

Abstract
It has been reported previously that keratin 8 (K8)-deficient mice of one strain die from a liver defect at around E12.5, while those of another strain suffer from colorectal hyperplasia. These findings have generated considerable confusion about the function of K8, K18 and K19 that are co-expressed in the mouse blastocyst and internal epithelia. To resolve this issue, we produced mice doubly deficient for K18 and K19 leading to complete loss of keratin filaments in early mouse development. These embryos died at around day E9.5 with 100% penetrance. The absence of keratins caused cytolysis restricted to trophoblast giant cells, followed by haematomas in the trophoblast layer. Up to that stage, embryonic development proceeded unaffected in the absence of keratin filaments. K18/19-deficient mouse embryos die earlier than any other intermediate filament knockouts reported so far, suggesting that keratins, in analogy to their well established role in epidermis, are essential for the integrity of a specialized embryonic epithelium. Our data also offer a rationale to explore the involvement of keratin mutations in early abortions during human pregnancies.
Keywords: embryonic lethal/intermediate filaments/keratin/knockout/trophoblast giant cell
Introduction
The formation of a blastocyst, consisting of an epithelium (trophectoderm) and an inner cell mass, is one of the earliest differentiation steps during mammalian embryogenesis. Like in adult epithelia, the formation of embryonic epithelia relies on the interaction of cytoskeletal proteins with specialized membrane attachment sites. Among the first cytoskeletal genes differentially expressed in the trophectoderm are keratins (Brulet et al., 1980; Jackson et al., 1980; Paulin et al., 1980; Oshima et al., 1983; Herrmann and Harris, 1998), desmoplakin and plakoglobin of desmosomes (Schwarz et al., 1990) and E-cadherin and catenins, which are components of adhaerens junctions (Takeichi, 1995). In order to study the function of these proteins, null mutations have been introduced in the corresponding genes. E-cadherin- and α-catenin-deficient embryos fail to form a trophectoderm and die before implantation (Larue et al., 1994). β-catenin-null mice were unable to form the mesoderm layer and anterior–posterior structures, leading to their death at around E6.5 (Haegel et al., 1995; Huelsken et al., 2000). Notably, endogenous plakoglobin replaced β-catenin at adhaerens junctions, arguing that their death was probably caused by deficient wnt signalling and not by a cytoskeletal deficiency (Huelsken et al., 2000). The targeted deletion of desmoplakin, the major cytoplasmic attachment site of keratin intermediate filaments (IF) to desmosomes, results in embryonic death at around E6.5 and has been interpreted as a failure in cell–cell adhesion followed by a collapse of keratin IF (Gallicano et al., 1998). Plakoglobin-null mice die primarily from heart rupture at mid-gestation, due to disturbed adhesion of myocardial cells (Bierkamp et al., 1996;Ruiz et al., 1996). Collectively, these results show that proteins responsible for the membrane attachment of the cytoskeleton are required for the normal development of the mouse embryo.
Keratins form the IF of all epithelia and are assembled from heterodimeric subunits of acidic type I [keratin 9 (K9) to K20] and basic type II (K1–K8) proteins (Moll et al., 1982). K8 and K18 are the first keratins expressed during mouse development, starting at the eight-cell stage of the mouse embryo (Brulet et al., 1980; Jackson et al., 1980; Paulin et al., 1980; Oshima et al., 1983), followed by K19 and K7 (M.Hesse and T.M.Magin, unpublished). At later stages, they are expressed in the trophectoderm and its derivatives, and in the embryonic and extra-embryonic endoderm (Jackson et al., 1981). Because of their early expression it has been assumed that these keratins have an important function during embryogenesis. However, knockout mice for K18 (Magin et al., 1998) and K19 (Harada et al., 1999) do not display early embryonic defects. This was shown to result from the mutual compensation of these type I keratins, in agreement with previous data showing that both keratins formed IF with K8 in vitro and in transfected cells (Hatzfeld and Weber, 1990; Bader et al., 1991). The knockout of K8 (Baribault et al., 1993, 1994), on the other hand, has generated considerable confusion regarding the function of keratins during embryonic development and in internal epithelia. In C57BL/6 mice, 98.4% of all K8-deficient embryos died at around E12.5 (Baribault et al., 1993), while in FVB/N mice, the embryonic lethality was overcome in 55% of the offspring. Surviving mice in this genetic background developed colorectal hyperplasia and females had reduced reproductive capacity (Baribault et al., 1994). Remark ably, the analysis of transgenic mice expressing human K18 with a mutation similar to that found in K14, where it caused the severe skin disorder epidermolysis bullosa simplex (Corden and McLean, 1996), resulted in very minor fragility of hepatocytes (Ku et al., 1995, 1996). In the latter mice, no phenotypic abnormalities were noted in embryos. Taken together, these data seemed to suggest that keratins were not essential during embryonic development. Moreover, there appeared to be a general consensus that keratin IF of internal epithelia are not required primarily as cytoskeletal proteins, but to provide other functions like binding of 14-3-3 proteins and mediating tumour necrosis factor (TNF) signalling (Ku et al., 1999; Caulin et al., 2000). These findings are in strong contrast to the phenotype arising from point mutations of epidermal keratins in humans (Corden and McLean, 1996) or corresponding knockout mice, both of which lead to severe skin fragility syndromes (Lloyd et al., 1995).
Given the complexity of keratin expression in embryonic epithelia including the trophectoderm, we were intrigued by the potential of certain keratins to replace each other (Magin et al., 1998), which could explain the absence of a phenotypic abnormality in some keratin knockout mice. This could be of functional significance in trophoblast giant cells, which express K7, K8, K18 and K19 (this manuscript). Trophoblast giant cells are derivatives of the mural trophectoderm (primary giant cells) and the ectoplacental cone (EC), the latter giving rise to the spongiotrophoblast layer and the chorionic ectoderm (Cross et al., 1994). The outermost cells of the EC differentiate into secondary trophoblast giant cells, which, together with primary trophoblast giant cells, finally surround the whole conceptus, establishing contact with maternal decidual cells. Together with the parietal endodermal cells that form Reichert’s membrane, they build the earliest placental structure (Cross et al., 1994). The importance of trophoblast giant cells for embryonic development is underlined by the phenotype of basic helix–loop–helix transcription factor knockout mice. While hand-1 deficiency arrests embryonic development at E7.5 due to a deficiency of trophoblast giant cells and a smaller EC (Firulli et al., 1998; Riley et al., 1998), Mash-2 knockout mice die at E10.5 from a decrease of the spongiotrophoblast layer (Guillemot et al., 1994).
In order to investigate the function of keratins during early embryonic development and to resolve some of the inconsistencies resulting from the previous K8 knockout, we have generated mice doubly deficient for K18 and K19 by mating K18–/– with K19–/– mice. In these animals, all embryonic type I keratins have been abolished. This left only the type II keratins, K7 and K8, which, like all other keratins of the same subfamily, are unable to form homopolymeric IF (Steinert et al., 1976; Kulesh et al., 1989; Magin et al., 1990). As a consequence, the formation of keratin IF (Steinert et al., 1976) was abolished (Hatzfeld and Franke, 1985). Here we show that K18/19 doubly deficient mice suffer from early embryonic lethality with 100% penetrance due to the fragility of trophoblast giant cells, demonstrating a primary cytoskeletal function of these keratins.
