

Abstract
Changes in activity-dependent calcium flux through voltage-gated calcium channels (CaVs) drive two self-regulatory calcium-dependent feedback processes that require interaction between Ca2+/calmodulin (Ca2+/CaM) and a CaV channel consensus isoleucine-glutamine (IQ) motif: calcium-dependent inactivation (CDI) and calcium-dependent facilitation (CDF). Here, we report the high-resolution structure of the Ca2+/CaM–CaV1.2 IQ domain complex. The IQ domain engages hydrophobic pockets in the N-terminal and C-terminal Ca2+/CaM lobes through sets of conserved ‘aromatic anchors.’ Ca2+/N lobe adopts two conformations that suggest inherent conformational plasticity at the Ca2+/N lobe–IQ domain interface. Titration calorimetry experiments reveal competition between the lobes for IQ domain sites. Electrophysiological examination of Ca2+/N lobe aromatic anchors uncovers their role in CaV1.2 CDF. Together, our data suggest that CaV subtype differences in CDI and CDF are tuned by changes in IQ domain anchoring positions and establish a framework for understanding CaM lobe–specific regulation of CaVs.
Voltage-gated calcium channels are the ion channels that define excitable cells1. These channels control cellular calcium entry in response to changes in membrane potential and are pivotal in the generation of cardiac action potentials, excitation-contraction coupling, hormone and neurotransmitter release and activity-dependent transcription initiation1,2. CaVs are multisubunit complexes composed of three essential channel subunits2, CaVα1, CaVβ and CaVα2δ, plus the ubiquitous intracellular calcium sensor calmodulin (CaM)3. An additional subunit, CaVγ, is associated with skeletal muscle channels, but its general importance in other tissues is unsettled4.
The CaVα1 subunits are single polypeptide chains of ∼1,800–2,200 residues in which the ion-conducting pore is formed from four homologous repeats that each bear six transmembrane segments2. There are three CaV subfamilies, which have diverse physiological and pharmacological properties that depend largely on the CaVα1-subunit: CaV 1.x (L-type), CaV2.x (2.1, P/Q-type; 2.2, N-type; 2.3, R-type) and CaV3.x (T-type)1. Large interdomain intracellular loops bridge the four transmembrane repeats of the CaVα1 subunit and serve as docking sites for auxiliary subunits and regulatory molecules that control channel activity and connect CaV channels to larger macromolecular complexes and cellular signaling pathways5,6.
Calcium influx is a potent activator of intracellular signaling pathways but is toxic in excess1,7. Because CaVs are major sources of calcium influx, CaV activity is strongly controlled by both self-regulatory and extrinsic mechanisms that tune channel action in response to electrical excitation, neurotransmitter stimulation and hormonal cues1,2,8. This intense regulation uses multiple mechanisms and classes of intracellular signaling molecules. The bi-lobed calcium sensor CaM has a preeminent role in the intrinsic mechanisms of CaV calcium-dependent regulation9–15. Activity-dependent changes in intracellular calcium levels from flux through CaV channel pores drive two self-regulatory calcium-dependent feedback mechanisms that limit or enhance calcium entry through the channel: CDI and CDF8. Both require the interaction of Ca2+/CaM with a consensus IQ motif in the CaVα1 C-terminal cytoplasmic tail9,11–16.
Multiple lines of evidence suggest that CaM is constitutively tethered to the channel to detect calcium in the vicinity of the channel pore12,17,18 and to permit calcium ions to exert control over their own entry through CaVs. The nature of the tethering site remains unclear. It may comprise a region immediately adjacent to the IQ domain on the N-terminal side, termed the Pre-IQ region17,19,20, or a composite of the Pre-IQ and IQ domain18,20.
The IQ domain is believed to be the site of action of Ca2+/CaM. Numerous studies show that Ca2+/CaM binds IQ peptides from different CaV channels in vitro9,11–14,17,20–22 and that IQ domain mutations in full-length channels can eliminate CDI9,13–15,23 and CDF9,14.
Although CDI and CDF are well documented, the details are complex, differ among channel subtypes and have not been readily predictable given the primary-sequence similarities among the CaV CaM-binding domains. Elegant experiments have demonstrated that CDI and CDF are controlled independently by the abilities of the N-terminal and C-terminal CaM lobes (the N lobe and C lobe) to bind calcium9,10,12,16,23. CDI is present in both L-type (CaV1.2) and non–L-type (CaV2.1, CaV2.2 and CaV2.3) channels but arises from the action of opposite CaM lobes in these two cases9,23. In the L-type channel CaV1.2, CDI is governed by the binding of calcium ions to the C lobe12, whereas in non–L-type channels (CaV2s), CDI is triggered by the binding of calcium ions to the N lobe9,11,23. In the non–L-type channel CaV2.1, CDF arises from interactions between calcium ions and the C lobe. Despite the fact that the C lobe–mediated processes elicit different channel behaviors in different channel subtypes, CDI in L-type and CDF in non–L-type channels, both C lobe processes share insensitivity to the fast calcium chelator BAPTA23. This shared resistance suggests that in both cases the C lobe captures calcium that is local to the channel pore23. In contrast, CaV2 N lobe–mediated CDF is sensitive to the slow calcium chelator EGTA, suggesting that it detects global changes in calcium concentration9,23. These lobe-specific calcium sensitivities have been suggested to provide mechanisms for CaVs to sense, decode and distinguish local calcium changes due to calcium permeation through the channel from global calcium changes due to aggregate cellular signals9,23.
