

Abstract
Our global goal is that of synthesizing complex polypeptides and glycopeptides in homogeneous form. Chemistry-derived access to homogeneous biologics could well have useful consequences in the discovery of drugs and vaccines. The key finding in this report is that thio acids can become highly competent acyl donors following even trace levels of oxidative activation, thereby undergoing amide bond formation upon reaction with N-terminal peptides. Though our data set does not establish the specific mechanism of this reaction, a framework to account for the fact that minute levels of oxidation actuate amide bond formation with high turnover is offered. An apparently general coupling of thio acids (including complex peptide thio acids with N-termini of complex peptides) has thus been realized. These ligations are conducted with minimal α-epimerization in the C-terminal group and allow for the coupling of N-terminal and C-terminal glycopeptides en route to homogeneous glycoproteins.
Introduction
There is a sharp division in current modalities of development between small molecule-based drugs and large molecule agents, which are often referred to as “biologics.” Small molecule prospects are seen to arise from “chemistry.” By contrast, biologics (cf. vaccines, antibodies, enzymes, factors) are perceived to be derivable from strictly biological means. It is our view that recent advances in the scope and depth of organic chemistry raise the possibility that chemical synthesis could well play a valuable role in fashioning biologic level candidate structures.1 For such a goal to be feasible in the molecular space of biologics, complex issues associated with the assembly of key biolevel repeating building blocks must be mastered. Biologically active glycopeptides and glycoproteins are of particular interest to our laboratory.2 A formidable challenge in reaching such compounds via synthesis is that of joining and managing two differing biolevel domains (polysaccharides3 and polypeptides), each with their own chemical personalities and vulnerabilities. Since target glycopeptides or glycoproteins tend to arise in nature as horrific mixtures of glycoforms, chemical synthesis might well provide the best prospect for reaching and evaluating homogeneous glycopeptides for SAR studies. We have described strategies and enabling methodologies for assembling complex oligosaccharides with high levels of convergence and stereocontrol.4 These advances have, for instance, been used in the building of fully synthetic vaccines, thereby establishing the accessibility of a class of highly complex biologics to chemical synthesis.5
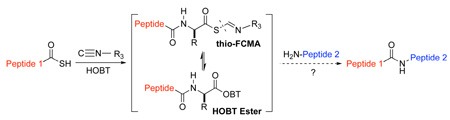
A massive advance in the capacity to synthesize homogeneous polypeptides, and even modestly sized proteins, arose from the seminal discovery of Native Chemical Ligation (NCL) by Kent and colleagues.6 In NCL, a C-terminal acyl donor is initially joined to the SH group of an N-terminal cysteine site. Following S→N acyl transfer, a peptide bond is fashioned (Figure 1a). Our laboratory has extended the inherent logic of NCL by exploiting metal-free chemospecific de-thiolation of SH groups, thereby allowing Ala ligation to become a practical option via an N-terminal Cys.7 By installing thiol groups into otherwise proteogenic amino acids through chemical synthesis, the elegant concept of NCL has been extended to enable ligations at N-terminal Phe, Val, Thr and Leu sites.8 Helpful as such advances have been, there is still a huge unmet need for a broadly based method to enable the ligation of peptides, including glycopeptides, independent of the logic of NCL. It is to this goal that the research described in this paper is addressed.
Figure 1.

(a) Native chemical ligation; (b) thio-FCMA ligation; (c) Challenge: thio-FCMA ligation in peptide couplings.
Results and Discussion
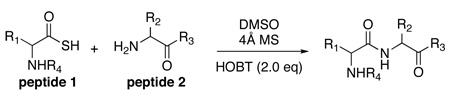
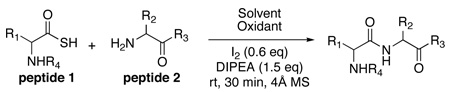
Before describing our findings, it is well to relate the etiology of the discovery progression. It will thus be appreciated that happenstance played no small role in mediating our advances. The organizing concept started with the reaction of a thio acid, 1, with an isonitrile, 2, in the presence of an amine-based acyl acceptor, of the type 4 (Figure 1b),9 giving rise to a presumed thio-formimidate carboxylate mixed anhydride (termed as a thio-FCMA, 3). The latter can be interdicted by amines to generate even complex amides. However, attempts to extend the scope of this coupling to a C-terminal thio acid of even a dipeptide failed to provide useful yields of desired product. It was surmised that the dipeptide thio-FCMA intermediate suffers rapid conversion to its corresponding oxazolone, which is not a competent acyl donor under these conditions (Figure 1c). We asked whether the presumed thio-FCMA (3) could be diverted to produce a more functional acyl donor than the presumed oxazolone. For instance, peptide bond formation via –HOBT esters tends to result in markedly reduced levels of C-terminal, oxazolone-promoted epimerization.10 Accordingly, we investigated the possible formation of peptidic bonds via the reaction of a C-terminal thio acid with cyclohexylisonitrile and N-terminal peptide in the presence of HOBT. The hope was that this FCMA would give way to an HOBT ester. The results, shown in Table 1, seemed quite encouraging.