Results
Embryonic lethality in K18/19 double-null mice
Doubly deficient mice were generated by crossing K18–/– mice in a 129/Sv background (Magin et al., 1998) with K19–/– mice in a C57BL/6 × FVB/N background (Y.Tamai, T.Ishikawa, M.R.Bösl, M.Mori, M.Nozaki, H.Baribault, R.G.Oshima and M.M.Taketo, in preparation). Inbreeding of the resulting F1 generation resulted in a strong prevalence of K18+/–K19–/– over K18–/–K19+/– animals in a ratio of 3:1, which we have not analysed further. As a result of intercrossing K18+/–K19–/– mice, no doubly deficient mice were born, suggesting an embryonic lethal phenotype with 100% penetrance. To exclude effects of the mixed genetic background on the phenotype of doubly deficient mice, K18+/–K19–/– matings were observed for five generations; no changes in the Mendelian distribution were seen (Table I). Based on the observation that K8–/– mice died at around E12.5 in the C57BL/6 background (Baribault et al., 1993), concepti of K18+/–K19–/– intercrosses were dissected at E13; this revealed that several embryos were resorbed, as indicated by blood-filled implantation sites. Later on, those were confirmed to be doubly deficient. Genotyping of embryos dissected from K18+/–K19–/– intercrosses on days 9.5 and 10.5 post-coitum showed a Mendelian distribution (Table I). Therefore, embryonic death must have occurred between E9.5 and 10.5. The deviation from the mean seen with E10.5 K18–/–K19–/– embryos was due to a small number of embryos in the state of absorption. These could not be genotyped because of contamination with maternal tissue and blood.
Table I. Genotype analysis of newborn and E9.5 and E10.5 embryos from K18–/+K19–/– matings.
| Genotype | Newborn | E9.5 embryos | E10.5 embryos | |||
|---|---|---|---|---|---|---|
| |
Total No. |
Experimental ratio |
Total No. |
Experimental ratio |
Total No. |
Experimental ratio |
| K18–/+K19–/– | 257 | 2 | 74 | 2 | 33 | 1.9 |
| K18+/+K19–/– | 126 | 1 | 37 | 1 | 17 | 1 |
| K18–/–K19–/– | 0 | 0 | 32 | 0.9 | 13 | 0.8 |
| Average litter size | 6.5 | 9.6 | 10.3 | |||
The deviation from the mean seen with E10.5 K18–/–K19–/– embryos was due to a small number of embryos in the state of absorption. These could not be genotyped because of contamination with maternal tissue and blood. Adult animals were genotyped by PCR analysis. E9.5 and E10.5 embryos were dissected from the decidua and the extra-embryonic tissues and lysed for PCR analysis.
Excessive bleeding in extra-embryonic tissues of doubly deficient mice
Starting on about day 11, peaking on day 12 and vanishing on day 14 post-coitum, strong bleeding from the vaginas of pregnant females of K18+/–K19–/– intercrosses was noted (data not shown). While bleeding on day 12 or 13 of gestation is natural (Rugh, 1968), there was more bleeding than usual in these mice.
Dissection revealed haematomas at some implantation sites of E10.5 uteri (Figure 1A). Further dissection of the uterine wall and parts of the decidua revealed blood between the maternal decidual tissue and the embryonic yolk sac without penetrating the latter (Figure 1B). These embryos were later identified as doubly deficient. The yolk sac of K18–/–K19–/– mice on E9.5 appeared paler than the wild-type yolk sac; 1 day later (E10.5) it remained pale and imploded (Figure 1C and D). E9.5 embryos doubly deficient for K18 and K19 were slightly smaller than their littermates but otherwise appeared normally developed (Figure 1E). As double knockout embryos showed necrosis and their growth was significantly retarded at E10.5 (Figure 1F), we conclude that the absence of keratin IF caused embryonic death around E9.5. Extensive analysis of many embryos revealed that bleeding usually started between E9 and E9.5; in a small number of cases it began at E10 (data not shown).
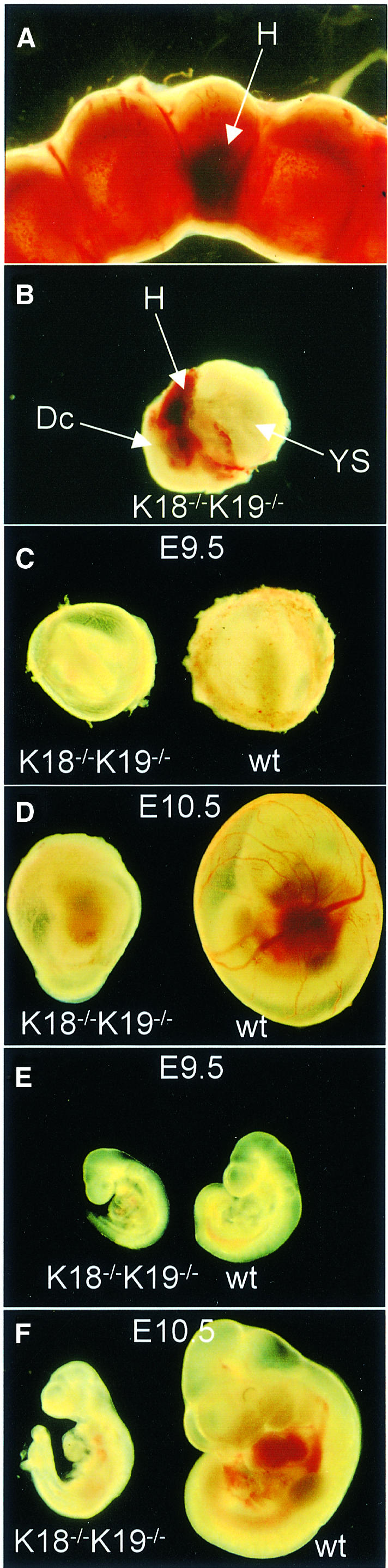
Fig. 1. Appearance of K18–/–K19–/– extra-embryonic tissues and embryos. (A) An E10.5 doubly deficient embryo (middle) with its littermates adjacent in its implantation site in utero. Note the decreased size and the haematoma (H) located on the anti-mesometrial side (arrow). (B) Dissected E10.5 doubly deficient embryo in its implantation site. The uterine wall and parts of the decidua were dissected. Bleeding occurred between maternal decidual tissue (Dc) and the yolk sac (YS). (C and D) Comparison between yolk sacs of wild-type (wt) and doubly deficient mice. (C) At E9.5, yolk sacs of K18–/–K19–/– mice were slightly smaller and of pale appearance. (D) Yolk sacs of E10.5 K18–/–K19–/–-mice were significantly smaller, pale and deformed. (E and F) Comparison of wild-type and doubly deficient embryos. (E) Doubly deficient embryos on E9.5 were slightly growth retarded but otherwise appeared normal. (F) One day later, double mutant embryos were significantly growth retarded, necrotic and in the process of absorption.
Formation of K8-positive aggregates in trophoblast giant cells, yolk sac and gut
In contrast to previous findings (Baribault et al., 1993, 1994; Ku et al., 1995, 1996; Magin et al., 1998), our morphological findings established that keratins are essential during mouse embryogenesis. To study whether embryos showed an altered distribution of other cytoskeletal proteins known to interact with K18 and K19, indirect immunofluorescence with antibodies against several keratins and desmoplakin was performed. We also analysed whether the type III IF proteins, vimentin and desmin, were induced or whether actin became reorganized to rescue the keratin deficiency.