To delineate the ways in which CDI and CDF arise, we set out to determine the molecular basis for Ca2+/CaM interactions. Here, we report the high-resolution crystal structure of the human Ca2+/CaM–CaV1.2 IQ domain complex. By using the structural data to inform functional experiments, we uncover an unexpected competition between the Ca2+/CaM lobes for a common site on the IQ domain and demonstrate a previously unknown role for the Ca2+/N lobe anchors in CaV1.2 CDF. This structure provides the first insight into the molecular machinery that underlies Ca2+/CaM regulation of CaVs.
Results
Structure of the Ca2+/CaM–CaV1.2 IQ domain complex
We solved the crystal structure of the Ca2+/CaM–CaV1.2 IQ domain complex at 2.00-Å resolution using a three-wavelength MAD experiment on a selenomethionine (SeMet)-substituted protein crystal. The asymmetric unit contains three independent 1:1 Ca2+/CaM–IQ domain complexes (complexes A, B and C) that have two principal conformations, represented by complexes A and C (Fig. 1a,b). Complex B is similar to complex A but has poorly defined density for the C-terminal half of the IQ domain (Supplementary Fig. 1 online). Thus, our analysis focuses on the well-defined structures of complexes A and C. Both are compact structures in which Ca2+/CaM embraces the largely α-helical IQ domain in a parallel orientation (Fig. 1a,b) through extensive interactions that involve twenty-two (complex A) or twenty-one (complex C) contiguous residues in the IQ domain, and both bury ∼3,100 Å2 total surface area of which roughly 1,650 Å2 is hydrophobic (Fig. 1c). The observed parallel orientation in which Ca2+/N lobe binds the N-terminal portion of the target helix and Ca2+/C lobe binds the C-terminal portion is unusual and known in only one other Ca2+/CaM peptide structure, the Ca2+/CaM-dependent kinase peptide complex24,25. A high density of positively charged side chains project from the CaV1.2 IQ helix near the C terminus and make electrostatic interactions with negatively charged residues that ring the Ca2+/CaM exit tunnel (Fig. 2). These interactions are consistent with the proposal that the distribution of positive charges on Ca2+/CaM-binding peptides is an important determinant of binding orientation24.
Figure 1.
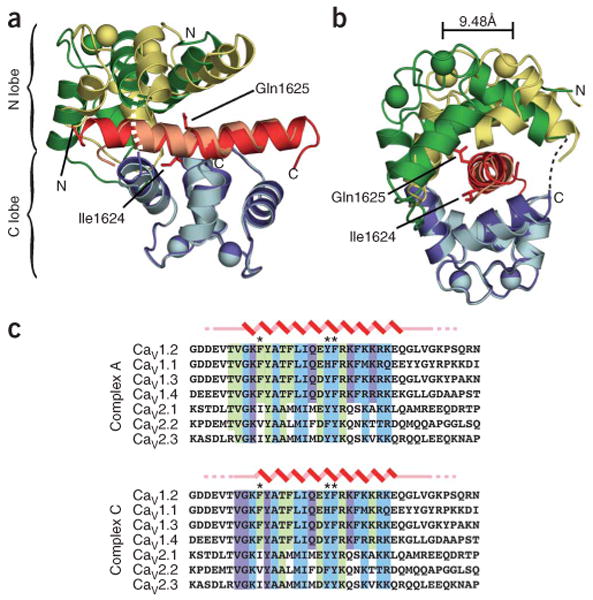
Structure of the Ca2+/CaM–CaV1.2 IQ domain complex. (a) Ribbon diagram of the complex. Green, CaM Ca2+/N lobe; blue, Ca2+/C lobe; red, IQ domain, with residues Ile1624 and Q1625 from complex A in stick representation; darker shades, complex A; lighter shades, complex C. The complexes were superposed using the Ca2+/C lobes and IQ domains. Labels indicate termini of components in complex A. Dashed lines indicate regions absent from the structures. (b) 90° rotation of a. The shift in Ca2+/N lobe Ca2+ position in EF-hand 2 is indicated. (c) Sequence alignment of IQ regions from each CaV1 and CaV2 isoform. Zigzag, α-helical regions; straight line, nonhelical residues; dashed lines, residues present in the crystallized construct but absent from the electron density. Residues contacting CaM (≤4 Å) for conformation A and conformation C are highlighted as follows: green, contacts to Ca2+/N lobe; cyan, Ca2+/C lobe; purple, Ca2+/N lobe and Ca2+/C lobe. Asterisks indicate principal aromatic anchor positions. Sequences are human CaV1.2 1609–1647, human CaV1.1 1514–1552, rat CaV1.3 1641–1679, human CaV1.4 1563–1602, human CaV2.1 1947–1985, human CaV2.2 1845–1883 and rat CaV2.3 1811–1850.
