modifies one or more of the NMDA receptor subunits at the cell surface. NMDA receptors expressed in hippocampal neurons are composed of at least one NR1 subunit and two or more NR2A and NR2B subunits (52, 53). The NR2A subunit increased developmentally with age in cultured hippocampal neuron whole cell lysates (Fig. 4, A and C). Furthermore, both the NR1 and NR2A content in cell membranes increased in an age-dependent manner (Fig. 4, B and D). Quantitative immunoblotting experiments showed no changes in the NR2A or NR2B levels when the content of cholesterol was reduced in mature hippocampal neurons (Fig. 5A). However, decreasing cholesterol in these cells significantly decreased the NR1 subunit content as compared with untreated controls (Fig. 5A). In contrast, immunoblotting analysis of membrane fractions detected no change in NR1, NR2A, or NR2B content in cholesterol-treated young neurons as compared with controls (Fig. 5B).
FIGURE 4.

NMDA receptor content changed in hippocampal neurons as they aged in culture. A and B, 7 and 21 days in culture neuron whole cell lysates (A) and membrane fractions (B) were immunoblotted using antibodies against NMDA receptor (NR) subunits as follows: NR1, NR2A, and NR2B. Samples were loaded using tubulin as an internal control (A) or by total protein (B). C and D, NMDA receptor subunit bands were quantified and compared between young (white bars) and mature (black bars) cells. Mature neuron whole cell fractions (C) contained more NR2A, whereas their membranes fractions (D) revealed more NR1 and NR2A immunoreactivity than young neurons. Values in the graphs represent the mean ± S.E. from five independent experiments per condition. Quantification from 7 days in culture lysates was considered 100%. * differs from young neurons, p < 0.01.
FIGURE 5.

Membrane cholesterol modifications induced changes in NMDA receptor content in mature but not young hippocampal neurons. A and B, membrane fractions obtained from mature (A) and young (B) neurons in culture treated with (+) or without (−) MBCD (2 mm) or cholesterol (30 μm) immunoblotted with NMDA receptor (NR) subunit antibodies as follows: NR1, NR2A, and NR2B. Receptor subunit band densities were quantified and compared between control (white bars) and treated (black bars) cells. NR1 content at the membrane was decreased after MBCD treatment in mature cells, whereas no significant change in NMDA receptor subunit content was observed in cholesterol-treated young cells. Samples were loaded by total protein. Values in the graphs represent the mean ± S.E. from five independent experiments per condition. Untreated age-matched controls were considered 100%. * differs from control, p < 0.05. CH, cholesterol.
We next performed immunocytochemical and biochemical experiments to determine to what extent membrane cholesterol changes affected NMDA receptor subunit localization at the cell surface. For immunolabeling experiments, neurons were fixed with paraformaldehyde or methanol and labeled with NR1-, NR2A-, or NR2B-specific antibodies that recognize extracellular epitopes. Untreated and cholesterol-modified mature neurons displayed disperse, bright NMDA subunit receptor-immunoreactive punta along the neuronal processes (Fig. 6, A–C). Quantification of the total number of puncta per cell showed that NR1 and NR2B values were unchanged in cholesterol-reduced mature neurons as compared with their age-matched controls (Table 1). In contrast, the total number of NR2A puncta per cell in MBCD-treated neurons was reduced by nearly 45% (Table 1). In addition, many of the NR1, NR2A, and NR2B immunoreactive puncta were of a noticeably larger size in MBCD-treated mature neurons (Fig. 6, A–C). Therefore, we examined the relative size of the NMDA receptor subunit puncta in control and MBCD-treated cells using MetaMorph Image Analysis software. The area of the majority of the observed puncta for each receptor subunit ranged from 0.3 to 3 μm2 under all experimental conditions analyzed. However, the percentage of larger NR1, NR2A, and NR2B puncta (area ranging from 3 to 6 μm2) nearly doubled in cholesterol-reduced mature neurons as compared with untreated ones (Table 1). There was also a 2-fold increase in the largest puncta of NR1 (6–10 μm2) when MBCD-treated cells were compared with controls (Table 1). To determine whether the larger (>3 μm2) NMDA receptor subunit puncta were located at places of synaptic contact, we double-labeled mature neurons treated with or without MBCD with NR1, NR2A, or NR2B and synaptophysin antibodies. Under normal conditions, mature neuron NR1 and synaptophysin immunoreactive spots colocalized along the neuronal processes (Fig. 6D). In contrast, the majority (∼60%) of the larger puncta for each NMDA receptor subunit observed in cholesterol-reduced hippocampal neurons was not juxtaposed to synaptophysin-labeled presynaptic sites (Fig. 6D). A similar pattern was detected when neurons were stained using NR2A or NR2B antibodies and the anti-synaptophysin one (data not shown).