Embryos were cryosectioned in their implantation sites in the uterus and analysed by immunofluorescence. K18, which in wild-type mice is strongly expressed in trophoblast giant cells, EC, parietal and visceral yolk sac, amnion, surface ectoderm and primitive gut, was completely absent in double knockout mice (Figure 2E and F). The staining of uterine glands was due to the maternal K18+/–K19–/– genotype, which allowed the formation of sparse but normal K8/18 filaments. Most surprisingly, K8, the typical partner of K18, was not completely absent in K18/19 doubly deficient embryos. In those tissues that contained large amounts of K8 in normal mice (Jackson et al., 1981; see also Figure 2C) we detected fine K8 aggregates in doubly deficient embryos (Figure 2D). These aggregates were found mainly in trophoblast giant cells, cytotrophoblast, primitive gut and, most prominently, in the yolk sac (Figure 3A and G). They were located apically in the cell periphery and were reminiscent of K8 aggregates in K18–/– (Magin et al., 1998, 2000) or K1 aggregates in K10T knockout mice (Porter et al., 1996). Double staining with desmoplakin antibodies revealed a localization close to but not at desmosomes (Figure 3C and G). We noted that intact trophoblast giant cells contained numerous desmosomes, as in the wild type (Figure 3E), but showed large K8 aggregates in a predominantly apical distribution, while in the controls a dense meshwork of keratin filaments was observed (Figure 3D). In contrast, trophoblast giant cells cytolysing due to the absence of keratins showed a drastically reduced number of desmosomes. This demonstrates that desmosomes in trophoblast giant cells are not sufficient to protect them from mechanical stress.

Fig. 2. Lack of K7, K18 and K19 in K18–/–K19–/– mice. (A and B) Immunofluorescence analysis of K7. (A) Strong staining of trophoblast giant cell layer in wild-type implantation sites. Maternal uterine glands and epithelia were stained weakly (arrows). (B) Positive staining of uterine epithelia and glands (arrows) but absence in doubly deficient extra-embyronic and embryonic compartments. (C and D) Staining for K8. (C) In wild-type animals, trophoblast giant cells, early placenta and maternal uterine glands and epithelia (arrows) were strongly positive. Parietal and visceral yolk sac, amnion, embryonic surface ectoderm, notochord and gut were also positive for K8. (D) Implantation site containing a doubly deficient embryo displayed strong staining in maternal uterine epithelia and glands (arrows). Weak staining was noted in the trophoblast giant cell layer and embryonic gut corresponding to K8 aggregates. (E and F) Staining for K18. (E) In wild type, the trophoblast giant cell layer and visceral yolk sac were strongly positive for K18. The amnion, embryonic ectoderm, gut and notochord were also positive. Maternal uterine glands and epithelia were stained. (F) Only maternal epithelia and glands were positive in doubly deficient implantation sites (arrows). (G and H) Staining for K19. (G) The trophoblast giant cell layer and maternal uterine epithelia were strongly positive. Weaker staining was observed in amnion, surface ectoderm, gut and notochord. (H) No staining was noted in doubly deficient embryos. The lack of staining in maternal tissues was due to the K19–/– genotype of parental mice. Bar, 1 mm.

Fig. 3. K8 aggregates in extra-embryonic compartments of K18–/–K19–/– (–/–) mice. (A–F) Indirect immunofluorescence staining of E9.5 cryosections of wild-type and K18–/–K19–/– extra-embryonic compartments. (A) Staining for K8 in the visceral yolk sac (VYS) and giant trophoblast (gT) cell layer. Note the apically located aggregates in endodermal cells of the visceral yolk sac and in the trophoblast giant cells. (B) Staining for desmoplakin of the same section as in (A). Note the large number of desmosomes in the trophoblast giant cells not altered compared with wild-type. (C) Double staining for K8 and desmoplakin. (D) Staining for K8 in wild-type (+/+) trophoblast giant cells and visceral yolk sac. Both compartments were strongly positive for K8. (E) Staining of the same section as in (D) for desmoplakin. (F) Double staining for K8 and desmoplakin. Desmosomes co-localized with the keratin filaments. (G–J) Immunofluorescence analysis of fragile trophoblast giant cells at the edges of tissue separation and haematoma 9.5 days post-coitum. The area displayed here was similiar to that shown in Figure 4C. (G) Immunofluorescence staining of giant trophoblast cells at the border of a split between decidual tissue (Dc) and parietal yolk sac, partly in the process of cytolysis. Triple immunofluorescence (K8, desmoplakin, DAPI) revealing aggregates in the cytoplasm of trophoblast giant cells (K8, red fluorescence) and showing colocalization between some but not all K8 aggregates and desmosomes (yellow fluorescence). Cytolysing trophoblast giant cells lacked desmoplakin staining (green fluorescence) at the edge of the split. Note the enormous size of giant trophoblast nuclei (blue fluorescence). (H–J) Immunofluorescence analysis of desmosomes in trophoblast giant cells (n = nucleus) of doubly deficient embryos. (H) Note the large amount of apically located desmosomes in intact trophoblast giant cells. (I and J) Trophoblast giant cells at the edge of maternal blood with cytolysis and loss of contact. Desmoplakin staining revealed a discontinous distribution or missing of desmosomes (arrows in I). Bar, 100 µm.
The expression of K7 in K18–/–K19–/– mice was restricted to uterine epithelia and glands, while, in normal littermates, trophoblast giant cells, EC, surface ectoderm and primitive gut were stained (Figure 2A and B). In line with the maternal K19–/– genotype, no K19 was detectable (Figure 2H). In wild-type mice, trophoblast giant cells, EC, amnion, surface ectoderm and primitive gut expressed K19 (Figure 2G). The expression or distribution of desmin, vimentin and of F-actin was not altered in doubly deficient mice (data not shown).
Absence of a keratin cytoskeleton leads to cytolysis and necrosis of trophoblast giant cells
In order to address the functional consequences of the absence of keratin IF, we performed histological analysis of K18/19 doubly deficient embryos. The results showed that individual trophoblast giant cells were cytolytic (Figure 4B), allowing maternal blood to penetrate into the extra-embryonic tissues where it formed large haematomas. These reached but did not penetrate Reichert’s membrane, where it deformed the parietal and visceral yolk sacs. Analysis of many doubly deficient embryos revealed that bleeding occurred mainly on the anti-mesometrial side (decidua capsularis). At higher magnification, several trophoblast giant cells (arrows in Figure 4C) displayed a ruptured cytoplasm or were even missing and therefore must have been cytolysed earlier. The infiltration site clearly resulted from maternal blood and contained non-nucleated erythrocytes, fibrin aggregations and infiltrated granulocytes. The lack of lymphocytes confirmed the absence of a maternal immune reaction. Serial sectioning of implantation sites of several embryos confirmed that the blood always originated from capillaries in the decidual tissues surrounding the embryo (Figure 4C). The visceral and parietal yolk sacs showed no morphological abnormalities. Reichert’s membrane was formed and in the visceral yolk sac blood vessels and nucleated embryonic erythrocytes had developed normally (Figure 4C).