Table 1a.
 | ||||
|---|---|---|---|---|
| entry | peptide 1 | product | reaction | yield |
| peptide 2 | time (h) | |||
| 1 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 |  |
20 | 60% |
| H2N-FGPELWP 8 | ||||
| 2 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 | 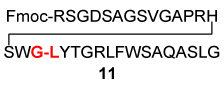 |
48 | 73% |
| H2N-LYTGRLFWSAQASLG 10 | ||||
| 3 | Fmoc-ESHRGWITAP-COSH 12 | 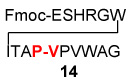 |
48 | 80% |
| H2N-VPVWAG 13 | ||||
| 4 | Fmoc-ESHRGWITAP-COSH 12 | 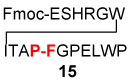 |
48 | 78% |
| H2N-FGPELWP 8 | ||||
| 5 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 |  |
48 | 82% |
| H2N-VPVWAG 13 | ||||
Key: DMSO, 5–8 eq cyclohexyl isonitrile, 2.0 eq HOBT, 4Å MS, rt. HOBT: 1-hydroxy-1H-benzotriazole.
In a control experiment, we asked whether C-terminal thio acids11 could couple with N-terminal peptides in the presence of HOBT, but in the absence of “throwaway” isonitrile. It was indeed fortunate that this control experiment was conducted. As seen in Table 2, coupling had occurred even more effectively than was the case with the isonitrile present (compare Table 1 and Table 2, entries 1–5). Though chromatographic criteria suggest coupling efficiencies of ca 90%, Table 2 reports only yields of purified isolated products. As seen, both C-terminal Gly and Pro residues were readily accommodated. Moreover, coupling of a range of hindered N-terminal amino acid residues, including Phe, Leu, and Val was accomplished smoothly. Even Pro-Val ligations could be achieved in excellent yield (Table 2, entry 3, 87%). Ligations of large peptide partners can be readily realized. Although significantly longer reaction times were required (48h), product yields were not compromised. The method is also applicable to macrolactamization (Table 2, entries 6 and 7).
Table 2.
HOBT-Mediated Peptide Coupling.a
 | ||||
|---|---|---|---|---|
| entry | peptide 1 | product | reaction | yield |
| peptide 2 | time (h) | |||
| 1 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 | 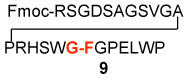 |
12 | 82% |
| H2N-FGPELWP 8 | ||||
| 2 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 | 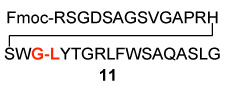 |
12 | 80% |
| H2N-LYTGRLFWSAQASLG 10 | ||||
| 3 | Fmoc-ESHRGWITAP-COSH 12 | 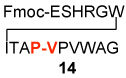 |
12 | 87% |
| H2N-VPVWAG 13 | ||||
| 4 | Fmoc-ESHRGWITAP-COSH 12 | 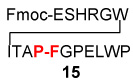 |
20a | 70% |
| H2N-FGPELWP 8 | ||||
| 5 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 |  |
20a | 77% |
| H2N-VPVWAG 13 | ||||
| 6 | H2N-GWPLILG-COSH |  |
48 | 81% |
| 17 | ||||
| 7 | H2N-FGPELWP-COSH |  |
48 | 80% |
| 19 | ||||
Key: DMSO, 2 eq HOBT, 4Å MS, air, rt;
DMSO, 0.3 eq HOBT, 4Å MS, air, rt.
Clearly, the finding that the isonitrile component is not required for ligation does not rule out the validity of the pathway and mechanism advanced above, when isonitrile is present. However, it does establish the existence of a pathway to amide bond formation from the reaction of thio acids and amines independent of isonitrile chemistry.