Figure 2.
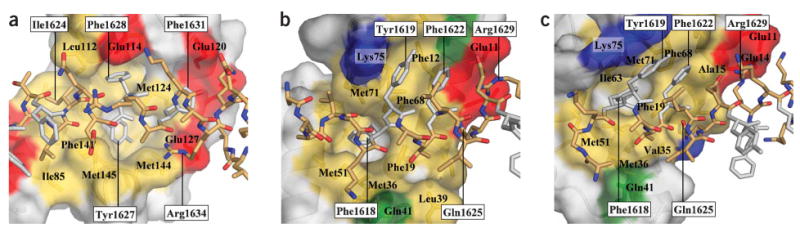
Lobe-specific Ca2+/CaM–CaV1.2 IQ domain interactions. (a) Ca2+/CaM C lobe from complex A bound to the IQ domain. Buried surface area = 1,819 Å2 (965 Å2 hydrophobic). (b) Ca2+/CaM N lobe from complex A bound to the IQ domain. Buried surface area = 1,450 Å2 (743 Å2 hydrophobic). (c) Ca2+/CaM N lobe from complex C bound to the IQ domain. Buried surface area = 1,491 Å2 (500 Å2 hydrophobic). IQ domain is shown in stick representation with aromatic anchor residues in white. CaM lobes are shown in surface representation with residues that contribute hydrophobic (yellow), negatively charged (red), positively charged (blue) and polar (green) side chain contacts (≤4 Å) to the IQ domain indicated. Select residues are labeled to orient the reader. IQ domain residue labels are boxed.
The Ca2+/C lobes of complexes A and C are nearly identical (r.m.s. deviation for Cα atoms is 0.475) and are anchored similarly to the IQ domain helix (Figs. 1 and 2). The Ca2+/N lobes are also similar (r.m.s. deviation for Cα atoms is 0.97) and have only small changes in the positions of side chains involved in direct contacts with the IQ peptide (Supplementary Fig. 2 online). Superposition of Ca2+/C lobes and IQ helices shows that the Ca2+/N lobe adopts two distinct binding modes (Fig. 1a,b). In complex A, the Ca2+/N lobe is positioned further along the N-terminal end of the IQ domain, whereas in complex C, the Ca2+/N lobe is tilted and shifted toward the C-terminal end of the IQ domain, leading to a more compact conformation. The conformational differences cause relative displacements in the positions of the two Ca2+ ions in the Ca2+/N lobe EF-hands of ∼8.6 Å and ∼9.5 Å and increase the bend present in both IQ domain helices at Ile1624 by ∼10° in complex C.
Aromatic anchors mediate IQ domain–Ca2+/CaM contacts
The CaV1.2 IQ helix engages Ca2+/CaM through a set of ‘aromatic anchor’ residues. The C-terminal portion of the IQ helix displays three aromatic anchors (Tyr1627, Phe1628 and Phe1631) that bind hydrophobic Ca2+/C lobe pockets. Two anchors (Tyr1627 and Phe1628) are deeply buried (Fig. 2a). The N-terminal part of the IQ helix presents three aromatic residues (Phe1618, Tyr1619 and Phe1622) that make hydrophobic interactions with the Ca2+/N lobe; these residues reside on the opposite helical face from the C-terminal anchors. Phe1618 makes the most extensive contacts and binds a deep hydrophobic pocket (Fig. 2b,c). Despite the alternative positions of the Ca2+/N lobe, Phe1618 remains buried in the same Ca2+/N lobe hydrophobic pocket in both complexes by adopting different side chain rotamers (Fig. 2b,c and Supplementary Fig. 2).
Ca2+/CaM interactions with IQ domain consensus residues
IQ domains are defined by the consensus sequence (I/L/V) QXXXRXXXX(R/K) (where X is any residue)26,27. The Ca2+/CaM–CaV1.2 structure reveals the ways in which the hallmark residues of the IQ domain interact with the Ca2+/CaM lobes. The Ile1624 side chain is completely buried (solvent-exposed area = 0.7 Å2) and contacts the hydrophobic surface of the C lobe (Fig. 2a and Supplementary Figs. 2 and 3 online). The Gln1625 side chain has many contacts to the Ca2+/N lobe. This residue also makes side chain and main chain water-mediated hydrogen bonds to the Ca2+/C lobe (Supplementary Figs. 2 and 3). The central arginine, Arg1629, and terminal basic residue, Arg1634, form salt bridges with Glu11 and Glu14 of the Ca2+/N lobe (Supplementary Figs. 2 and 3) and Glu127 of the Ca2+/C lobe (Supplementary Fig. 3), respectively. Thus, all IQ hallmark residues seem important for IQ domain–Ca2+/CaM interactions. These residues, together with the aromatic anchors and other positions throughout the IQ domain, establish the extensive network of contacts in the complex (Supplementary Fig. 3).