FIGURE 6.
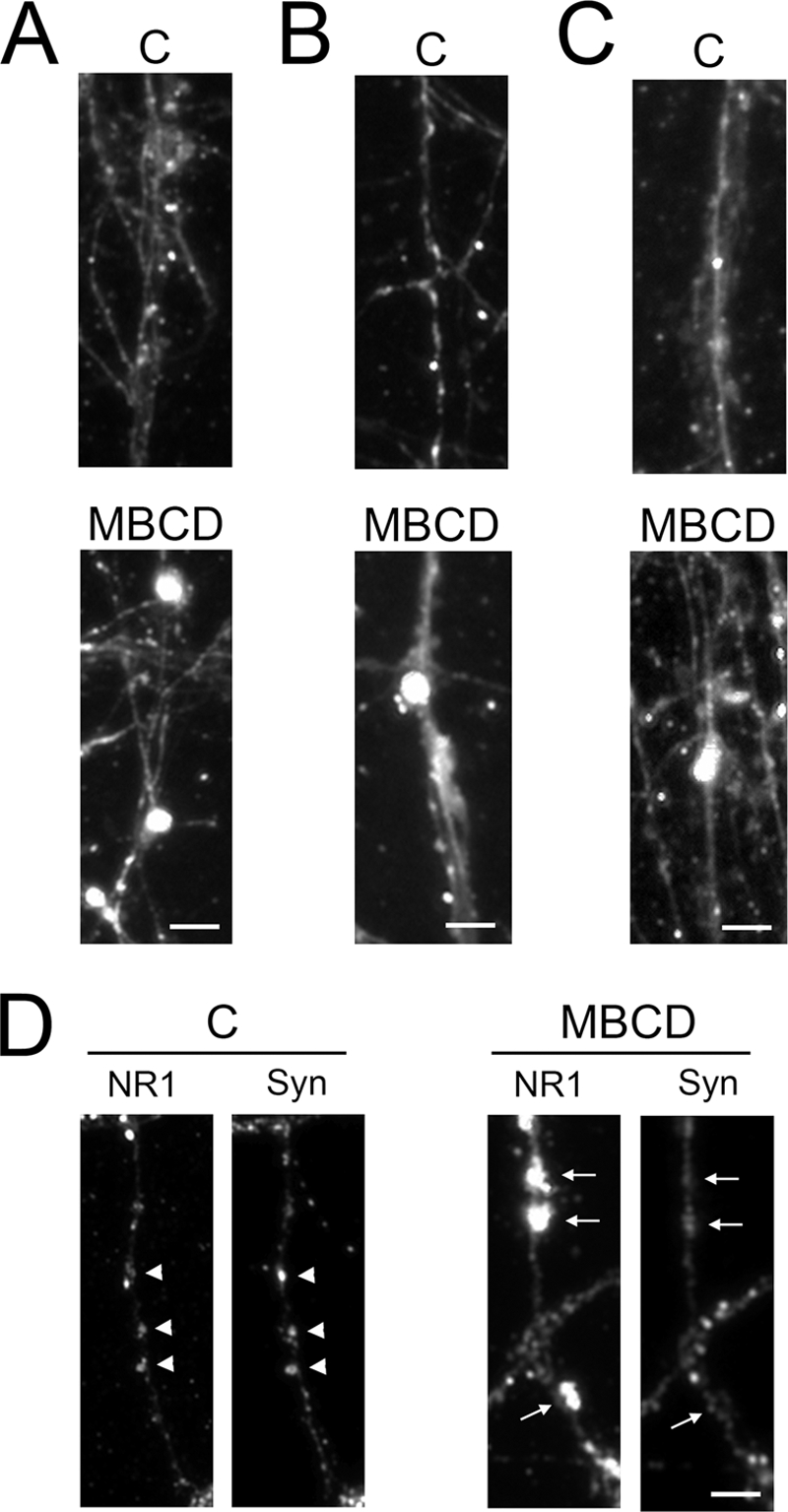
Decreasing membrane cholesterol altered NMDA receptor cluster size and synaptic localization. A–C, control and MBCD-treated mature neurons were fixed and labeled with antibodies against the NMDA receptor (NR) subunits as follows: NR1 (A), NR2A (B), and NR2B (C). Labeling with antibodies against each of these subunits revealed larger puncta in the neuron processes in MBCD-treated cells as compared with controls. D, mature neurons were incubated in the absence or presence of MBCD and double-labeled with NR1 and synaptophysin antibodies to determine whether the large NR1 puncta were located in sites juxtaposed (arrowheads) or away from (arrows) presynaptic terminals. Control neuron NR1 puncta were at synaptic sites, whereas the large NR1 puncta observed in MBCD-treated cells were not. Scale bars, 5 μm. C, control; NR1, NR1 subunit; Syn, synaptophysin.