Fig. 4. Bleeding and giant trophoblast fragility in K18–/–K19–/– mice (A and B) Sagittally sectioned wild-type and doubly deficient E9.5 embryos. (A) In the wild-type embryos the barrier between embryonic and maternal compartments is formed by giant trophoblast (gT) cells (arrows). The visceral yolk sac (VYS), placenta (P) and amnion (A) are clearly visible. (B) Note the haematoma (H) in the mutant embryo, which is deforming the parietal and visceral yolk sacs. In the region with the haematoma, trophoblast giant cells surrounding the conceptus (arrows) were destroyed by cytolysis. Bar, 500 µm. (C) Higher magnification of (B). The layer of trophoblast giant cells is disrupted, with trophoblast giant cells in the process of cytolysis (arrows). Cytolysed trophoblast giant cells were surrounded by granulocytes. The origin of blood was a maternal vessel in the decidual tissue (Dcv). After breakdown of the trophoblast giant cell layer, maternal blood entered between the parietal yolk sac (PYS) and decidual tissue (Dc) unimpaired. The haematoma consisted of maternally derived erythrocytes, fibrin (Fi) and infiltrated granulocytes. Note that Reichert’s membrane (basal lamina in PYS) was not penetrated by blood. Em, embryo; ∗, visceral yolk sac. Bar, 200 µm.
At E9.5, the embryo proper of the doubly deficient mouse was slightly smaller than that of its littermates, but histological analysis showed no morphological abnormalities (Figure 4B and data not shown). Doubly deficient embryos at E10.5 were growth retarded compared with wild-type mice (Figure 5A and B) and completely necrotic (Figure 5C). This was possibly due to the inability of the yolk sac to provide the embryo with nutrients. The visceral yolk sac displayed normal vascularization and contained embryonal, nucleated blood cells (data not shown). In E10.5 mutant embryos, decidual cells adjacent to cytolysed trophoblast giant cells exhibited necrosis accompanied by fibrin clots and granulocytes (Figure 5D). A careful inspection of K18/19 doubly deficient embryos revealed no morphological abnormalities in other keratin-expressing extra-embryonic or embryonic tissues. Placentas of doubly deficient embryos were smaller in diameter but nevertheless appeared normally developed (Figure 6).
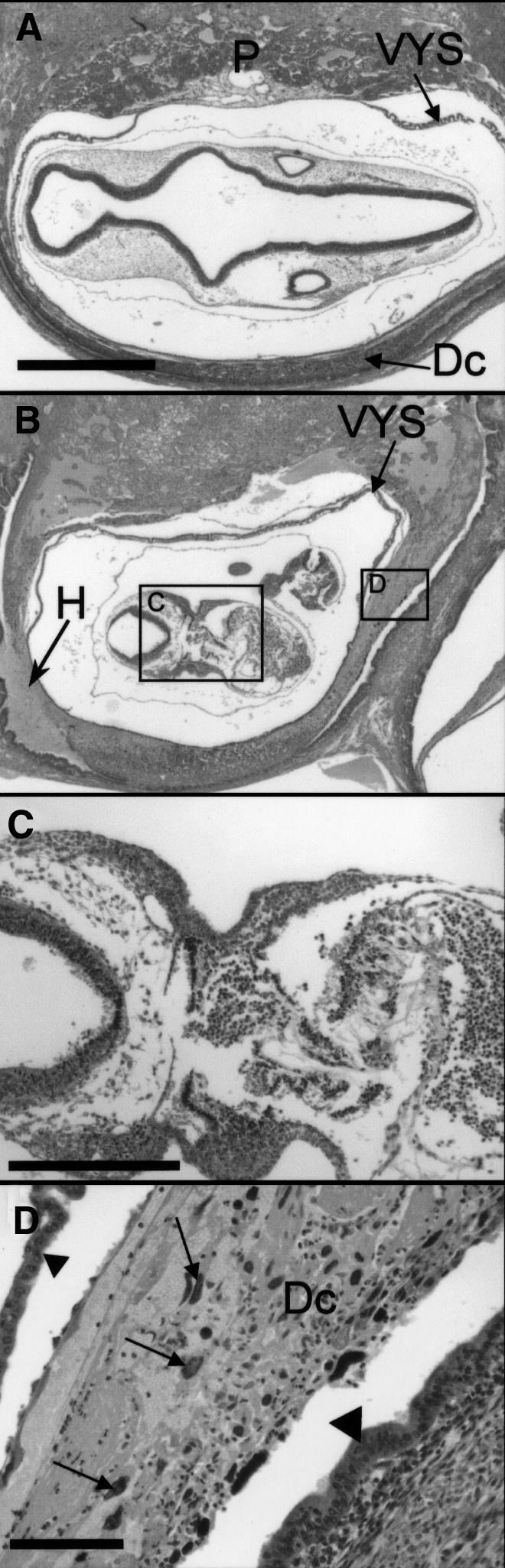
Fig. 5. Histological analysis of E10.5 double-null and wild-type embryos. (A) Wild-type embryo with intact extra-embryonic membranes, like visceral yolk sac (VYS), placenta (P) and decidual tissue (Dc). The embryo was sectioned through the head, showing rhombencephalon and otic vesicles. (B) Same magnification as (A). The K18–/–K19–/– embryo showed intact parietal and visceral yolk sac, but severe necrosis of giant trophoblast and decidual cells accompanied by bleeding (H) into the uterine lumen. The placenta, which is not visible in this section, was smaller than that of wild-type embryos. Bar, 1 mm. (C) Higher magnification of (B). The embryo showed necroses and died on ∼E10. Bar, 300 µm. (D) Higher magnification of (B). Necrosis of giant trophoblast (arrows) and decidual cells (Dc). The small triangle marks the visceral yolk sac and the large triangle marks the uterine epithelium. Fibrin and infiltrated granulocytes were noted between the dead cells. Bar, 100 µm.
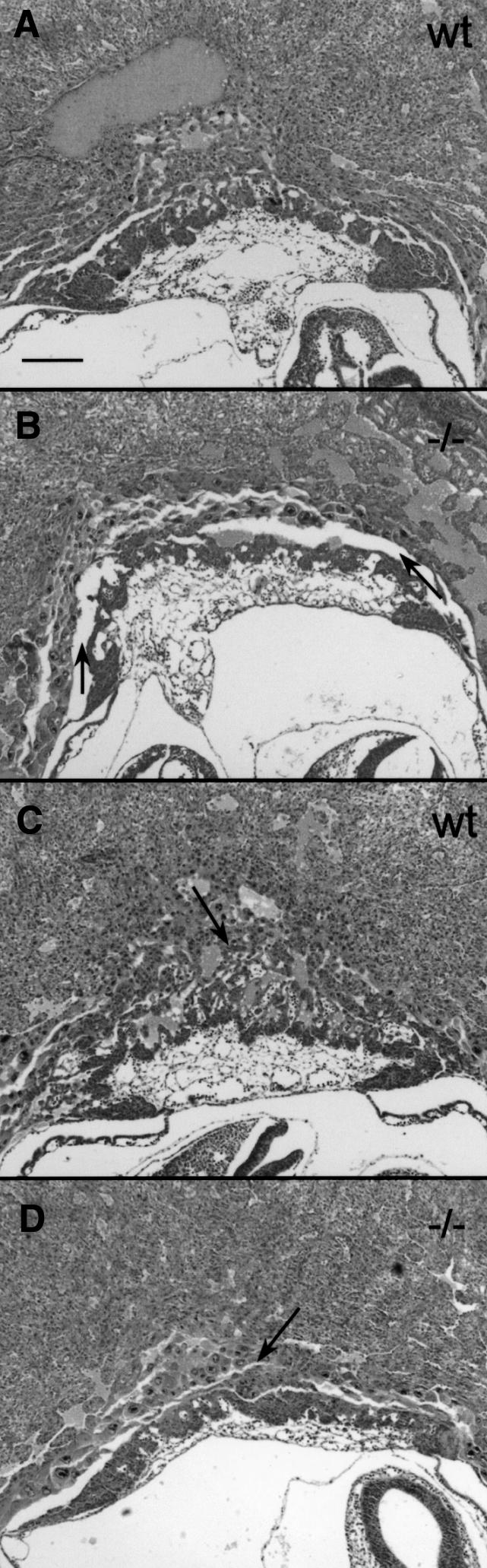
Fig. 6. Placentas of K18–/–K19–/– (–/–) and wild type (wt) mice 9.5 days post-coitum. (A and C) Placentas of wild-type mice. Note the well developed labyrinthian layer and the deeply invaded giant trophoblast cells (arrows). (B and D) Placentas of K18–/–K19–/– mice. Note the cleft between the trophoblast giant cells and the spongiotrophoblast (arrows in B) and the poorly developed labyrinth (arrow in D). Bar, 200 µm.
