

Abstract
Programmed cell death (PCD) in mammals has been implicated in several disease states including cancer, autoimmune disease, and neurodegenerative disease. In Caenorhabditis elegans, PCD is a normal component of development. We find that Salmonella typhimurium colonization of the C. elegans intestine leads to an increased level of cell death in the worm gonad. S. typhimurium-mediated germ-line cell death is not observed in C. elegans ced-3 and ced-4 mutants in which developmentally regulated cell death is blocked, and ced-3 and ced-4 mutants are hypersensitive to S. typhimurium-mediated killing. These results suggest that PCD may be involved in the C. elegans defense response to pathogen attack.
Several human pathogens, including Pseudomonas aeruginosa, Salmonella typhimurium, Serratia marcescens, and Burkholderia pseudomallei, kill the nematode Caenorhabditis elegans when supplied as a food source, and a variety of bacterial virulence factors have been shown to play a role in both nematode and mammalian pathogenesis (1–6). In addition, C. elegans mutants that are either more susceptible or more resistant to bacterial killing can be readily identified (7, 8). These facts make C. elegans an attractive host for dissecting the molecular basis of bacterial pathogenesis.
An important feature of a variety of mammalian–pathogen interactions is programmed cell death (PCD) of both immune and somatic cells. PCD in mammals has also been implicated in several disease states, including cancer, autoimmune disease, and neurodegenerative disease (9). Because many of the key components of the mammalian cell death machinery were first identified by genetic studies in C. elegans (10, 11), it was of interest to determine the role, if any, of PCD in C. elegans in response to pathogen attack.
Mutations in at least three C. elegans genes, ced-3, ced-4, and ced-9, affect the 131 somatic cell deaths that occur during the development of the C. elegans hermaphrodite (12, 13). Loss-of-function mutations in ced-3 or ced-4 or a gain-of-function mutation in the gene ced-9 result in the survival of cells that normally die. CED-3 and CED-4 proapoptotic activity is antagonized by the Bcl-2 family member CED-9 (14). In cells fated to die, EGL-1 binds to and directly inhibits the activity of CED-9 (15).
The 131 cells that undergo PCD during the somatic development of C. elegans represent about 12% of the cells in an adult worm. In contrast to somatic cells, germ cells do not have a fixed lineage or population of cells. In normal adult hermaphrodites, over half of all potential oocytes are eliminated by PCD that is mediated by the same core machinery (ced-3, ced-4, and ced-9) responsible for somatic PCD during development (16). Germ cell deaths are more abundant in starving and old worms than in well fed young animals (17), suggesting that PCD in the germ line is regulated by environmental conditions.
P. aeruginosa and S. typhimurium kill C. elegans by at least three distinct mechanisms when provided to C. elegans as the sole source of food. At least in the case of P. aeruginosa, the mechanism of killing depends on the strain of P. aeruginosa used and the medium used to grow the bacteria. Typically, C. elegans are propagated in the laboratory by feeding them on Escherichia coli strain OP50 grown on NG medium. When fed on P. aeruginosa strain PA14 grown on the relatively low osmolarity NG medium, PA14 accumulates within the lumen of the C. elegans intestine, killing worms relatively slowly over the course of two–three days (slow killing; ref. 5). In contrast, PA14 grown in a rich and high osmolarity medium kills worms more quickly through a toxin-mediated mechanism (fast killing; refs. 5 and 8). P. aeruginosa strain PAO1 grown on brain-heart infusion medium kills by a third mechanism involving the generation of one or more neurotoxins (7). A variety of S. enterica serovars, including S. typhimurium, grown on NG medium, kill C. elegans over the course of several days by a mechanism that involves the establishment of a persistent infection in the C. elegans intestine (1).
We tested whether germ-line cell death plays a role in the C. elegans defense response to pathogen attack by examining the interaction between P. aeruginosa or S. typhimurium and C. elegans mutants deficient in PCD. Our results show that S. typhimurium elicits germ-line cell death, and that C. elegans ced-3 and ced-4 mutants are more susceptible to S. typhimurium-mediated killing. In contrast, germ-line cell death does not seem to play a significant role in the C. elegans—P. aeruginosa interaction.