TABLE 1.
NMDA receptor subunit puncta size was increased after cholesterol reduction in mature hippocampal neurons
Twenty one days in culture hippocampal neurons were treated with or without MBCD, fixed, and labeled with antibodies against the NMDA receptor subunits. Cells were imaged using a Nikon inverted microscope connected to a PhotoSnap HQ2 camera. Puncta number and size were analyzed using MetaMorph software; n = 5 per condition. The results represent the mean ± S.E.
| Receptor subunit | Treatment | Puncta/cell | Puncta size (% of total) |
||
|---|---|---|---|---|---|
| 0.3–3 μm2 | 3–6 μm2 | 6–10 μm2 | |||
| NR1 | None | 212 ± 33 | 91.1 ± 0.6 | 6.5 ± 0.4 | 2.3 ± 0.3 |
| MBCD | 211 ± 9 | 82.6 ± 0.6a | 13 ± 0.3a | 4.4 ± 0.4a | |
| NR2A | None | 335 ± 49 | 94.3 ± 0.8 | 4.2 ± 0.6 | 1.5 ± 0.1 |
| MBCD | 185 ± 37b | 90.6 ± 0.9a | 7.5 ± 0.9a | 1.9 ± 0.3 | |
| NR2B | None | 149 ± 30 | 93.5 ± 0.9 | 4.9 ± 0.6 | 2 ± 0.3 |
| MBCD | 241 ± 55 | 92.5 ± 0.2 | 6.2 ± 0.3b | 1.3 ± 0.2a | |
a Values differ from untreated cells, p < 0.01.
b Values differ from untreated cells, p < 0.05.
Finally, we assessed whether cholesterol content modulated the distribution of each NMDA receptor subunit in DRM and DSM domains. For these experiments, each fraction was immunoblotted using antibodies against the NMDA receptor subunits, and a ratio was established between subunit expression in the DRM and DSM microdomains. As shown in Table 2, the DRM/DSM distribution ratio of the NR1 subunits in MBCD-treated mature neurons was increased nearly 70% as compared with control cells. This ratio change was due in part to an increase in NR1 content in DRMs and an even greater decrease in NR1 subunit content in the DSM fractions (data not shown). Contrastingly, the distribution of the NR2A and NR2B subunits remained unchanged with respect to untreated age-matched control neurons (Table 2). No changes in the overall distribution of NR1, NR2A, or NR2B subunits between DRM and DSM fractions were detected in cholesterol-treated young neurons when compared with untreated controls (Table 2).
TABLE 2.
NMDA receptor distribution in membrane microdomains after cholesterol modifications
Hippocampal neurons were cultured for 21 or 7 days and treated with or without either MBCD (2 mm) or cholesterol (30 μm), respectively. DRM and DSM fractions were isolated and immunoblotted with NMDA receptor subunit antibodies. Band densities were determined and expressed as a ratio between the DRM and DSM fractions. The results represent the mean ± S.E., n = 5 per condition. Untreated age-matched control values were considered 100%.
| Days in culture | Treatment | Receptor subunit | DRM:DSM Ratio |
|---|---|---|---|
| % control | |||
| 21 | MBCD | NR1 | 167 ± 26a |
| 21 | MBCD | NR2A | 99 ± 8 |
| 21 | MBCD | NR2B | 90 ± 5 |
| 7 | Cholesterol | NR1 | 90 ± 8 |
| 7 | Cholesterol | NR2A | 107 ± 20 |
| 7 | Cholesterol | NR2B | 99 ± 11 |
a Values differ from age-matched controls, p < 0.01.