The notion that keratins play an essential role in trophoblast giant cells was supported by immunofluorescence analysis of trophoblast giant cells in affected E9.5 K18–/–K19–/– embryos adjacent to maternal blood (Figure 3I) or at the region where trophoblast cells began to separate from maternal tissue as a consequence of cytolysis (Figure 3G). Cytolysing trophoblast giant cells were devoid of a keratin cytoskeleton but contained K8 aggregates (Figure 3G, red fluorescence). Remarkably, desmoplakin staining on the cytoplasmic site adjacent to the region of tissue separation was missing but maintained at the sites of cell–cell contact (Figure 3G, green fluorescence). The nuclei of trophoblast giant cells, easily detectable due to their enormous size, showed no apparent signs of apoptosis (Figure 3G, blue fluorescence). Triple immunofluorescence revealed that some of the K8-positive aggregates co-localized with desmoplakin (Figure 3G). In intact trophoblast giant cells, numerous desmosomes were apparent and showed an apical distribution (Figure 3H), similar to the situation in the wild-type (Figure 3E). Adjacent to the rupture of a blood vessel, some trophoblast giant cells were still intact while others showed cytolysis (Figure 3I and J). In a minor trophoblast giant cell population, desmoplakin staining was noted in the cytoplasm (Figure 3G and J). Disturbance of the desmosomal layer corresponded to cytolysis (arrows in Figure 3I). We concluded that, in analogy to keratins in stratified epithelia, which are required to maintain an intact epidermis (Fuchs and Weber, 1994), those of internal epithelia are crucial for supporting the integrity of trophoblast giant cells under conditions of mechanical stress.
Western blot analysis of K7/8/18/19 expression in wild-type and mutant E9.5 embryos
In order to investigate whether a large amount of protein might coincide with a structural requirement for keratins during embryonic development, we prepared protein extracts from dissected E10.5 wild-type and mutant embryos and yolk sacs. In support of this idea, large amounts of K8 and K18 were detected by western blotting in wild-type yolk sacs. A lower but detectable amount was found in embryo preparations (Figure 7A and B). In agreement with our immunofluorescence data, K7 and K19 were absent in yolk sac extracts (Figure 7C and D). K7, K18 and K19 were absent in mutant embryos and yolk sacs. A very small amount of K8 in both mutant embryo and yolk sac corresponded to the K8 aggregates found by indirect immunofluorescence analysis (Figure 7A).

Fig. 7. Western blot analysis of total proteins from K18–/–K19–/– (–/–) and wild-type (+/+) embryos (E) and yolk sacs (Y) separated by SDS–PAGE. Detection of (A) K8, (B) K18, (C) K19 and (D) K7. (A) Note the large amount of K8 in wild-type yolk sacs and embryos compared with the small amount in K18–/–K19–/– embryos and yolk sacs. K8 detectable by western blotting probably corresponded to K8 aggregates detected by immunofluorescence. (B) K18 was absent in doubly deficient yolk sacs and embryos but present in normal amounts in wild-type embryos. There was much more K8 and K18 in wild-type yolk sacs than in wild-type embryos (A and B). K7 and K19 were only detectable in wild-type embryos and not in doubly deficient embryos (C and D); they were absent in wild-type and doubly deficient yolk sacs.
Our analysis has clarified the role of keratins during mouse development. It has also helped to dissect the different role of desmoplakin and keratin IF during early mouse development: we have shown that the presence of an intact keratin cytoskeleton is crucial in a subset of embryonic epithelia, namely the trophoblast giant cell layer of the mouse embryo. Based on the normal development of K18/19 doubly deficient embryos up to E9, we conclude that the collapse of keratin IF occurring in desmoplakin-deficient mice, which die at E6.5, does not exacerbate their phenotypic abnormalities (Gallicano et al., 1998). Remarkably, cell fragility resulting from the ablation of one of these proteins, which physically interact (Kouklis et al., 1994) and are co-expressed during development, results in distinct phenotypes.
Discussion
Protection against mechanical stress is a unified function for all keratins
Previous work, including gene ablations for IF such as desmin (Li et al., 1996; Milner et al., 1996), vimentin (Colucci-Guyon et al., 1998), glial fibrillary acidic protein (Gomi et al., 1995; Pekny et al., 1995), neurofilaments (Elder et al., 1998a,b; Rao et al., 1998; Zhu et al., 1998) and lamin A (Sullivan et al., 1999), had suggested that IF proteins may not be required during embryogenesis (Magin et al., 2000). The lack of phenotypic abnormalities in embryos of mice deficient for K18 or K19 was explained by the ability of these two keratins to replace one another (Magin et al., 1998; Harada et al., 1999). In strong contrast, K8-deficient mice were reported to die from a liver defect at around E12.5 in one mouse strain and to suffer from colorectal hyperplasia in another (Baribault et al., 1993). These findings, the molecular basis of which has not been resolved up to now, have generated considerable confusion about the function of keratins during mouse embryogenesis (Baribault et al., 1994; Magin et al., 1998; Harada et al., 1999). Following the deletion of K18 and K19, we show here that the lack of a keratin cytoskeleton makes trophoblast giant cells fragile and causes embryonic lethality with 100% penetrance at around E9.5. At the same time, embryonic epithelia appeared unaffected. This suggests that the extent of mechanical stress occurring during folding of embryonic epithelia is not sufficient to cause cytolysis. The trophoblast phenotype is reminiscent of the fragility of epidermal keratinocytes caused by dominant-negative keratin mutations (Fuchs and Weber, 1994) and strongly suggests that all keratins, including those of embryonic and internal epithelia, are bona fide and essential cytoskeletal proteins. While this does not preclude additional functions emerging in recent studies (Ku et al., 1999; Caulin et al., 2000; Stumptner et al., 2000), it re-emphasizes the need for future studies using conditional gene ablations.
What is the mechanism of cytolysis of trophoblast giant cells?