Isothermal titration calorimetry
We used isothermal titration calorimetry (ITC) to gain insight into the thermodynamics of lobe-specific Ca2+/CaM–IQ domain interactions (Fig. 3). Ca2+/C lobe binds the IQ domain with 1:1 stoichiometry and a high affinity (Kd = 2.63 × 10−9 M) that is driven by favorable enthalpic and entropic contributions (Fig. 3a and Table 1). Similar favorable components drive Ca2+/N lobe binding; however, the isotherms revealed that there are two different binding sites, a medium-affinity (Kd = 5.76 × 10−8 M) and low-affinity (Kd = 1.92 × 10−5 M) site (Fig. 3b and Table 1). Both Ca2+/N lobe–IQ domain interactions are substantially weaker than the Ca2+/C lobe–IQ domain interaction. Titration of Ca2+/N lobe into a solution containing Ca2+/C lobe–IQ domain complexes yielded no further binding energy (Fig. 3c). These data directly demonstrate that the high-affinity Ca2+/C lobe–IQ domain interaction occludes both measurable Ca2+/N lobe–binding sites.
Figure 3.
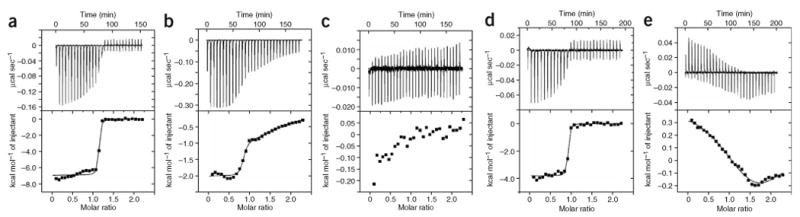
ITC characterization of Ca2+/CaM–CaV1.2 IQ domain interactions. (a) 70 μM IQ domain into 7 μM Ca2+/C lobe. (b) 500 μM Ca2+/N lobe into 50 μM IQ domain. (c) 200 μM Ca2+/N lobe into a solution of 20 μM IQ domain and 37 μM C lobe. (d) 60 μM IQ domain F1628A mutant into 6 μM Ca2+/C lobe. (e) 500 μM Ca2+/N lobe into a solution of 5 μM IQ domain F1628A mutant. Isotherms are fit to a single binding site model for a and d and a double binding site model for b and e. Panels show addition of 10 μl aliquots of titrant into the target solution (top) and binding isotherms (bottom).
Table 1. Thermodynamic parameters for CaV1.2 IQ domain–Ca2+/CaM lobe interactions.
| WT–Ca2+/C lobe | WT–Ca2+/N lobe | F1628A–Ca2+/C lobe | F1628A–Ca2+/N lobe | |
|---|---|---|---|---|
| N1 | 0.98 ± 0.18 | 0.85 ± 0.05 | 0.92 ± 0.04 | 0.96 ± 0.13 |
| Kd1 (nM) | 2.63 ± 0.07 | 57.6 ± 35.5 | 2.59 ± 0.24 | 1,003 ± 567 |
| ΔH1 (kcal mol−1) | −6.77 ± 0.21 | −1.91 ± 0.14 | −3.69 ± 0.17 | 0.45 ± 0.02 |
| ΔS1 (cal mol−1 K−1) | 15.75 ± 0.78 | 26.70 ± 0.85 | 26.45 ± 0.78 | 29.20 ± 1.13 |
| ΔG1 (kcal mol−1) | −11.31 ± 0.01 | −9.60 ± 0.38 | −11.31 ± 0.06 | −7.96 ± 0.35 |
| N2 | 1.04 ± 0.0 | 1.0 ± 0.0 | ||
| Kd2 (nM) | 19,190 ± 3,600 | 9,812 ± 8,220 | ||
| ΔH2 (kcal mol−1) | −1.42 ± 0.13 | −0.67 ± 0.22 | ||
| ΔS2 (cal mol−1 K−1) | 16.60 ± 0.84 | 21.00 ± 2.69 | ||
| ΔG2 (kcal mol−1) | −6.20 ± 0.11 | −6.71 ± 0.55 |
Thermodynamic parameters of Ca2+/C lobe and Ca2+/N lobe binding to wild-type (WT) and F1628A CaV1.2 IQ domain at pH 7.4 in the presence of 1 mM CaCl2. Each value corresponds to the mean of two separate experiments with different batches of both components (± s.d.).