DISCUSSION
The data described above suggest that age-dependent differences in membrane cholesterol content modulated the susceptibility of hippocampal neurons to Aβ-induced Tau cleavage by modifying synaptic sites. In addition, our results provide insights into a dual mechanism by which cholesterol regulates this Aβ-induced neurodegenerative pathway. Pharmacological lowering of membrane cholesterol in mature neurons reduced NMDA receptor content and changed their localization in membrane microdomains. On the other hand, increasing membrane cholesterol in young neurons caused a significant increase in synaptic number and hence the functional NMDA receptors in these cells. Both cholesterol-mediated changes at the synaptic level altered, at least in part, the ability of Aβ to induce Ca2+ influx leading to calpain activation and cleavage of Tau into a neurotoxic 17-kDa fragment.
Recently, we have shown that membrane cholesterol played a critical role in determining the age-dependent susceptibility of cultured hippocampal neurons to Aβ-induced Tau toxicity. Those results indicated that Aβ treatment caused a significant increase in intracellular Ca2+ levels leading to the activation of calpain and subsequent cleavage of the Tau protein into a neurotoxic 17-kDa fragment only in mature neurons (17). This increase in susceptibility of mature neurons to Aβ-induced neurotoxicity occurred concomitantly with an age-dependent increase in membrane cholesterol levels. Furthermore, decreasing membrane cholesterol levels in mature neurons to the levels detected in their young counterparts attenuated their susceptibility to Aβ-dependent Tau toxicity. On the other hand, increasing membrane cholesterol in young neurons to levels similar to those of mature neurons made them more susceptible to the detrimental effects of Aβ (17).
The mechanisms by which membrane cholesterol modulates Aβ-induced Ca2+ influx leading to calpain activation and Tau dysfunction remain poorly understood. The data presented here provide insights into such mechanisms. Our results showed that the binding of Aβ to neuronal membranes was developmentally regulated. Furthermore, the amount of membrane-bound Aβ correlated with the extent of Aβ toxicity mediated by Tau cleavage in these neurons (17). Based on these results, it is tempting to speculate that cholesterol might modulate the susceptibility of developing hippocampal neurons to Aβ-induced toxicity by determining the extent of Aβ-membrane interactions. Previous reports have shown that Aβ binds specifically to lipid raft areas of the plasma membrane, although it remains controversial whether cholesterol is necessary for this interaction (36–40, 54–56). In our model system, changes in membrane cholesterol levels did not affect the amount of membrane-bound peptide either in mature or young neurons. These results suggested that Aβ-membrane binding was cholesterol-independent in hippocampal neurons. These data are in agreement with previous reports identifying raft components other than cholesterol as the mediator in Aβ-membrane interactions (36, 56).
Alternatively, cholesterol might mediate developmental changes in the susceptibility of hippocampal neurons to Aβ-induced neurotoxicity by regulating the molecular mechanisms involved in Ca2+ homeostasis. We have previously shown that Aβ induced a sustained increase in Ca2+ levels only in mature neurons (17). This increase in Ca2+ levels was the result of enhanced extracellular Ca2+ influx and not of its mobilization from intracellular storage sites (42). Furthermore, the NMDA glutamate receptors seemed to be involved in this mechanism (42). A growing body of evidence implicates glutamate receptors as key players in the excitotoxic events that underlie AD and other neurodegenerative diseases (reviewed in Refs. 57–59). However, little is known about their role in determining the age-dependent susceptibility of central neurons to these neurodegenerative processes. The results presented in this study showed an increase in NMDA receptor content at the cell surface in mature neurons as compared with young ones. These data extended previous reports indicating that the expression of the different NMDA receptor subunits was developmentally regulated in the brain (30, 52, 53, 60–71). In addition, this study provides more direct evidence of the role of NMDA receptors in Aβ-dependent neurodegeneration mediated by Tau cleavage into the neurotoxic 17-kDa fragment in hippocampal neurons. Pretreatment of mature neurons with the NMDA receptor antagonist memantine inhibited the aforementioned cellular cascade triggered by Aβ treatment. Memantine also attenuated Aβ-dependent Ca2+ influx, calpain activity, and Tau cleavage in cholesterol-enriched young neurons.