Materials and Methods
Bacterial Strains and Growth Conditions.
E. coli OP50 (18), S. typhimurium SL1344 (19), S. typhimurium 14028 (20), or S. typhimurium LH954 (20) were grown overnight at 37°C in Luria–Bertani medium. Bacterial lawns used for C. elegans killing assays were prepared by spreading 10 μl of the bacterial strains on modified nematode growth agar medium (0.35% instead of 0.25% peptone) in 3.5-cm diameter plates. Plates were incubated at 37°C for 12 h and then allowed to equilibrate to room temperature for 3 h before seeding them with worms.
C. elegans Killing Assay.
Ten worms were placed on a bacterial lawn and the plates were incubated
at 20°C. Each independent assay was carried out in duplicate. During
the reproductive period, adults were transferred daily to fresh plates
and, thereafter, were transferred approximately every 5 days. Worm
mortality was scored over time, and a worm was considered dead when it
failed to respond to touch. The time for 50% of the nematodes to die
(time to death 50, TD50) was calculated by using
the prism V. 2.00 computer program using the equation:
Y = Bottom + (Top − Bottom)/(1 +
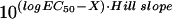 ),
where X is the logarithm of days and Y is the
average of dead worms. The relative mortality of worms feeding on
S. typhimurium was calculated with the equation: Relative
mortality = (TD50 wild-type C.
elegans on S. typhimurium/TD50
C. elegans mutant on S.
typhimurium)/(TD50 wild-type C.
elegans on E. coli/TD50
C. elegans mutant on E. coli).
),
where X is the logarithm of days and Y is the
average of dead worms. The relative mortality of worms feeding on
S. typhimurium was calculated with the equation: Relative
mortality = (TD50 wild-type C.
elegans on S. typhimurium/TD50
C. elegans mutant on S.
typhimurium)/(TD50 wild-type C.
elegans on E. coli/TD50
C. elegans mutant on E. coli).
Nematode Strains.
All C. elegans strains were maintained as hermaphrodites at 20°C, grown on modified NG agar plates (0.35% peptone instead of 0.25%) and fed with E. coli strain OP50 as described (18). The C. elegans mutants used were derived from the wild-type variety Bristol N2. ced-3(n717) and ced-9(n1653ts) mutants were kindly provided by Michael Hengartner, Cold Spring Harbor Laboratory. ced-3(n1286), ced-3(n2433), ced-4(n1162), ced-4(n1894), egl-1(n1084n3082), ces-1(n703), and ces-2(n732) were obtained from the Caenorhabditis Genetics Center, University of Minnesota, St. Paul.
Cell Corpse Assay.
To quantify the number of apoptotic germ cells, the animals were stained with SYTO 12 (Molecular Probes) as previously described (16). Briefly, the worms were incubated in 50 μM SYTO 12 for 3–4 h at room temperature and then seeded on bacterial lawns to reduce the amount of stained bacteria in the gut. After 20–30 min, animals were mounted in a drop of M9 salt solution containing 30 mM NaN3 and observed by using a Leica TCS SP confocal microscope. Only animals that were brightly and equally stained were scored.
Results
S. typhimurium but Not P. aeruginosa Elicits Germ-Line Cell Death.
Both L4 and one-day-old C. elegans adults died more quickly when fed on S. typhimurium strain SL1344 than when fed on E. coli strain OP50, the usual food source for growing C. elegans in the laboratory. Because we also observed that S. typhimurium infection results in an approximately 30% ± 10% decrease in brood size (data not shown), we determined whether S. typhimurium affects the rate of germ-line cell death by identifying germ-line cell apoptotic corpses with Nomarski optics or by staining with the nucleic acid stain SYTO 12 as described in Materials and Methods. SYTO 12 specifically stains condensed structures in the gonad of adult hermaphrodites that are apparently the corpses of apoptotic germ-line cells that have undergone PCD (16).