To investigate further whether the Ca2+/N lobe– and Ca2+/C lobe–binding sites depend on interactions observed in the crystal structure, we examined the consequence of a mutation in the IQ domain's aromatic anchor for Ca2+/C lobe, F1628A. The F1628A mutant binds Ca2+/C lobe with an affinity that is identical to the wild-type domain (Kd = 2.59 × 10−9 M), but with a reduced enthalpy (Fig. 3d and Table 1). This type of enthalpy-entropy compensation is a common feature in protein-protein interactions28 and reflects a loss of key interactions in the bound state that is compensated by increased disorder. In contrast, the F1628A mutation causes a substantial perturbation of Ca2+/N lobe medium-affinity binding (Kd = 1.003 × 10−6 M for F1628A compared to Kd = 5.76 × 10−8 M for the wild-type domain) and causes an unfavorable binding enthalpy. These data directly demonstrate that the medium-affinity Ca2+/N lobe–binding site requires interactions with residues that comprise the crystallographically observed Ca2+/C lobe site.
N lobe anchors have a role in CDF
Although Ca2+/C lobe has an established role in CaV1.2 CDI12,16,17, no role has yet been defined for Ca2+/N lobe. Therefore, we tested whether the crystallographically observed Ca2+/N lobe interface has a role in channel function by using two-electrode voltage clamp to interrogate mutant channels that were heterologously expressed in Xenopus laevis oocytes. A triple mutant lacking the three aromatic anchors for Ca2+/N lobe (F1618A Y1619A F1622A), ‘TripleA,’ showed no appreciable difference in CaV1.2 inactivation when either Ca2+ or Ba2+ was the charge carrier (Fig. 4a,b). Thus, Ca2+/N lobe–aromatic anchor interactions do not seem important for CaV1.2 CDI. This result agrees with the observation that overexpression of the CaM mutant CaM12, in which the N lobe EF-hands have diminished calcium binding, has little effect on CDI12,16. TripleA channels consistently gave lower current amplitudes compared to wild-type channels. Previous reports have suggested a role for the IQ domain in CaV trafficking29; our observation raises the possibility that aromatic anchors for Ca2+/N lobe may be important in this process.
Figure 4.
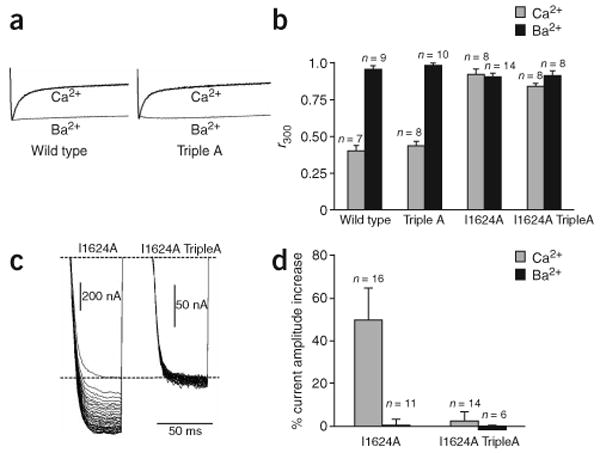
Lobe-specific interactions affect CDI and CDF. (a) Voltage-activated Ba2+ and Ca2+ currents from wild-type (WT) and TripleA channels during a 600-ms depolarizing step from −90 mV to +20 mV. Traces are normalized to the peak current to facilitate comparison. Tail currents are not shown. (b) r300 values (current fraction 300 ms after depolarization) in Ba2+ (black) and Ca2+ (grey). Error bars show s.d. (c) I1624A and I1624A TripleA Ca2+ currents in a 3-Hz 40-pulse train (50-ms steps to +20-mV from −90-mV holding potential). The first pulse currents are scaled to the same level for comparison. (d) Relative current increase between last and first pulses for I1624A and I1624A TripleA. Error bars show s.d.
Because the aromatic anchors for Ca2+/N lobe are not crucial for CaV1.2 CDI, we asked whether they might have a role in CDF. CDF is a prominent property of CaV2.1 channels9,10,30. Although CDF is not readily detectable in wild-type CaV1.2 channels, robust CDF is unmasked by the I1624A mutation14,15. The I1624A mutation in the TripleA background (I1624A TripleA) results in channels lacking both CDI (Fig. 4b) and CDF (Fig. 4c,d). Lowering expression of a I1624A-only mutant such that current magnitudes were equivalent to those of the I1624A TripleA mutant still produced clear CDF (data not shown). This result excludes the possibility that CDF loss in I1624A TripleA is a consequence of the lower current magnitudes. Together, these experiments suggest that the aromatic anchors for the Ca2+/N lobe have a previously unappreciated role in CaV1.2 CDF.
Discussion
Calcium ions are potent chemical effectors of many cellular processes1,7. The opening of CaVs in response to changes in membrane potential is a major source of calcium influx1 and thus couples two forms of biological signals, electrical and chemical. The CaV activity that drives this powerful signaling combination is subject to a variety of control mechanisms. A diverse set of proteins that includes auxiliary channel subunits, G-proteins, synaptic vesicle components, kinases, phosphatases and calcium sensors interact with and modify the behavior of the pore-forming subunit to limit or enhance calcium influx1,2.