One possible mechanism by which cholesterol could modulate NMDA receptor activity could involve the regulation of synaptogenesis and hence the formation of clusters of NMDA receptors juxtaposed to presynaptic terminals. Previous reports have suggested that cholesterol is a necessary component for synaptic development and maintenance (44–48, 72). This seems to be the case in young hippocampal neurons with pharmacologically enhanced membrane cholesterol content. In these cells, quantitative analysis showed a significant increase in the number of synaptic sites per cell as compared with untreated controls. On the other hand, no changes in the number of synaptic contacts were detected when cholesterol content was decreased by MBCD in mature neurons. These results suggested that an alternative mechanism(s) might underlie cholesterol regulation of NMDA receptor-mediated Ca2+ influx in mature neurons treated with Aβ. This mechanism(s) could involve changes in the overall levels or the localization of NMDA receptor in the membrane potentially leading to altered excitability. Our findings indicated that the reduction of membrane cholesterol caused a significant decrease in NR1 content at the cell surface in MBCD-treated mature neurons. This change in NR1 content was not accompanied by modifications of the amount of NR2 subunits in the membrane. Nevertheless, the decrease in membrane NR1 alone could be enough to lessen NMDA receptor-dependent Ca2+ influx because this subunit is essential for NMDA receptor function (73–75). Our data also revealed a decrease in the number of NR2A puncta in cholesterol-reduced neurons as compared with controls. Because the NR1 subunit predominantly assembles with NR2A at synaptic sites, a reduction in this NR2 subunit clusters might decrease overall synaptic current induced by the Aβ peptide. The changes in the membrane content of NR1 and number of NR2A immunoreactive clusters described above were accompanied by changes in size of NMDA receptor subunit spots. Our results showed a significant increase in the subset of NMDA receptor immunoreactive subunit clusters of a size larger than 3 μm2 in cholesterol-reduced mature neurons when compared with untreated controls. Larger NMDA receptor clusters have been implicated in enhanced synaptic activity (76–78). If that were the case under the experimental conditions used in this study, the reduction of cholesterol would have resulted in enhanced Ca2+ influx and calpain activation in mature neurons, an opposite phenotype to the one described above. A possible explanation for this discrepancy could arise from the lack of colocalization of these larger NMDA receptor clusters and the presynaptic marker in cholesterol-reduced neurons. These results suggested that the larger NMDA clusters observed in MBCD-treated mature neurons were not at synaptic sites and thus were unresponsive to synaptic activation.
Changes in the localization of NMDA receptor clusters upon membrane cholesterol reduction in mature neurons could be the result, at least in part, of their redistribution between DSM and DRM microdomains. Previous studies have demonstrated that cholesterol reduction alters the membrane localization of these complexes and their related proteins to and from raft-like areas under other experimental conditions (26, 79). In addition, it has been shown that cholesterol reduction by MBCD alters the detergent solubility of non-raft membrane-bound proteins (80, 81). Therefore, the increase in the NR1 DRM/DSM ratio might be due, at least in part, to a loss of NR1 observed in the DSM domain of cholesterol-reduced neurons as reflected by our biochemical analyses. Finally, it is worth mentioning that the described cholesterol-induced changes in the localization of NMDA receptor subunits could be due, at least in part, to alterations in other NMDA receptor-associated components, such as synaptic receptor scaffolding proteins as described previously (26, 44, 64, 82).
Regardless, our results provide insights into the mechanism underlying the role of cholesterol as a regulator of age-dependent susceptibility of central neurons to Aβ-induced neurodegeneration. The identification of some of the key molecular components of these mechanisms, such as NMDA receptors, calpain, and the 17-kDa neurotoxic Tau fragment, opens interesting new avenues of drug discovery for the treatment of not only AD but also other neurodegenerative disorders associated with excitotoxicity.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 NS39080. This work was also supported byAlzheimer's Association Grant 57869 (to A. F.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
- AD
- Alzheimer disease
- Aβ
- β-amyloid
- NMDA
- N-methyl-d-aspartic acid
- MBCD
- methyl-β-cyclodextrin
- DRM
- detergent-resistant membrane
- DSM
- detergent-soluble membrane.