A higher rate of apoptosis was observed in C. elegans germ cells when the worms were fed S. typhimurium SL1344 than when fed E. coli OP50 (Fig. 1A and Table 1). The higher percentage of apoptotic corpses in infected worms persisted throughout the reproductive life of the animals. The condensed structures identified as apoptotic corpses observed in worms feeding on E. coli (Fig. 1B) or S. typhimurium (Fig. 1D) were located in a region of the germ line occupied by syncytial germ cells undergoing meiosis and corresponded to the structures stained by SYTO 12 (Fig. 1 E and C).
Figure 1.

S. typhimurium induces PCD in the C. elegans germ line. (A) Cell corpses were counted over time in one gonad arm, starting with young adult animals fed on E. coli OP50 or S. typhimurium SL1344. Data (mean ± SD) are from at least three independent experiments; more than 15 animals were scored at each time point. (B–E) Confocal images showing 1-day-old hermaphrodite worms fed on E. coli OP50 (B and C) or S. typhimurium SL1344 (D and E) for 12 h. In the transmission images (B and D), the cell corpses are indicated with arrows. Merged images (C and E) show corpses stained with SYTO 12. (Bar = 30 μm.)
Table 1.
Relationship between germ cell death and Salmonella-mediated killing
| Condition | Germ cell corpses* | n† | TD50‡ |
|---|---|---|---|
| N2 + E. coli OP50 | 29 ± 1.8 | 20 | 16.8 ± 0.3 |
| N2 + S. typhimurium SL1344 | 91 ± 2.5 | 20 | 13.5 ± 1.5 |
| ced-3(n717) + E. coli OP50 | 0.0 | 20 | 18.8 ± 0.9 |
| ced-3(n717) + S. typhimurium SL1344 | 0.2 ± 0.4 | 20 | 5.8 ± 0.3 |
| ced-4(n1162) + E. coli OP50 | 0.0 | 15 | 13.5 ± 1.3 |
| ced-4(n1162) + S. typhimurium SL1344 | 0.0 | 19 | 6.9 ± 0.3 |
| ced-9(n1950) + E. coli OP50 | 25 ± 1.4 | 20 | 16.1 ± 0.2 |
| ced-9(n1950) + S. typhimurium SL1344 | 29 ± 1.8 | 20 | 5.6 ± 0.2 |
| egl-1(n1084n3082) + E. coli OP50 | 28 ± 0.9 | 20 | 16.9 ± 1.9 |
| egl-1(n1084n3082) + S. typhimurium SL1344 | 36 ± 1.4 | 20 | 7.5 ± 1.1 |
| N2 + S. typhimurium SL14028 (wild type) | 75 ± 2.3 | 20 | 13.0 ± 1.0 |
| N2 + S. typhimurium LH954 (phoP phoQ) | 31 ± 1.7 | 20 | 16.5 ± 0.5 |
| N2 + P. aeruginosa PA14 | 30 ± 0.4 | 15 | 3.4 ± 0.6 |
The germ cell corpses were scored 3 h after the infection of young adult animals by using the vital dye Syto12 as described in Materials and Methods. The number of germ corpses per gonad arm was scored only in equally stained animals.
n, number of animals counted.
TD50s were calculated as described in Materials and Methods and data (mean ± SD) were from duplicates.
To evaluate whether the mere presence of S. typhimurium in the C. elegans intestine is capable of inducing PCD or whether PCD requires the expression of specific virulence factors, we analyzed the role of the S. typhimurium PhoP/PhoQ signal transduction system, a key regulator of virulence-related genes (21). We took advantage of an S. typhimurium mutant (LH 954) with a phoP/phoQ/purB deletion that we previously showed causes significantly less killing of C. elegans (1). The results shown in Table 1 indicate that the phoP/phoQ/purB (LH 954) mutant kills C. elegans at a slower rate than the parental S. typhimurium strain (SL14028) and does not elicit an apoptotic response.
In contrast to S. typhimurium, we did not observe an elevated rate of germ-line cell death when C. elegans was fed P. aeruginosa strain PA14 under either the so-called slow (Table 1) or fast (data not shown) killing conditions. Surprisingly, only approximately 5% of the worms that had been in contact with P. aeruginosa for 3 h under the slow killing conditions were capable of taking up the SYTO 12 stain. This latter result suggests that despite the lipidic nature of the dye, some kind of active transport may be required for the uptake of the dye, and that P. aeruginosa blocks this process.