We next addressed the question of C-terminal epimerization during the course of ligation in more discriminating settings. A series of thio acid peptides, presenting C-terminal residues such as Phe, Ala, Leu, and Val, were evaluated as acyl donors (Table 3). In the simplest case, thio acid 21 readily undergoes amino acid extension with 22, to provide 23 in 89% yield with no detectable loss of stereointegrity (< 3%). This result is quite encouraging, since Phe is particularly prone to C-terminal epimerization.12 An even more discriminating case, involving coupling of the C-terminal Phe-bearing thio acid 24 with peptide 13, furnished adduct 25 in 82% yield with only a modest degree of epimerization at the Phe site (Table 3, entry 2). By contrast, C-terminal Ala-presenting thio acids seemed to be markedly more resistant to epimerization (Table 3, entries 3–5). Even when peptide thio acids bearing C-terminal Leu and Val residues were employed, epimerization levels were only 4% and 5%, respectively (Table 3, entries 6 and 7). We noted some improvement in the suppression of C-terminal epimerization via the use of HOOBT (seen in Table 3, entries 2 and 7).
Table 3.
HOBT-Mediated Peptide Coupling.a
 | ||||
|---|---|---|---|---|
| peptide 1 peptide 2 |
product | yield | L/D ratio |
|
| 1 | Boc-VF-COSH 21 H2N-F-OtBu 22 |
89%a | 97:3 | |
| 2 | Boc-AFGF-COSH 24 H2N-VPVWAG 13 |
82%b | 94:6 | |
| 3 | Boc-AFGA-COSH 26 H2N-VPVWAG 13 |
80% | 97:3 | |
| 4 | Ac-GRFSWGA-COSH 28 H2N-VPVWAG 13 |
 |
82%c | >99:1 |
| 5 | Ac-GRFSWGA-COSH 28 H2N-FGPELWP 8 |
 |
82% | >99:1 |
| 6 | Ac-GRFSWGL-COSH 31 H2N-VPVWAG 13 |
 |
80% | 96:4 |
| 7 | Ac-GRFSWGV-COSH 33 H2N-VPVWAG 13 |
 |
86%b | 95:5 |
Key: general procedure, DMSO, 2 eq HOBT, 4Å MS, rt, 12h;
CH2Cl2, 2 eq HOBT, 4Å MS, rt, 12h;
DMSO, 2 eq HOOBT, 4Å MS, rt, 12h;
DMSO, 0.3 eq HOBT, 4Å MS, rt, 20h. HOOBT: hydroxy-3,4-dihydro-4-oxo-1,2,3-benzotriazine.
The thio acid/HOBT method was shown to be effective in ligating a glycopeptide with a C-terminal thio acid, as demonstrated in the synthesis of 18-mer 36, containing a hexasaccharide of the high-mannose type. As shown in Figure 2, in the presence of 2.0 equiv. HOBT, peptide thio acid 12 and glycopeptide 35 undergo condensation to provide adduct 36 in 80% yield. Moreover, in the presence of 2.0 equiv. HOBT, glycopeptide thio acid 37 and glycopeptide 38 readily undergo fragment condensation to provide adduct 39 in 81% yield (Figure 2). The capability of ligating peptides to glycopeptides and glycopeptides to glycopeptides – so critical to synthesizing homogeneous glycoproteins in the laboratory – has thus been realized.
Figure 2.

We now turn to experiments which were aimed toward gaining some mechanistic insight into this amide bond forming reaction between thio acids and amines. Indeed, in retrospect, there could be found early harbingers of such a reaction in very simple systems.13 A later paper by Rosen, et al,14 also described such a reaction in a simple setting. Of course, the most obvious interpretation of such findings would be to suppose that, unlike carboxylic acids, their thio counterparts have strong inherent acyl donor character. While we could not rule this out, we began to suspect that there could be an oxidative component in these ligations. We first probed this possibility by conducting the couplings in an argon atmosphere rather than in air, but with no other precautions to ensure exclusion of air. Entries 1 and 2 in Table 4 are instructive. Under these “less oxidizing” conditions, the yield of coupling was clearly reduced. Not surprisingly, suppression of ligation was even more successful when HOBT was also omitted (see Table 4, entry 2). The same trend is also supported by entries 3 and 4 (Table 4). Attempted suppression of oxygen in the absence of HOBT still provided a 28% yield of tetrapeptide (see Table 4, entry 4). We note that in larger peptide-peptide settings, omission of HOBT, or conduct of the reaction under argon results in, at best, trace amounts of peptide bond formation with simple unhindered substrates (Table 4, entry 5). It is not clear whether the suppressed couplings under attempted anaerobic conditions reflect residual acyl donor competence of the thioacid itself. In principle, in a non-oxidative coupling, the sulfur should formally emerge at the H2S oxidation level, rather than as elemental sulfur, which would be reflective of oxidation (vide infra).