Two types of feedback regulation in which calcium ions affect their own entry through CaVs and control local calcium levels have been intensively studied for more than two decades8. CDI, which limits calcium flux through CaVs, and CDF, which enhances calcium flux through CaVs, both result from interactions between calcium ions, a channel-resident CaM and the CaVα1 subunit's C-terminal cytoplasmic IQ domain9,11–16,31. Further dissection of the molecular origins of CDI and CDF has shown that each process arises from the binding of specific calcium-bound CaM lobes to the IQ domain10,12–14; however, the precise details have remained unknown.
IQ domains are protein segments that contain the 11-residue consensus sequence (I/L/V)QXXXRXXXX(R/K) and comprise a family of Ca2+/CaM– and apo CaM–binding motifs found in diverse proteins including molecular motors, voltage-gated calcium channels, voltage-gated sodium channels and phosphatases26,27. The Ca2+/CaM–IQ domain structure presented here offers the first high-resolution picture of the molecular details of a Ca2+/CaM–IQ domain interaction. A previous work has proposed that Ile1624 is exposed in the Ca2+/CaM–CaV1.2 complex to provide hydrophobic contacts to other parts of the channel-gating machinery32. Contrary to this prediction, the structure shows that Ile1624 is completely buried (solvent-exposed area = 0.7 Å2) and contacts the hydrophobic surface of the C lobe (Fig. 2a and Supplementary Figs. 2 and 3). Other hallmark residues of the IQ domain engage in extensive hydrophobic and polar interactions with the CaM lobes that establish the importance of these residues for the Ca2+/CaM–IQ domain interaction (Fig. 1 and Supplementary Figs. 2 and 3).
The aromatic anchors that form the IQ domain's principal contacts to the Ca2+/CaM lobes were not anticipated from prior studies. Comparison of the CaV IQ domain defined by the crystal structure with other IQ motifs reveals that many anchor positions are unique to CaVs. Such an array of aromatic anchors, particularly the N-terminal anchors, is lacking in the C-terminal cytoplasmic IQ domains of the closely related family of voltage-gated sodium channels (NaVs)33–35 (Supplementary Fig. 4 online). This observation suggests that CaM binding to CaV and NaV IQ domains and subsequent modulatory effects may be fundamentally different despite overall similarities in NaV and CaV archictecture1.
Examination of the Ca2+/CaM–CaV1.2 IQ domain complexes shows interesting differences in the ways Ca2+/N lobe and Ca2+/C lobe interact with the IQ domain: the number of binding conformations differs, with two for Ca2+/N lobe versus one for Ca2+/C lobe; the number of deep anchor positions differs, with one for Ca2+/N lobe (Phe1618) versus two for Ca2+/C lobe (Tyr1627 and Phe1628); and the buried surface areas are not equivalent, as Ca2+/C lobe buries more total and more hydrophobic surface than Ca2+/N lobe (Fig. 2). These differences suggest that the Ca2+/C lobe binds more tightly to the IQ domain. As it is not simple to infer directly the energetic importance of molecular interactions from structural data alone36, we pursued experiments to determine whether these observed differences in modes of interaction have functional consequences.
We used ITC to probe the binding between the individual CaM lobes and the IQ domain. ITC experiments directly determine the thermodynamic parameters that underlie binding reactions and provide a degree of resolution for studying macromolecular interactions that is unmatched by other methods37. The experiments showed that the Ca2+/C lobe binds the IQ domain with high affinity (Kd = 2.6 nM). To our surprise, we found multiple binding sites for the Ca2+/N lobe on the IQ domain (Fig. 5a). The lack of appreciable Ca2+/N lobe binding to the Ca2+/C lobe–IQ domain complex (Fig. 3c), together with the large reduction in Ca2+/N lobe binding affinity by the aromatic-anchor mutation F1628A (Fig. 3e), indicates that the medium-affinity Ca2+/N lobe–binding site overlaps directly with the Ca2+/C lobe–binding site (Fig. 5). The competition experiment (Fig. 3c) also showed that the binding of Ca2+/C lobe to the IQ domain lowers the affinity of the second Ca2+/N lobe–binding site to an undetectable level (Kd > 200 μM, given the concentrations used in the experiment37). The dominance of Ca2+/C lobe in IQ domain binding energetics agrees with the eminent role of the Ca2+/C lobe in CDI of L-type channels12,16,17. Given that the medium-affinity Ca2+/N lobe site overlaps with the high-affinity Ca2+/C lobe site and that Ca2+/C lobe binding to its high-affinity site weakens the avidity of Ca2+/N lobe for its low-affinity site, the thermodynamic data suggest that the crystallographically observed Ca2+/N lobe interactions correspond to the Ca2+/N lobe low-affinity site. These interactions arise from the high effective concentration38 of the Ca2+/N lobe relative to the peptide caused by Ca2+/C lobe binding and the CaM interlobe linker (Fig. 5d).