S. typhimurium-Induced Germ-Line Cell Death Depends on the ced-3/ced-4 Cell Death Pathway.
We used a variety of C. elegans mutants that affect PCD in somatic and germ-line cells to determine whether the S. typhimurium-induced apoptosis in the germ line requires previously identified apoptotic machinery and whether this apoptotic machinery is involved in S. typhimurium-mediated killing. The proximal cause of apoptosis in C. elegans is the activation of CED-3, which is required for germ cell death (16). As shown in Table 1, no germ-line cell deaths were observed in a ced-3 mutant feeding on E. coli or S. typhimurium. ced-3 encodes a prototypical caspase (22), and CED-4 is similar to mammalian Apaf-1, an activator of caspases (23) that binds and activates CED-3 (24–26). Thus, if activated CED-3 is required for S. typhimurium-induced cell death, ced-4 mutants should also impair cell death in Salmonella-infected worms. As observed in ced-3 animals, no germ-line cell deaths were observed in ced-4 mutants feeding on S. typhimurium lawns (Table 1).
C. elegans ced-3 and ced-4 Mutants Are Hypersusceptible to S. typhimurium-Mediated Killing.
As shown in Fig. 2 A and B, ced-3 mutants died much more quickly than wild-type worms when feeding on S. typhimurium, but died at the same rate as wild-type worms when feeding on E. coli. The TD50 for 1-day-old hermaphrodite nematodes when fed at 20°C on S. typhimurium was 14 days for wild-type worms compared with 5.9, 6.6, and 6.5 days (respectively) for three different ced-3 mutant alleles, whereas the TD50 for the three ced-3 mutants feeding on E. coli was 19, 16, and 16 days (respectively) compared with 17 days for wild-type worms feeding on E. coli. In addition, the rate at which wild-type worms died when feeding on S. typhimurium (TD50 = 14 days) was faster than the rate at which both wild-type worms (TD50 = 17) and ced-3 mutant worms (TD50 = 19, 16, 16 days) died when feeding on E. coli (for example, see Fig. 2A). Two different ced-4 mutants were also more susceptible to S. typhimurium-mediated killing (Fig. 2C). The fact that three ced-3 alleles and two ced-4 alleles exhibited the same phenotype when feeding on S. typhimurium makes it unlikely that enhanced susceptibility is caused by secondary mutations or the effect of a particular allele on a process unrelated to cell death. Consistent with the observation that P. aeruginosa strain does not induce a high level of germ-line PCD, ced-3 and ced-4 mutants were not more susceptible to P. aeruginosa-mediated fast or slow killing (data not shown). These experiments, therefore, are consistent with the conclusion that CED-3 and CED-4 are involved in a C. elegans defense response to S. typhimurium but not to P. aeruginosa.
Figure 2.

ced-3 and ced-4 mutants are more susceptible to S. typhimurium-mediated killing. One-day-old adult animals were fed either E. coli or S. typhimurium. (A) Wild type and ced-3(n717). (B) Wild type, ced-3(n1286), and ced-3(n2433). (C) Wild type, ced-4(n1162), and ced-4(n1894). More than 10 animals were used in each case and data (mean ± SD) were from duplicates. The results are representative of at least three experiments.
The C. elegans Upstream Regulators of Cell Death CED-9 and EGL-1 Are Also Involved in S. typhimurium-Induced Germ-Line Cell Death.
CED-9 is a member of the Bcl-2 family of cell death regulators (14) that directly inhibits CED-4, apparently by sequestering CED-4 and proCED-3 in an inactive ternary complex called the apoptosome (27). The ced-9(1950) gain-of-function mutant has a similar phenotype to ced-3 and ced-4 loss-of-function mutants with respect to PCD in developing worms (14). EGL-1 is thought to activate the PCD cascade by inhibiting CED-9 (15). Consistent with the results described in the previous section, Table 1 shows that Salmonella-induced apoptosis is substantially reduced in the ced-9(n1950) mutant. Similarly, the egl-1(n1084n3082) loss-of-function mutant also exhibited a reduction in Salmonella-induced germ-line apoptosis compared with wild-type N2 worms (Table 1). Moreover, both the ced-9(n1950)and the egl-1(n1084n3082) mutants were more susceptible to Salmonella-mediated killing (Fig. 3 A and C).