Although keratins form a cytoskeleton beginning at the eight-cell stage (Oshima et al., 1983) and are present in all embryonic epithelia, our findings demonstrate that early embryonic development can proceed through implantation in their absence. Why does the loss of keratin IF lead to cell fragility exclusively in trophoblast giant cells between E9.0 and E9.5 of mouse development? Because of their large size, trophoblast cells seem to rely upon their keratin cytoskeleton to withstand mechanical stress. This would be compatible with the interpretation of keratin phenotypes in epidermal keratinocytes resulting either from point mutations (Corden and McLean, 1996) or gene deletions (Lloyd et al., 1995; Porter et al., 1996). It has been argued that, in keratinocytes, an intact cytoskeleton is required at sites where the epidermis has to sustain increased mechanical stress (Fuchs and Cleveland, 1998). In an analogous way, trophoblast giant cells need to withstand the pressure of maternal blood. In keeping with this function, they are equipped with numerous desmosomes and a dense array of keratin IF (see Figure 3D–F). Following the loss of keratin IF, the large trophoblast cells seem to rupture. Does this observation exclude a structural function of keratins of internal epithelia in smaller, ‘normal sized’ cells? There is evidence from K18 and K19 single-knockout mice suggesting no other essential function of these keratins during early embryonic stages. As K8 and K18 are the only keratins expressed in the visceral yolk sac, there was a complete loss of keratin filaments in the K18 and K8 knockout mice, which are both viable in the appropriate genetic background (Baribault et al., 1993; Magin et al., 1998). It remains important to explain why K8–/– mice of the FVB/N strain (Baribault et al., 1994) are much more viable than those of the C57BL/6 strain (Baribault et al., 1993).
Another possibility is that, in trophoblast giant cells, keratins have an additional non-cytoskeletal function. Rodent trophoblast giant cells produce prolactin-like hormones to sustain production of progesterone from the corpora lutea in the ovary (Cross et al., 1994). Trophoblast keratins may be indirectly involved in the synthesis or secretion of hormones by providing attachment sites for associated proteins or enzymes (Suzuki et al., 1998; Ku et al., 1999; Tang et al., 2000). Consequently, prolactin synthesis or secretion could be modified even before cytolysis of trophoblast giant cells. In fact, several keratins have been shown to have other functions besides structural ones (Ku et al., 1998). Recently, it was reported that K8 and K18 can affect TNF signalling and that K8- and K18-deficient mice are more susceptible to TNF-dependent apoptotic liver damage (Caulin et al., 2000). K8 and K18 are both capable of binding the cytoplasmic domain of TNFR2 and moderate TNF-mediated NF-κB activation. Embryonic death could also result from a maternal immune reaction. Our histological data, however, have not revealed the presence of lymphocyte infiltrates. Therefore, we conclude that the phenotype was not caused by a maternal immune response.
Collectively, the data on available keratin gene knockouts suggest that those embryonic compartments that express relatively low levels of keratins are not affected by their absence, while extra-embryonic tissues like the trophoblast giant cells, which are characterized by a dense desmosome keratin array, suffer defects of structural origin. This is reminiscent of human skin disorders, like epidermolysis bullosa simplex or epidermolytic hyperkeratosis (Corden and McLean, 1996). The double-null mutation in K18/19 mice may therefore be considered a functional equivalent of a dominant-negative point mutation. In light of our histological data which reveal close similarity to keratinocyte fragility resulting from keratin mutations, we interpret the phenotype of our keratin-deficient embryos as resulting from mechanical fragility.
Comparison of embryos deficient for keratins and desmosomal proteins
Keratin IF interact with desmosomes via desmoplakin and other constituent proteins (Jackson et al., 1981). They establish the IF cytoskeletal architecture in epithelia; this is already formed in the trophectoderm layer at E3.5 in the mouse (Jackson et al., 1980, 1981). Both of them are encoded by gene families that are differentially expressed in various epithelia (Kowalczyk et al., 1999). Remarkably, deletion of certain constituent proteins of either gene family leads to embryonic lethality, whereas inactivation/mutation of others allows development to term.
Desmoplakin knockout mice show mechanical fragility in the endoderm and EC due to a weakening of cell–cell junctions, dying at about E6.5 (Gallicano et al., 1998). Desmoplakin-null embryos not only lacked typical desmosomes but suffered from disrupted keratin filaments. This left open the possibility that their defect could have resulted not only from the inability to form proper desmosomes but also from a collapse of the keratin cytoskeleton. As we have shown here, the latter seems dispensable before E9.5 when it becomes crucial in trophoblast giant cells. Investigation of whether desmoplakin and keratins depend on each other in functional terms during embryonic development has to await the combination of both knockouts. Such an experiment might help to understand whether there is a common mechanism underlying cell fragility.
Could keratin mutations affect embryonic development in humans?
Spontaneous abortion in human pregnancy occurs during the peri-implantation period with a frequency of ∼30% (Wilcox et al., 1988) and has been attributed mainly to failures in implantation and placental development. Before the analysis of K18/19 doubly deficient mice, it was been argued that, in contrast to epidermal keratins, those of internal epithelia are not essential as cytoskeletal proteins. This was in line with the lack of mutations in inherited disorders of internal epithelial keratins among adult patients analysed so far (Corden and McLean, 1996). Our findings of trophoblast fragility resulting from a cytoskeletal defect make K7, K8, K18 and K19 genes candidates for future studies.
The human trophoblast cell lineage does not differentiate into trophoblast giant cells, but forms a syncytium which invades the uterus and breaches maternal vessels, thus staying in direct contact with maternal blood (Cross et al., 1994). Therefore, it cannot be predicted whether the giant trophoblast phenotype in K18–/–K19–/– mice would occur in a similar fashion in humans. Given that mouse trophoblast giant cells in mice are the functional equivalent of the human syncytiotrophoblast, keratins of human internal epithelia (Daya and Sabet, 1991), e.g. K7, K8, K18 and K19, may be essential for providing mechanical strength to those tissues. The analogy to the deletion of two keratins in mice (this study) could be the presence of a dominant-negative mutation in one of the orthologous keratins in humans. This could lead to fragility of the syncytiotrophoblast and to loss of contact between maternal and embryonic tissues, resulting in early abortion.
Materials and methods
Genotyping
Genotyping of the normal and targeted K18 loci in adult mice was performed by PCR as described (Magin et al., 1998). The respective K19 loci of adult mice were genotyped by PCR using primers as follows: 5′ region of exon 1 of K19, 5′-TGGCGGAGTCCGCGGTGGAAGTT-3′; downstream in intron 2 of K19, 5′-CCTGACTAGATTCAAGTT AACTG-3′; primer for the phosphoglycerate kinase promoter, driving the neomycin resistance gene, 5′-CTAAAGCGCATGCTCCAGACT-3′. This PCR reaction gave two products of 958 bp (wild-type allele) and 739 bp (altered allele) with the following time/temperature parameters: 25 s at 94°C, 25 s at 60°C, 1 min at 72°C for 35 cycles.
DNA preparation of paraffin-embedded and sectioned embryos for genotyping was carried out by scraping embryonic tissue from two 20 µm sections into lysis buffer (Magin et al., 1998) containing 0.5 µg/µl proteinase K, and incubating overnight at 55°C. K18 PCR with DNA from paraffin sections was performed with the following primers and conditions: 5′-GCTTCACAACTCGCTCCACCAC-3′, 5′-AGCCTACC CTCTGGTAGATTGTCG-3′, 5′-CGGTCTGGATTCCACCCATTC-3′; 25 s at 94°C, 25 s at 60°C, 1 min at 72°C for 35 cycles. The PCR reaction gave a product of 0.2 kb specific for the wild-type and a product of 0.4 kb specific for the altered allele. Taq polymerase (MBI Fermentas, Walldorf) was used for all applications.
Histology
Uteri were dissected from pregnant mice, fixed for 2 h in Carnoy’s fixative at room temperature, embedded in paraffin, sectioned in their implantation sites (5 µm) and stained with Mayer’s haematoxylin and eosin (Bancroft and Cook, 1994).