REFERENCES
- 1. Mayeux R., Ottman R., Maestre G., Ngai C., Tang M. X., Ginsberg H., Chun M., Tycko B., Shelanski M. (1995) Neurology 45, 555–557 [DOI] [PubMed] [Google Scholar]
- 2. Robakis N. K., Ramakrishna N., Wolfe G., Wisniewski H. M. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 4190–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yankner B. A., Lu T., Loerch P. (2008) Annu. Rev. Pathol. 3, 41–66 [DOI] [PubMed] [Google Scholar]
- 4. Chartier-Harlin M. C., Crawford F., Houlden H., Warren A., Hughes D., Fidani L., Goate A., Rossor M., Roques P., Hardy J., et al. (1991) Nature 353, 844–846 [DOI] [PubMed] [Google Scholar]
- 5. Nunan J., Small D. H. (2000) FEBS Lett. 483, 6–10 [DOI] [PubMed] [Google Scholar]
- 6. Higuchi M., Iwata N., Saido T. C. (2005) Biochim. Biophys. Acta 1751, 60–67 [DOI] [PubMed] [Google Scholar]
- 7. Harkany T., Abrahám I., Kónya C., Nyakas C., Zarándi M., Penke B., Luiten P. G. (2000) Rev. Neurosci. 11, 329–382 [DOI] [PubMed] [Google Scholar]
- 8. Blurton-Jones M., Laferla F. M. (2006) Curr. Alzheimer Res. 3, 437–448 [DOI] [PubMed] [Google Scholar]
- 9. Green K. N., LaFerla F. M. (2008) Neuron 59, 190–194 [DOI] [PubMed] [Google Scholar]
- 10. Bharadwaj P. R., Dubey A. K., Masters C. L., Martins R. N., Macreadie I. G. (2009) J. Cell. Mol. Med. 13, 412–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee V. M. (1996) Ann. N.Y. Acad. Sci. 777, 107–113 [DOI] [PubMed] [Google Scholar]
- 12. Tsai L. H., Lee M. S., Cruz J. (2004) Biochim. Biophys. Acta 1697, 137–142 [DOI] [PubMed] [Google Scholar]
- 13. Pevalova M., Filipcik P., Novak M., Avila J., Iqbal K. (2006) Bratisl. Lek. Listy 107, 346–353 [PubMed] [Google Scholar]
- 14. Arioka M., Tsukamoto M., Ishiguro K., Kato R., Sato K., Imahori K., Uchida T. (1993) J. Neurochem. 60, 461–468 [DOI] [PubMed] [Google Scholar]
- 15. Park S. Y., Ferreira A. (2005) J. Neurosci. 25, 5365–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cutler R. G., Kelly J., Storie K., Pedersen W. A., Tammara A., Hatanpaa K., Troncoso J. C., Mattson M. P. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2070–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nicholson A. M., Ferreira A. (2009) J. Neurosci. 29, 4640–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goslin K., Asmussen H., Banker G. (1998) in Culturing Nerve Cells (Banker G., Goslin K. eds) 2nd Ed., pp. 339–370, MIT Press, Cambridge, MA [Google Scholar]
- 19. Bottenstein J. E., Sato G. H. (1979) Proc. Natl. Acad. Sci. U.S.A. 76, 514–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferreira A., Lu Q., Orecchio L., Kosik K. S. (1997) Mol. Cell. Neurosci. 9, 220–234 [DOI] [PubMed] [Google Scholar]
- 21. van der Bliek A. M., Redelmeier T. E., Damke H., Tisdale E. J., Meyerowitz E. M., Schmid S. L. (1993) J. Cell Biol. 122, 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Damke H., Baba T., Warnock D. E., Schmid S. L. (1994) J. Cell Biol. 127, 915–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 24. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 25. Bensadoun A., Weinstein D. (1976) Anal. Biochem. 70, 241–250 [DOI] [PubMed] [Google Scholar]
- 26. Abulrob A., Tauskela J. S., Mealing G., Brunette E., Faid K., Stanimirovic D. (2005) J. Neurochem. 92, 1477–1486 [DOI] [PubMed] [Google Scholar]
- 27. Towbin H., Staehelin T., Gordon J. (1979) Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yakunin A. F., Hallenbeck P. C. (1998) Anal. Biochem. 258, 146–149 [DOI] [PubMed] [Google Scholar]
- 29. Murphy S. N., Miller R. J. (1989) Mol. Pharmacol. 35, 671–680 [PubMed] [Google Scholar]