Figure 3.

Mutants deficient in stress-induced germ-line cell death are more susceptible to S. typhimurium-mediated killing. One-day-old adult animals were exposed to either E. coli or S. typhimurium. (A) Wild type, ced-9(n1950) (gf), and egl-1(n1084n3082) (lf) (gf, gain-of-function; lf, loss-of-function). B, wild type and ced-9(n1653ts) (lf). (C) Wild type, ced-3 (n717), ced-4(n1162), ced-9(n1653ts) (lf), ced-9(n1950) (gf), egl-1(n1084n3082), ces-1(n703), and ces-2(n732). The TD50s were calculated in each case and the relative mortality was calculated as described in Materials and Methods. More than 10 animals were used in each case; data (mean ± SD) were from three to five experiments.
Consistent with previous observations that ced-9 loss-of-function mutants exhibit a much higher rate of spontaneous cell death than wild-type worms (16), the ced-9(n1653ts) loss-of-function mutant exhibited elevated PCD in the gonads (data not shown). Fig. 3B shows that the ced-9(n1653ts) mutant had a short lifespan when feeding on S. typhimurium. On the other hand, it also had a short lifespan when feeding on E. coli. Therefore, the relative mortality of ced-9 worms feeding on S. typhimurium [defined as (TD50 of wild-type worms feeding on S. typhimurium/TD50 ced-9 on S. typhimurium)/(TD50 wild-type worms on E. coli/TD50 ced-9 on E. coli)] was not significantly different from control wild-type worms as illustrated in Fig. 3C. The short lifespan of the ced-9 mutant is most likely related to the fact that loss-of-function mutations in ced-9 cause sterility and maternal-effect lethality as a consequence of ectopic cell death (14).
Mutations in the genes ces-1 and ces-2 affect a specific subset of somatic PCDs (28) but have not been shown to be involved in germ-line cell death (16). As expected, these mutants were not more susceptible than wild-type worms to Salmonella-mediated killing (Fig. 3C).
Discussion
In vertebrates, PCD is used to eliminate cells that have been produced in excess, have already served their purpose, or are potentially detrimental to the organism. In C. elegans, most cell death seems to belong to the first two categories. However, the data presented in this paper show that S. typhimurium colonization of the C. elegans intestine leads to an increased level of cell death in the worm gonad and that C. elegans PCD mutants are more susceptible to S. typhimurium-mediated killing. These results suggest that S. typhimurium virulence factors trigger somatic signals that induce the PCD pathway and that induction of the PCD pathway serves a protective role when C. elegans encounters an adverse environmental stimulus such as the attack of a potentially pathogenic bacterium. These conclusions are consistent with the following observations. First, when C. elegans are colonized by S. typhimurium, the intestinal lumen of infected animals is distended and full of intact bacteria, but no bacteria are found beyond this region or in contact with the gonads (1). Second, C. elegans PCD-related mutants, including several loss-of-function alleles of ced-3 and ced-4 and a gain-of-function allele of ced-9, do not exhibit S. typhimurium-elicited PCD (Table 1), and are more susceptible to S. typhimurium-mediated killing (Figs. 2 and 3). Third, a pleiotropic S. typhimurium phoP/phoQ mutant that fails to synthesize a variety of virulence-related factors does not elicit germ-line cell death (Table 1).