Immunofluorescence microscopy
Implantation sites were cut as whole mounts (10 µm) and processed as described (Magin et al., 1998). The following primary antibodies were used: OVTL 12-30 (against K7; Eurodiagnostics, Maastricht, The Netherlands), Ks 18.04 and Ks 8.07 (against K18 and K8, respectively; Progen, Heidelberg, Germany), Troma-1 and Troma-3 (against K8 and K19, respectively), phalloidin toxin (Molecular Probes Europe BV, Leiden, The Netherlands), DP-1 (against desmoplakin; Progen, Heidelberg, Germany) and 4′,6-diamidino-2-phenylindole (Sigma, Munich, Germany).
Western blot analysis
Western blotting was performed as previously described (Magin et al., 1998).
Acknowledgments
Acknowledgements
This work is dedicated to W.W.Franke on the occasion of his 60th birthday. We thank G.Eversloh, R.Klockzin and M.Michels for excellent technical assistance. We are most grateful to Dr K.Zatloukal (University of Graz, Austria) for stimulating discussion. We acknowledge support by the Deutsche Forschungsgemeinschaft (SFB 284; C7), the Bonner Forum Biomedizin and the Fonds der Chemischen Industrie to T.M.M.
References
- Bader B.L., Magin,T.M., Freudenmann,M., Stumpp,S. and Franke,W.W. (1991) Intermediate filaments formed de novo from tail-less cytokeratins in the cytoplasm and in the nucleus. J. Cell Biol., 115, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft J.D. and Cook,H.C. (1994) Manual of Histological Techniques and their Diagnostic Application. Churchill Livingstone, Edinburgh. [Google Scholar]
- Baribault H., Price,J., Miyai,K. and Oshima,R.G. (1993) Mid-gestational lethality in mice lacking keratin 8. Genes Dev., 7, 1191–1202. [DOI] [PubMed] [Google Scholar]
- Baribault H., Penner,J., Iozzo,R.V. and Wilson,H.M. (1994) Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev., 8, 2964–2973. [DOI] [PubMed] [Google Scholar]
- Bierkamp C., Mclaughlin,K.J., Schwarz,H., Huber,O. and Kemler,R. (1996) Embryonic heart and skin defects in mice lacking plakoglobin. Dev. Biol., 180, 780–785. [DOI] [PubMed] [Google Scholar]
- Brulet P., Babinet,C., Kemler,R. and Jacob,F. (1980) Monoclonal antibodies against trophectoderm-specific markers during mouse blastocyst formation. Proc. Natl Acad. Sci. USA, 77, 4113–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulin C., Ware,C.F., Magin,T.M. and Oshima,R.G. (2000) Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J. Cell Biol., 149, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci-Guyon E., Gimenez,Y.R., Maurice,T., Babinet,C. and Privat,A. (1998) Cerebellar defect and impaired motor coordination in mice lacking vimentin. Glia, 25, 33–43. [PubMed] [Google Scholar]
- Corden L.D. and McLean,W.H. (1996) Human keratin diseases: hereditary fragility of specific epithelial tissues. Exp. Dermatol., 5, 297–307. [DOI] [PubMed] [Google Scholar]
- Cross J.C., Werb,Z. and Fisher,S.J. (1994) Implantation and the placenta: key pieces of the development puzzle. Science, 266, 1508–1518. [DOI] [PubMed] [Google Scholar]
- Daya D. and Sabet,L. (1991) The use of cytokeratin as a sensitive and reliable marker for trophoblastic tissue. Am. J. Clin. Pathol., 95, 137–141. [DOI] [PubMed] [Google Scholar]
- Elder G.A., Friedrich,V.L.,Jr, Bosco,P., Kang,C., Gourov,A., Tu,P.H., Lee,V.M. and Lazzarini,R.A. (1998a) Absence of the mid-sized neurofilament subunit decreases axonal calibers, levels of light neurofilament (NF-L) and neurofilament content. J. Cell Biol., 141, 727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder G.A., Friedrich,V.L.,Jr, Kang,C., Bosco,P., Gourov,A., Tu,P.H., Zhang,B., Lee,V.M. and Lazzarini,R.A. (1998b) Requirement of heavy neurofilament subunit in the development of axons with large calibers. J. Cell Biol., 143, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli A.B., McFadden,D.G., Lin,Q., Srivastava,D. and Olson,E.N. (1998) Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nature Genet., 18, 266–270. [DOI] [PubMed] [Google Scholar]
- Fuchs E. and Cleveland,D.W. (1998) A structural scaffolding of intermediate filaments in health and disease. Science, 279, 514–519. [DOI] [PubMed] [Google Scholar]
- Fuchs E. and Weber,K. (1994) Intermediate filaments: structure, dynamics, function, and disease. Annu. Rev. Biochem., 63, 345–382. [DOI] [PubMed] [Google Scholar]
- Gallicano G.I., Kouklis,P., Bauer,C., Yin,M., Vasioukhin,V., Degenstein,L. and Fuchs,E. (1998) Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J. Cell Biol., 143, 2009–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomi H., Yokoyama,T., Fujimoto,K., Ikeda,T., Katoh,A., Itoh,T. and Itohara,S. (1995) Mice devoid of the glial fibrillary acidic protein develop normally and are susceptible to scrapie prions. Neuron, 14, 29–41. [DOI] [PubMed] [Google Scholar]
- Guillemot F., Nagy,A., Auerbach,A., Rossant,J. and Joyner,A.L. (1994) Essential role of Mash-2 in extraembryonic development. Nature, 371, 333–336. [DOI] [PubMed] [Google Scholar]
- Haegel H., Larue,L., Ohsugi,M., Fedorov,L., Herrenknecht,K. and Kemler,R. (1995) Lack of β-catenin affects mouse development at gastrulation. Development, 121, 3529–3537. [DOI] [PubMed] [Google Scholar]
- Harada N., Tamai,Y., Ishikawa,T., Sauer,B., Takaku,K., Oshima,M. and Taketo,M.M. (1999) Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J., 18, 5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld M. and Franke,W.W. (1985) Pair formation and promiscuity of cytokeratins: formation in vitro of heterotypic complexes and intermediate-sized filaments by homologous and heterologous recombinations of purified polypeptides. J. Cell Biol., 101, 1826–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld M. and Weber,K. (1990) Tailless keratins assemble into regular intermediate filaments in vitro. J. Cell Sci., 97, 317–324. [DOI] [PubMed] [Google Scholar]
- Herrmann H. and Harris,J.R. (1998) Intermediate Filaments. Plenum Press, New York, NY. [Google Scholar]
- Huelsken J., Vogel,R., Brinkmann,V., Erdmann,B., Birchmeier,C. and Birchmeier,W. (2000) Requirement for β-catenin in anterior–posterior axis formation in mice. J. Cell Biol., 148, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson B.W., Grund,C., Schmid,E., Burki,K., Franke,W.W. and Illmensee,K. (1980) Formation of cytoskeletal elements during mouse embryogenesis. Intermediate filaments of the cytokeratin type and desmosomes in preimplantation embryos. Differentiation, 17, 161–179. [DOI] [PubMed] [Google Scholar]
- Jackson B.W., Grund,C., Winter,S., Franke,W.W. and Illmensee,K. (1981) Formation of cytoskeletal elements during mouse embryo genesis. II. Epithelial differentiation and intermediate-sized filaments in early postimplantation embryos. Differentiation, 20, 203–216. [DOI] [PubMed] [Google Scholar]
- Kouklis P.D., Hutton,E. and Fuchs,E. (1994) Making a connection: direct binding between keratin intermediate filaments and desmosomal proteins. J. Cell Biol., 127, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk A.P., Bornslaeger,E.A., Norvell,S.M., Palka,H.L. and Green,K.J. (1999) Desmosomes: intercellular adhesive junctions specialized for attachment of intermediate filaments. Int. Rev. Cytol., 185, 237–302. [DOI] [PubMed] [Google Scholar]