Figure 5.
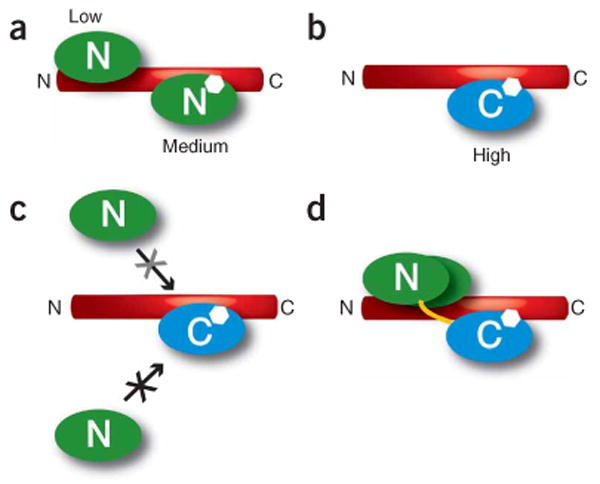
Schematic of Ca2+/CaM lobe multiple binding modes on the CaV1.2 IQ domain. (a) Ca2+/N lobe (green) has medium-affinity and low-affinity binding sites on the IQ domain. (b) Ca2+/C lobe (blue) has a high-affinity binding site on the IQ domain. (c) Binding of Ca2+/C lobe to the IQ domain blocks Ca2+/N lobe access to the Ca2+/N lobe medium-affinity site (black X) and reduces Ca2+/N lobe binding to the Ca2+/N lobe low-affinity site (grey X). (d) Representation of how Ca2+/C lobe binding to the IQ domain tethers Ca2+/N lobe near the Ca2+/N lobe low-affinity site. Yellow line indicates the CaM interlobe linker. In all panels, the approximate position of aromatic anchor Phe1628, which is shared by the Ca2+/N lobe medium-affinity site and the Ca2+/C lobe high-affinity site, is indicated by the white hexagon.
As Ca2+/C lobe binds tighter than Ca2+/N lobe to the site that includes Phe1628, Ca2+/N lobe is not expected to occupy this part of the IQ domain when both CaM lobes are loaded with calcium. However, Ca2+/N lobe may occupy this position in channel functional states in which the N lobe is loaded with calcium but the C lobe is in the apo state. This property may be particularly relevant for CaV2s, where functional experiments have demonstrated that CaM mutants that have diminished C lobe calcium binding but preserve N lobe calcium binding maintain CDI9,11,23.
Functional studies indicate lobe-specific tasks for CaM in CaV regulation9,12,16,17,23. The interactions that bring about lobe-specific regulation by CaM have remained obscure despite the investigation of an abundance of mutations to the IQ domain13–15,17,18,32. In the context of the structure, it is clear that most of the reported CaV1.2 mutants, many of which involve multiple residue changes, alter contacts to both lobes simultaneously and therefore cannot be used to decipher the roles of binding to each lobe individually. The exceptions are mutants having changes at two single positions that contact the Ca2+/C lobe, Ile1624 and Phe1628 (refs. 14,15). Ile1624 mutations that perturb the residue's hydrophobicity and size disrupt CDI15. F1628A changes channel inactivation properties15. Together, these results indicate that the crystallographically defined IQ domain interface that contacts the Ca2+/C lobe is important for channel inactivation.
Both CaM lobes have specific functional roles in modulation of CaV2 channels9,11,23. There is a clear role for the Ca2+/C lobe in CaV1 CDI. CaV1s and CaV2s are highly homologous in the region that interacts with CaM to mediate CDF and CDI. Despite the similarity, no role has yet been reported for the N lobe in CaV1 modulation. We tested whether the Ca2+/N lobe–anchoring residues observed in the Ca2+/CaM–IQ domain structure had a functional role. Simultaneous mutation of all three aromatic anchors for the N lobe to alanine did not affect CaV1.2 CDI (Fig. 4a,b). In contrast, these same mutations in the background of the I1624A mutation, which unmasks CaV1.2 CDF, completely disrupted CaV1.2 CDF, demonstrating a clear role for this Ca2+/N lobe interface in CDF (Fig. 4c,d). These experiments define a previously unknown role for the N lobe interface and suggest that the Ca2+/N lobe conformations seen in the structure are important for CDF. Apo CaM can also bind the IQ domain18,20. It remains possible that the functional effects we observed have more complex origins that arise from interplay between Ca2+/CaM– and apo CaM–IQ domain binding.
The dual binding mode of Ca2+/N lobe observed in the structure suggests a level of conformational plasticity in Ca2+/N lobe–channel interactions. Regions on the N-terminal side of the IQ domain bind Ca2+/CaM in vitro17,19–21,39, but with lower affinity than the IQ domain does17,20. Anchoring of the Ca2+/C lobe to the IQ domain may permit Ca2+/CaM to remain bound while other regions of the channel compete for Ca2+/N lobe or the Ca2+/N lobe aromatic anchors during various states of channel operation17,39,40. Alternatively, the observed conformational plasticity may provide a means for the complex to accommodate conformational changes that facilitate interactions with other channel domains while maintaining the Ca2+/N lobe–IQ interaction.