- 30. Brewer L. D., Thibault O., Staton J., Thibault V., Rogers J. T., Garcia-Ramos G., Kraner S., Landfield P. W., Porter N. M. (2007) Brain Res. 1151, 20–31 [DOI] [PubMed] [Google Scholar]
- 31. Sun D. A., Sombati S., Blair R. E., DeLorenzo R. J. (2004) Cell Calcium 35, 155–163 [DOI] [PubMed] [Google Scholar]
- 32. Foradori C. D., Werner S. B., Sandau U. S., Clapp T. R., Handa R. J. (2007) Neuroscience 149, 155–164 [DOI] [PubMed] [Google Scholar]
- 33. Resende R., Pereira C., Agostinho P., Vieira A. P., Malva J. O., Oliveira C. R. (2007) Brain Res. 1143, 11–21 [DOI] [PubMed] [Google Scholar]
- 34. Torp R., Head E., Milgram N. W., Hahn F., Ottersen O. P., Cotman C. W. (2000) Neuroscience 96, 495–506 [DOI] [PubMed] [Google Scholar]
- 35. Avdulov N. A., Chochina S. V., Igbavboa U., Warden C. S., Vassiliev A. V., Wood W. G. (1997) J. Neurochem. 69, 1746–1752 [DOI] [PubMed] [Google Scholar]
- 36. Williamson R., Usardi A., Hanger D. P., Anderton B. H. (2008) FASEB J. 22, 1552–1559 [DOI] [PubMed] [Google Scholar]
- 37. Morishima-Kawashima M., Ihara Y. (1998) Biochemistry 37, 15247–15253 [DOI] [PubMed] [Google Scholar]
- 38. Mizuno T., Nakata M., Naiki H., Michikawa M., Wang R., Haass C., Yanagisawa K. (1999) J. Biol. Chem. 274, 15110–15114 [DOI] [PubMed] [Google Scholar]
- 39. Oshima N., Morishima-Kawashima M., Yamaguchi H., Yoshimura M., Sugihara S., Khan K., Games D., Schenk D., Ihara Y. (2001) Am. J. Pathol. 158, 2209–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kawarabayashi T., Shoji M., Younkin L. H., Wen-Lang L., Dickson D. W., Murakami T., Matsubara E., Abe K., Ashe K. H., Younkin S. G. (2004) J. Neurosci. 24, 3801–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Christian A. E., Haynes M. P., Phillips M. C., Rothblat G. H. (1997) J. Lipid Res. 38, 2264–2272 [PubMed] [Google Scholar]
- 42. Kelly B. L., Ferreira A. (2006) J. Biol. Chem. 281, 28079–28089 [DOI] [PubMed] [Google Scholar]
- 43. Czogalla A., Sikorski A. F. (2005) Cell. Mol. Life Sci. 62, 1913–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hering H., Lin C. C., Sheng M. (2003) J. Neurosci. 23, 3262–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mauch D. H., Nägler K., Schumacher S., Göritz C., Müller E. C., Otto A., Pfrieger F. W. (2001) Science 294, 1354–1357 [DOI] [PubMed] [Google Scholar]
- 46. Thiele C., Hannah M. J., Fahrenholz F., Huttner W. B. (2000) Nat. Cell Biol. 2, 42–49 [DOI] [PubMed] [Google Scholar]
- 47. Mitter D., Reisinger C., Hinz B., Hollmann S., Yelamanchili S. V., Treiber-Held S., Ohm T. G., Herrmann A., Ahnert-Hilger G. (2003) J. Neurochem. 84, 35–42 [DOI] [PubMed] [Google Scholar]
- 48. Nägler K., Mauch D. H., Pfrieger F. W. (2001) J. Physiol. 533, 665–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fletcher T. L., Cameron P., De Camilli P., Banker G. (1991) J. Neurosci. 11, 1617–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qiu W. Q., Ferreira A., Miller C., Koo E. H., Selkoe D. J. (1995) J. Neurosci. 15, 2157–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fykse E. M., Takei K., Walch-Solimena C., Geppert M., Jahn R., De Camilli P., Südhof T. C. (1993) J. Neurosci. 13, 4997–5007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wenzel A., Fritschy J. M., Mohler H., Benke D. (1997) J. Neurochem. 68, 469–478 [DOI] [PubMed] [Google Scholar]
- 53. Wenzel A., Scheurer L., Künzi R., Fritschy J. M., Mohler H., Benke D. (1995) Neuroreport 7, 45–48 [PubMed] [Google Scholar]
- 54. Matsuzaki K., Horikiri C. (1999) Biochemistry 38, 4137–4142 [DOI] [PubMed] [Google Scholar]