Table 4.
Attempted Suppression of Possibilities for Oxidation.a
 | ||||
|---|---|---|---|---|
| entry | peptide 1 | product | conditions | conversion |
| peptide 2 | ||||
| 1 | Fmoc-Val-COSH 40 NH2-Val-OBn 41 |
Fmoc-Val-Val-OBn 42 | HOBT Argon DMF, 20 h |
42%a |
| 2 | Fmoc-Val-COSH 40 NH2-Val-OBn 41 |
Fmoc-Val-Val-OBn 42 | Argon DMF, 20 h |
10%b |
| 3 | Fmoc-ESHRGWITAP-COSH 12 H2N-VPVWAG 13 |
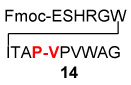 |
HOBT Argon DMSO, 4 h |
30%a |
| 4 | Fmoc-Gly-COSH 43 NH2-Phe-OEt 44 |
Fmoc-Gly-Phe-OEt 45 |
Argon DMSO, 6 h |
28%b |
| 5 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 H2N-VPVWAG 13 |
 |
Argon, DMSO, 20 h |
7%b |
Key:
Solvent, 2 eq HOBT, 4Å MS, Argon, rt;
Solvent, No HOBT, 4Å MS, Argon, rt.
Having shown that minimization of opportunities for adventitious oxidation does indeed attenuate coupling, we asked whether inclusion of an established oxidizing agent would promote amidation. The apparent validation of this expectation is shown in Table 5. While the yields of isolated peptide coupling are not substantially increased, the rates are enhanced (see conditions governing Table 5) and C-terminal epimerization is still suppressed (Table 5, entries 4 and 5). Remarkably (see Table 5, entries 1–3), several otherwise highly oxidation-vulnerable amino acids (Cys, Met, Trp, Tyr, and His) appear to survive nicely in the context of the peptides employed.
Table 5.
I2, HOBT-Mediated Peptide Coupling.a
 | ||||
|---|---|---|---|---|
| entry | peptide 1 peptide 2 |
product | yield | L/D ratio |
| 1 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 H2N-FGPELWP 8 |
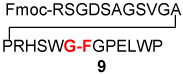 |
77%a | - |
| 2 | Fmoc-RSGDSAGSVGAPRHSWG-COSH 7 H2N-VPVWAG 13 |
 |
75%b | - |
| 3 | Ac-GC(Acm)MGWYP-COSH 46 H2N-FGPELWP 8 |
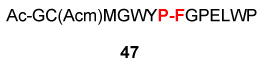 |
80%b | - |
| 4 | Boc-AFGF-COSH 24 H2N-VPVWAG 13 |
 |
80%c | 96:4 |
| 5 | Ac-GRFSWGL-COSH 31 H2N-FGPELWP 8 |
 |
82%c | 97:3 |
Key:
DMSO, 2 eq HOBT, 4Å MS, I2(0.6 eq), DIPEA(1.5 eq), rt, 30 min;
DMF, 2 eq HOBT, 4Å MS, I2(0.6 eq), DIPEA(1.5 eq), rt, 30 min;
DMF, 2 eq HOOBT, 4Å MS, I2(0.6 eq), DIPEA(1.5 eq), rt, 30 min.
In retrospect, it is well to note once again, that the concept of constructing amides from the coupling of thio acids and amines, albeit in very simple cases, goes back more than a century. In earlier work, when such amidation reactions were periodically described, the operating assumption had been that a thio acid is an inherent C-acyl donor.13,14 A provocative step forward was provided in 1952 by JC Sheehan, who showed that the rate of amide formation from an apparent thio acid acyl donor could be significantly increased by the use of an oxidizing agent (I2) and could be extended to a simple N-terminal amine.15 The notion of oxidation-mediated coupling of an amino acid to a thio acid was demonstrated more recently in conjectures about amide bond formation under prebiotic conditions by Orgel (using potassium ferricyanide as the oxidizing agent).,16 Formally, if stoichiometric level oxidation were to occur, elemental sulfur would be extruded. Hitherto, the oxidative version of the reaction15,16 had been predicated, quite reasonably, on the use of an established oxidizing agent at stoichiometric levels or in excess.