Despite the similarities in the regulation of somatic and germ-cell deaths in C. elegans, some differences have been found. For example, ced-9 gain-of-function and egl-1 loss-of-function mutations, which play key roles in PCD during hermaphrodite development, do not seem to be involved in the type of spontaneous germ-line cell deaths observed when C. elegans feeds on E. coli (16). However, a recent report demonstrates that they do have a role in stress-induced germ-line deaths. When worms were subjected to γ-irradiation to induce germ-line cell death, the ced-9 (n1950) (gain-of-function) mutation abolished radiation-induced germ-cell death, and the egl-1 (n1084n3082) mutation reduced it significantly (29). It is possible that both irradiation and Salmonella colonization may generate similar noxious stimuli, including oxidative stress, which triggers PCD. It would be interesting to know whether γ-irradiated PCD mutants die more quickly than irradiated wild-type worms. The fact that excess germ cells are eliminated by PCD in starving animals (17) raises the possibility that S. typhimurium infection could be causing nutritional stress that leads to germ-line cell death. However, the normal development of worms when feeding on S. typhimurium and the rapid induction of cell death by Salmonella (Fig. 1A) make this possibility unlikely.
In wild-type C. elegans, no somatic cell deaths were observed in the soma after the L2 stage and we did not observe any somatic PCD in worms feeding on S. typhimurium. However, we cannot rule out the possibility that some somatic cell deaths occur in response to bacterial attack and that these somatic cell deaths play an important role in the defense response to pathogens. It is also possible that S. typhimurium colonization activates a ced-3 ced-4-dependant signaling pathway that triggers cell death in germ-line cells but activates defense response pathways in somatic cells. Such a pathway, for example, could operate in the C. elegans intestine, which is in direct contact with potential bacterial pathogens. At this point in our investigation, we cannot distinguish between the possibilities that germ-line cell deaths per se play a direct protective role or that the ced-3 ced-4 signal pathway activates unknown defense responses. The mechanism by which germ-line cell death may protect the host from the deleterious effects of pathogen attack is not clear. Nevertheless, one could imagine that there is a selective advantage in regulating the rate of germ-line cell deaths in response to environmental signals.
In contrast to S. typhimurium, P. aeruginosa does not elicit PCD in C. elegans germ cells, and C. elegans PCD-related mutants do not exhibit an aberrant phenotype in response to P. aeruginosa compared with wild-type worms. A major difference between the P. aeruginosa and S. typhimurium infection models is that a small inoculum of S. typhimurium can proliferate in the intestine of C. elegans and establish a persistent and lethal infection even in the presence of a large excess of E. coli cells, whereas P. aeruginosa accumulates in the intestine only when it is the sole source of food. It is possible that the factors that allow S. typhimurium to proliferate in and colonize the intestine may correspond to specific C. elegans receptors that in turn may be involved in the elicitation of the PCD response. Consistent with this interpretation, it has been shown recently that PCD induced in lung epithelial cells by P. aeruginosa through the CD95/CD95 ligand pathway may protect the host from P. aeruginosa infection (30). The absence of C. elegans homologues for CD95 could explain why an increase in the PCD rate was not observed in P. aeruginosa-infected worms.
In conclusion, our results show that in response to S. typhimurium infection, PCD eliminates excess germ cells in the C. elegans gonad that could be detrimental to the worms, and that this PCD process may be involved in the C. elegans defense response to environmental insults, such as pathogen attack. These findings suggest that PCD is an ancient host defense mechanism that can be readily dissected by using the power of C. elegans genetic and genomic analyses.
Acknowledgments
We thank Michael Hengartner and Man-Wah Tan for their advice when we started this study, Sachiko Miyata for technical assistance, and Gary Ruvkun and Jonathan D. G. Jones for helpful comments on the manuscript. This work was funded by Aventis S.A. and by a National Institutes of Health Grant GM48707 (to F.M.A.).