- 55. Dante S., Hauss T., Dencher N. A. (2006) Eur. Biophys. J. 35, 523–531 [DOI] [PubMed] [Google Scholar]
- 56. Curtain C. C., Ali F. E., Smith D. G., Bush A. I., Masters C. L., Barnham K. J. (2003) J. Biol. Chem. 278, 2977–2982 [DOI] [PubMed] [Google Scholar]
- 57. Hynd M. R., Scott H. L., Dodd P. R. (2004) Neurochem. Int. 45, 583–595 [DOI] [PubMed] [Google Scholar]
- 58. Wenk G. L. (2006) J. Clin. Psychiatry 67, Suppl. 3, 3–7 [PubMed] [Google Scholar]
- 59. Lipton S. A. (2005) Curr. Alzheimer Res. 2, 155–165 [DOI] [PubMed] [Google Scholar]
- 60. Magnusson K. R. (1997) J. Gerontol. A. Biol. Sci. Med. Sci. 52, B291–B299 [DOI] [PubMed] [Google Scholar]
- 61. Magnusson K. R. (1998) Mech. Ageing Dev. 104, 227–248 [DOI] [PubMed] [Google Scholar]
- 62. Dunah A. W., Yasuda R. P., Wang Y. H., Luo J., Dávila-García M., Gbadegesin M., Vicini S., Wolfe B. B. (1996) J. Neurochem. 67, 2335–2345 [DOI] [PubMed] [Google Scholar]
- 63. Benke D., Wenzel A., Scheuer L., Fritschy J. M., Mohler H. (1995) J. Recept. Signal. Transduct. Res. 15, 393–411 [DOI] [PubMed] [Google Scholar]
- 64. Besshoh S., Chen S., Brown I. R., Gurd J. W. (2007) J. Neurosci. Res., 85, 1876–1883 [DOI] [PubMed] [Google Scholar]
- 65. Liu X. B., Murray K. D., Jones E. G. (2004) J. Neurosci. 24, 8885–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li J. H., Wang Y. H., Wolfe B. B., Krueger K. E., Corsi L., Stocca G., Vicini S. (1998) Eur. J. Neurosci. 10, 1704–1715 [DOI] [PubMed] [Google Scholar]
- 67. Monyer H., Burnashev N., Laurie D. J., Sakmann B., Seeburg P. H. (1994) Neuron 12, 529–540 [DOI] [PubMed] [Google Scholar]
- 68. Fritschy J. M., Weinmann O., Wenzel A., Benke D. (1998) J. Comp. Neurol. 390, 194–210 [PubMed] [Google Scholar]
- 69. Watanabe M., Inoue Y., Sakimura K., Mishina M. (1993) Ann. N.Y. Acad. Sci. 707, 463–466 [DOI] [PubMed] [Google Scholar]
- 70. Liu P., Smith P. F., Darlington C. L. (2008) Synapse 62, 834–841 [DOI] [PubMed] [Google Scholar]
- 71. Law A. J., Weickert C. S., Webster M. J., Herman M. M., Kleinman J. E., Harrison P. J. (2003) Eur. J. Neurosci. 18, 1197–1205 [DOI] [PubMed] [Google Scholar]
- 72. Goritz C., Mauch D. H., Pfrieger F. W. (2005) Mol. Cell. Neurosci. 29, 190–201 [DOI] [PubMed] [Google Scholar]
- 73. Ishii T., Moriyoshi K., Sugihara H., Sakurada K., Kadotani H., Yokoi M., Akazawa C., Shigemoto R., Mizuno N., Masu M., et al. (1993) J. Biol. Chem. 268, 2836–2843 [PubMed] [Google Scholar]
- 74. Kutsuwada T., Kashiwabuchi N., Mori H., Sakimura K., Kushiya E., Araki K., Meguro H., Masaki H., Kumanishi T., Arakawa M., et al. (1992) Nature 358, 36–41 [DOI] [PubMed] [Google Scholar]
- 75. Moriyoshi K., Masu M., Ishii T., Shigemoto R., Mizuno N., Nakanishi S. (1991) Nature 354, 31–37 [DOI] [PubMed] [Google Scholar]
- 76. Crump F. T., Dillman K. S., Craig A. M. (2001) J. Neurosci. 21, 5079–5088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yu W., De Blas A. L. (2008) J. Neurochem. 104, 830–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Carpenter-Hyland E. P., Woodward J. J., Chandler L. J. (2004) J. Neurosci. 24, 7859–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Frank C., Giammarioli A. M., Pepponi R., Fiorentini C., Rufini S. (2004) FEBS Lett. 566, 25–29 [DOI] [PubMed] [Google Scholar]
- 80. Lambert D., O'Neill C. A., Padfield P. J. (2005) Biochem. J. 387, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shah W. A., Peng H., Carbonetto S. (2006) J. Gen. Virol. 87, 673–678 [DOI] [PubMed] [Google Scholar]
- 82. Hou Q., Huang Y., Amato S., Snyder S. H., Huganir R. L., Man H. Y. (2008) Mol. Cell. Neurosci. 38, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.