However, there occurred to us an alternative explanation which could better account for our findings. We came to envision a chain reaction, involving low levels of oxidative initiation with turnover to reconstitute competent C-terminal acyl donor capacity (vide infra). The findings provided herein clearly point to a substoichiometric oxidative component in the amide construction, since couplings occur much more readily and efficiently when conducted in the presence of air as opposed to under attempted oxygen exclusion. Moreover, they occur somewhat more rapidly in the presence of an established oxidizing agent, such as iodine. In order to account for the excellent yields reported above, it is necessary to chart a scenario by which low levels of oxidative initiation are magnified through an efficient pathway for reconstituting acyl donor capacity.
The exact nature of the proposed oxidative initiation still awaits definition. Presently, we can only provide a type of framework, by which small levels of oxidation can be amplified to lead to what would otherwise, perhaps simplistically, be considered a simple acylation. Certainly, many variations of that projected in Figure 3 can be entertained. Among the possibilities which have been invoked for oxidative activation (as opposed to inherent acyl donor competence of a thio acid), is a diacyl disulfide of the type 50.17 While there is nothing in our data set which establishes 50 as the oxidatively induced acyl donor species, we suggest in Figure 3 a model for a type of high turnover pathway to recycle the competent acyl donor (i.e. 50). Thus, reaction of 50 with HOBT followed by amine 51 leads to amide and per-thio acid 52. Reaction of 52 with thio acid 49 reconstitutes acyl donor 50 (and H2S). Of course, reaction of extruded thio acid 49 with 52 at the SH group (see dotted line pathway) would constitute a non-productive (invisible) reaction.
Figure 3.

It would be expected that this type of trace oxidative pathway would be accelerated by enhanced opportunities for oxidation. As described above, this too was observed. Extensive experimentation will be necessary to pin down the actual operative details among a continuum of possibilities. Not to be excluded from such a continuum is the notion that thio acids themselves have some marginal intrinsic acyl donor capacity. While direct classical acylation has the virtue of simplicity (Occam’s Razor!), our data suggest that it is unlikely to be a major participant in the chemistry we have described above, particularly for the complex cases.
Conclusion
Mechanistic uncertainties notwithstanding, the coupling of C-terminal thio acids with N-terminal peptides in the presence of HOBT holds considerable promise for accomplishing new fragment coupling that is independent of the Cys–associated logic of Native Chemical Ligation. The method has been successfully demonstrated in a range of challenging peptide bond constructions. Ligation occurs with manageably low levels of C-terminal epimerization. The method has been demonstrated to work well in the contexts of macrolactamization, glycopeptide-peptide and glycopeptide-glycopeptide constructions. The C-terminal thio acid is cleanly differentiated from O-carboxy groups on side chains. At this stage, side chain amines do require protection.
Of course, much work will be required to establish how this method of peptide bond formation best augments the current menu of NCL based strategies,6–8 SPPS,18 and other non-cysteine related ligations.19 The optimal fitting of the protocols described above to particular sequences intended for ligation will also require considerable fact gathering and field-tested data sets. However, we are already confident that ligation of C-terminal thio acids with N-terminal peptides (in the presence of HOBT or HOOBT) will be a valuable method for building homogeneous biologics, including highly complex glycopeptides, by chemical synthesis. We expect that synergy between chemistry and biology will be of value in pharmaceutical level discovery research.
Supplementary Material
Acknowledgment
Support for this research was provided by the National Institutes of Health (CA28824 to SJD). We thank Drs. Jin Chen, Xuechen Li, Yu Yuan, and Jianglong Zhu for help and advice.
Footnotes
Supporting Information Available. General experimental procedures, including spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5792–5799. doi: 10.1021/ja809554x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5424–5431. doi: 10.1021/ja808704m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danishefsky SJ, Bilodeau MT. Angew. Chem. Int. Ed. 1996;35:1830–1419. [Google Scholar]
- 4.For reviews, see: Seeberger PH, Bilodeau MT, Danishefsky SJ. Aldrichimica Acta. 1997;30:75–92. Zhu J, Warren JD, Danishefsky SJ. Expert Rev. Vaccines. 2009;8:1399–1413. doi: 10.1586/erv.09.95.