Abbreviations
- PCD
programmed cell death
- TD50
time to death for 50% of worms
References
- 1.Aballay A, Yorgey P, Ausubel F M. Curr Biol. 2000;10:1539–1542. doi: 10.1016/s0960-9822(00)00830-7. [DOI] [PubMed] [Google Scholar]
- 2.Kurz C L, Ewbank J J. Trends Microbiol. 2000;8:142–144. doi: 10.1016/s0966-842x(99)01691-1. [DOI] [PubMed] [Google Scholar]
- 3.Labrousse A, Chauvet S, Couillault C, Leopold Kurz C, Ewbank J J. Curr Biol. 2000;10:1543–1545. doi: 10.1016/s0960-9822(00)00833-2. [DOI] [PubMed] [Google Scholar]
- 4.O'Quinn, A. L., Wiegand, E. M. & Jeddeloh, J. A. (2000) Cell. Microbiol., in press. [DOI] [PubMed]
- 5.Tan M W, Mahajan-Miklos S, Ausubel F M. Proc Natl Acad Sci USA. 1999;96:715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan M W, Rahme L G, Sternberg J A, Tompkins R G, Ausubel F M. Proc Natl Acad Sci USA. 1999;96:2408–2413. doi: 10.1073/pnas.96.5.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darby C, Cosma C L, Thomas J H, Manoil C. Proc Natl Acad Sci USA. 1999;96:15202–15207. doi: 10.1073/pnas.96.26.15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahajan-Miklos S, Tan M W, Rahme L G, Ausubel F M. Cell. 1999;96:47–56. doi: 10.1016/s0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 9.Thompson C B. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 10.Hedgecock E M, Sulston J E, Thomson J N. Science. 1983;220:1277–1279. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- 11.Horvitz H R, Shaham S, Hengartner M O. Cold Spring Harbor Symp Quant Biol. 1994;59:377–385. doi: 10.1101/sqb.1994.059.01.042. [DOI] [PubMed] [Google Scholar]
- 12.Sulston J E, Horvitz H R. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 13.Sulston J E, Schierenberg E, White J G, Thomson J N. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 14.Hengartner M O, Ellis R E, Horvitz H R. Nature (London) 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 15.Conradt B, Horvitz H R. Cell. 1998;93:519–529. doi: 10.1016/s0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- 16.Gumienny T L, Lambie E, Hartwieg E, Horvitz H R, Hengartner M O. Development (Cambridge, UK) 1999;126:1011–1022. doi: 10.1242/dev.126.5.1011. [DOI] [PubMed] [Google Scholar]
- 17.Sulston J E. In: The Nematode Caenorhabditis elegans. Wood W B, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. pp. 123–155. [Google Scholar]
- 18.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wray C, Sojka W J. Res Vet Sci. 1978;25:139–143. [PubMed] [Google Scholar]
- 20.Angelakopoulos H, Hohmann E L. Infect Immun. 2000;68:2135–2141. doi: 10.1128/iai.68.4.2135-2141.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia Vescovi E, Soncini F C, Groisman E A. Res Microbiol. 1994;145:473–480. doi: 10.1016/0923-2508(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 22.Yuan J, Shaham S, Ledoux S, Ellis H M, Horvitz H R. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 23.Zou H, Henzel W J, Liu X, Lutschg A, Wang X. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 24.Irmler M, Hofmann K, Vaux D, Tschopp J. FEBS Lett. 1997;406:189–190. doi: 10.1016/s0014-5793(97)00271-8. [DOI] [PubMed] [Google Scholar]
- 25.Spector M S, Desnoyers S, Hoeppner D J, Hengartner M O. Nature (London) 1997;385:653–656. doi: 10.1038/385653a0. [DOI] [PubMed] [Google Scholar]
- 26.Chinnaiyan A M, O'Rourke K, Lane B R, Dixit V M. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- 27.Hengartner M O. Nature (London) 1997;388:714–715. doi: 10.1038/41873. [DOI] [PubMed] [Google Scholar]
- 28.Ellis R E, Horvitz H R. Development (Cambridge, UK) 1991;112:591–603. doi: 10.1242/dev.112.2.591. [DOI] [PubMed] [Google Scholar]
- 29.Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner M O. Mol Cell. 2000;5:435–443. doi: 10.1016/s1097-2765(00)80438-4. [DOI] [PubMed] [Google Scholar]
- 30.Grassme H, Kirschnek S, Riethmueller J, Riehle A, von Kurthy G, Lang F, Weller M, Gulbins E. Science. 2000;290:527–530. doi: 10.1126/science.290.5491.527. [DOI] [PubMed] [Google Scholar]