- Ku N.O., Michie,S., Oshima,R.G. and Omary,M.B. (1995) Chronic hepatitis, hepatocyte fragility, and increased soluble phosphoglyco keratins in transgenic mice expressing a keratin 18 conserved arginine mutant. J. Cell Biol., 131, 1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.O., Michie,S.A., Soetikno,R.M., Resurreccion,E.Z., Broome,R.L., Oshima,R.G. and Omary,M.B. (1996) Susceptibility to hepatotoxicity in transgenic mice that express a dominant-negative human keratin 18 mutant. J. Clin. Invest., 98, 1034–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.O., Michie,S.A., Soetikno,R.M., Resurreccion,E.Z., Broome,R.L. and Omary,M.B. (1998) Mutation of a major keratin phosphorylation site predisposes to hepatotoxic injury in transgenic mice. J. Cell Biol., 143, 2023–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.O., Zhou,X., Toivola,D.M. and Omary,M.B. (1999) The cytoskeleton of digestive epithelia in health and disease. Am. J. Physiol., 277, G1108–G1137. [DOI] [PubMed] [Google Scholar]
- Kulesh D.A., Cecena,G., Darmon,Y.M., Vasseur,M. and Oshima,R.G. (1989) Posttranslational regulation of keratins: degradation of mouse and human keratins 18 and 8. Mol. Cell. Biol., 9, 1553–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue L., Ohsugi,M., Hirchenhain,J. and Kemler,R. (1994) E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc. Natl Acad. Sci. USA, 91, 8263–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Colucci,G.E., Pincon,R.M., Mericskay,M., Pournin,S., Paulin,D. and Babinet,C. (1996) Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev. Biol., 175, 362–366. [DOI] [PubMed] [Google Scholar]
- Lloyd C., Yu,Q.C., Cheng,J., Turksen,K., Degenstein,L., Hutton,E. and Fuchs,E. (1995) The basal keratin network of stratified squamous epithelia: defining K15 function in the absence of K14. J. Cell Biol., 129, 1329–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin T.M., Bader,B.L., Freudenmann,M. and Franke,W.W. (1990) De novo formation of cytokeratin filaments in calf lens cells and cytoplasts after transfection with cDNAs or microinjection with mRNAs encoding human cytokeratins. Eur. J. Cell Biol., 53, 333–348. [PubMed] [Google Scholar]
- Magin T.M., Schroder,R., Leitgeb,S., Wanninger,F., Zatloukal,K., Grund,C. and Melton,D.W. (1998) Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J. Cell Biol., 140, 1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin T.M., Hesse,M. and Schroder,R. (2000) New insights in intermediate filament function from studies of transgenic and knockout mice. Protoplasma, 211, 140–150. [Google Scholar]
- Milner D.J., Weitzer,G., Tran,D., Bradley,A. and Capetanaki,Y. (1996) Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol., 134, 1255–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll R., Franke,W.W., Schiller,D.L., Geiger,B. and Krepler,R. (1982) The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell, 31, 11–24. [DOI] [PubMed] [Google Scholar]
- Oshima R.G., Howe,W.E., Klier,F.G., Adamson,E.D. and Shevinsky,L.H. (1983) Intermediate filament protein synthesis in preimplantation murine embryos. Dev. Biol., 99, 447–455. [DOI] [PubMed] [Google Scholar]
- Paulin D., Babinet,C., Weber,K. and Osborn,M. (1980) Antibodies as probes of cellular differentiation and cytoskeletal organization in the mouse blastocyst. Exp. Cell Res., 130, 297–304. [DOI] [PubMed] [Google Scholar]
- Pekny M., Leveen,P., Pekna,M., Eliasson,C., Berthold,C.H., Westermark,B. and Betsholtz,C. (1995) Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J., 14, 1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter R.M., Leitgeb,S., Melton,D.W., Swensson,O., Eady,R.A. and Magin,T.M. (1996) Gene targeting at the mouse cytokeratin 10 locus: severe skin fragility and changes of cytokeratin expression in the epidermis. J. Cell Biol., 132, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao M.V., Houseweart,M.K., Williamson,T.L., Crawford,T.O., Folmer,J. and Cleveland,D.W. (1998) Neurofilament-dependent radial growth of motor axons and axonal organization of neurofilaments does not require the neurofilament heavy subunit (NF-H) or its phosphorylation. J. Cell Biol., 143, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley P., Anson-Cartwright,L. and Cross,J.C. (1998) The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nature Genet., 18, 271–275. [DOI] [PubMed] [Google Scholar]
- Rugh R. (1968) The Mouse: Its Reproduction and Development. Oxford University Press, Oxford, UK. [Google Scholar]
- Ruiz P. et al. (1996) Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J. Cell Biol., 135, 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M.A., Owaribe,K., Kartenbeck,J. and Franke,W.W. (1990) Desmosomes and hemidesmosomes: constitutive molecular components. Annu. Rev. Cell Biol., 6, 461–491. [DOI] [PubMed] [Google Scholar]
- Steinert P.M., Idler,W.W. and Zimmerman,S.B. (1976) Self-assembly of bovine epidermal keratin filaments in vitro. J. Mol. Biol., 108, 547–567. [DOI] [PubMed] [Google Scholar]
- Stumptner C., Omary,M.B., Fickert,P., Denk,H. and Zatloukal,K. (2000) Hepatocyte cytokeratins are hyperphosphorylated at multiple sites in human alcoholic hepatitis and in a Mallory body mouse model. Am. J. Pathol., 156, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan T., Escalante-Alcalde,D., Bhatt,H., Anver,M., Bhat,N., Nagashima,K., Stewart,C.L. and Burke,B. (1999) Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol., 147, 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N., Zara,J., Sato,T., Ong,E., Bakhiet,N., Oshima,R.G., Watson,K.L. and Fukuda,M.N. (1998) A cytoplasmic protein, bystin, interacts with trophinin, tastin, and cytokeratin and may be involved in trophinin-mediated cell adhesion between trophoblast and endometrial epithelial cells. Proc. Natl Acad. Sci. USA, 95, 5027–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. (1995) Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol., 7, 619–627. [DOI] [PubMed] [Google Scholar]
- Tang K., Finley,R.L.,Jr, Nie,D. and Honn,K.V. (2000) Identification of 12-lipoxygenase interaction with cellular proteins by yeast two-hybrid screening. Biochemistry, 39, 3185–3191. [DOI] [PubMed] [Google Scholar]
- Wilcox A.J., Weinberg,C.R., O’Connor,J.F., Baird,D.D., Schlatterer,J.P., Canfield,R.E., Armstrong,E.G. and Nisula,B.C. (1988) Incidence of early loss of pregnancy. N. Engl. J. Med., 319, 189–194. [DOI] [PubMed] [Google Scholar]
- Zhu Q., Lindenbaum,M., Levasseur,F., Jacomy,H. and Julien,J.P. (1998) Disruption of the NF-H gene increases axonal microtubule content and velocity of neurofilament transport: relief of axonopathy resulting from the toxin β,β′-iminodipropionitrile. J. Cell Biol., 143, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]