- 5.For two particularly interesting approaches to amide bond formation from non-obvious coupling partners, see: Carrillo N, Davalos EA, Russak JA, Bode JW. J. Am. Chem. Soc. 2006;128:1452–1453. doi: 10.1021/ja057706j. Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027–1033. doi: 10.1038/nature09125.
- 6.(a) Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]; (b) Dawson PE, Churchill MJ, Ghadiri MR, Kent SBH. J. Am. Chem. Soc. 1997;119:4325–4329. [Google Scholar]
- 7.Wan Q, Danishefsky SJ. Angew. Chem. Int. Ed. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 8.(a) Yang L, Dawson PE. J. Am. Chem. Soc. 2001;123:526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Banerjee A. J. Am. Chem. Soc. 2007;129:10064–10065. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]; (c) Haase C, Rohde H, Seitz O. Angew. Chem. Int. Ed. 2008;47:6807–6810. doi: 10.1002/anie.200801590. [DOI] [PubMed] [Google Scholar]; (d) Chen J, Wan Q, Yuan Y, Zhu J, Danishefsky SJ. Angew. Chem. Int. Ed. 2008;47:8521–8524. doi: 10.1002/anie.200803523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen J, Wang P, Zhu J, Wan Q, Danishefsky SJ. Tetrahedron. 2010;66:2277–2283. doi: 10.1016/j.tet.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Harpaz Z, Siman P, Kumar KSA, Brik A. ChemBioChem. 2010;11:1232–1235. doi: 10.1002/cbic.201000168. [DOI] [PubMed] [Google Scholar]
- 9.(a) Yuan Y, Zhu J, Li X, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:2329–2333. doi: 10.1016/j.tetlet.2009.02.205. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rao Y, Li X, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:12924–12926. doi: 10.1021/ja906005j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.König W, Geiger R. Chem. Ber. 1970;103:788–798. doi: 10.1002/cber.19701030319. [DOI] [PubMed] [Google Scholar]
- 11.The C-terminal thioacids were prepared by well known simple methods, as described in the Supporting Information. cf. inter alia: Crich D, Sharma I. Angew. Chem. Int. Ed. 2009;48:2355–2358. doi: 10.1002/anie.200805782. Crich D, Sharma I. Angew. Chem. Int. Ed. 2009;48:1–5. doi: 10.1002/anie.200903050. Vetter S. Synth. Commun. 1998;28:3219–3223.
- 12.Kovacs J. In: Racemization and Coupling Rates of N-a-Protected Amino Acid and Peptide Active Esters: Predictive Potential, In The Peptides. Gross E, Meienhofer J, editors. Vol. 2. New York: Academic; 1980. pp. 485–539. [Google Scholar]
- 13.A more complete review of the relevant literature is provided in the Supporting Information. Selected references: Pawlewski B. Chemische Berichte. 1898;31:661–663. Gunter H. Chemische Berichte. 1950;83:258–261.
- 14.Rosen T, Lico IM, Chu DTW. J. Org. Chem. 1988;53:1580–1582. [Google Scholar]
- 15.Sheehan JC, Johnson DA. J. Am. Chem.Soc. 1952;74:4726–4727. [Google Scholar]
- 16.Liu R, Orgel LE. Nature. 1997;389:52–54. doi: 10.1038/37944. [DOI] [PubMed] [Google Scholar]
- 17. Yamashiro D, Li. CH. Int. J. Peptide Prorein Res. 1988;31:322–334. doi: 10.1111/j.1399-3011.1988.tb00040.x. Høeg-Jensen T, Olsen CE, Holm A. J. Org. Chem. 1988;59:1257–1263. (c) For a different but related variation of the framework suggested in Figure 3, see: Liu C-F, Rao C, Tam JP. Tetrahedron Lett. 1996;37:933–936.
- 18.Merrifield RB. J. Am. Chem.Soc. 1963;85:2149–2154. [Google Scholar]
- 19.(a) Blake J, Li CH. Proc. Natl. Acad. Sci. 1981;78:4055–4058. doi: 10.1073/pnas.78.7.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen G, Wan Q, Tan Z, Kan C, Hua Z, Ranganathan K, Danishefsky SJ. Angew. Chem. Int. Ed. 2007;46:7383–7387. doi: 10.1002/anie.200702865. [DOI] [PubMed] [Google Scholar]; (c) Payne RJ, Ficht S, Greenberg WA, Wong CH. Angew. Chem. Int. Ed. 2008;47:4411–4415. doi: 10.1002/anie.200705298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.