CaV2s show lobe-specific Ca2+/CaM modulation that seems inverted relative to CaV1.2 (refs. 9,11,23). CaV2 CDI relies on the Ca2+/N lobe9,11,23, whereas the prominent CDF of CaV2.1 originates with the Ca2+/C lobe9,11. The ITC data show that the Ca2+/N lobe–binding sites in the CaV1.2 IQ domain overlap with the Ca2+/C lobe–binding site. It is notable that three aromatic anchors identified here, Phe1618, Phe1622 and Phe1631, including Ca2+/N lobe aromatic anchor Phe1618, are conserved among CaV1s but not between CaV1s and CaV2s (Fig. 1c). Amino acid substitutions at these positions, together with changes at Gln1625 and Lys1633 (Fig. 1c), may tilt the Ca2+/N lobe– and Ca2+/C lobe–IQ domain affinity differences in favor of the Ca2+/N lobe. Such changes, in concert with interactions to other parts of the channel, may underlie the fundamental differences in lobe-specific modulation between CaV1.2 and CaV2s in a way that exploits the prodigious adaptability of CaM to recognize varied targets41.
The Ca2+/CaM–CaV1.2 IQ domain structure presented here is the first high-resolution view of the components that transduce Ca2+-dependent CaV regulation, and it provides a necessary molecular framework for detailed dissection of the transitions that drive the rich regulation of CaVs by CaM.
Methods
Purification
The IQ domain of human Cav1.2 (CaVα1c77) (residues 1611–1644), human CaM N lobe (residues 1–78) and human CaM C lobe (residues 79–148) were cloned into a modified pET28 vector (Novagen) denoted HMT42 that contains, in sequence, a His6-tag, maltose-binding protein and a cleavage site for the tobacco etch virus (TEV) protease. Full-length CaM was cloned into pEGST43 without using any affinity tag. The F1628A mutation in the IQ domain was obtained using the QuikChange protocol (Stratagene).
All proteins were expressed in Escherichia coli BL21(DE3)pLysS grown in 2×YT media at 37 °C. Cells were lysed in 250 mM KCl, 10 mM K-HEPES (pH 7.4) and 1 mM CaCl2 (buffer A) supplemented with 1 mM PMSF. The complex of CaM and IQ domain was obtained by coexpression and was purified on a Poros20MC column (Perseptive Biosystems), washed with buffer A and eluted with buffer A plus 500 mM imidazole (buffer B). After cleavage with His-tagged TEV protease44 at room temperature for ∼ 12 h and dialysis against 100 mM KCl, 10 mM Tris-HCl (pH 8.8) and 1 mM CaCl2 (buffer C), the complex was purified on a Hiload HQ column (Amersham) in buffer C with a linear gradient to 30% buffer D (1 M KCl, 10 mM Tris-HCl (pH 8.8), 1 mM CaCl2). Trace amounts of residual maltose-binding protein were removed by collecting the flow-through from an additional passage of the purified material over a Poros20MC column in buffer A. Finally, the sample was dialyzed against 20 mM KCl, 10 mM K-HEPES (pH 7.4) and 1 mM CaCl2.
SeMet-substituted complex was expressed in BL21(DE3)pLysS cells in M9 minimal medium containing 20% (w/v) glucose, with the methionine biosynthesis pathway inhibited45. Purification was as described above with all buffers supplemented with 5 mM methionine and 10 mM β-mercaptoethanol.
The first steps of purifying HMT–CaM N lobe and HMT–CaM C lobe fusions were similar to the CaM–IQ domain purification. After TEV cleavage, the material was dialyzed against buffer E (10 mM Tris-HCl (pH 8.8), 10 mM KCl, 1 mM CaCl2) and purified on a Hiload HQ column with a gradient from 0 to 50% buffer F (1 M KCl, 10 mM Tris-HCl (pH 8.8), 1 mM CaCl2). The proteins were purified further on a Poros20MC column in buffer A, and the concentrated flow-through was applied to a TSK2000 column (Tosoh Biosep) run in buffer A.
To obtain free IQ domain we purified the complex with CaM as described above and separated the two components on a preparative C18 HPLC column (Vydac) with a linear gradient from 35 to 50% acetonitrile in 0.1% (v/v) trifluoroacetic acid over 15 column volumes. The purity was verified by MALDI-TOF mass spectrometry.
Crystallization and structure determination
Complexes of native and SeMet-substituted CaM–IQ domain were crystallized by sitting or hanging drop vapor diffusion46 at 20 °C by mixing equal volumes of protein (∼ 10 mg ml−1) and well solution containing 0.1 M Bis-Tris (pH 6.5) and 20–30% (w/v) PEG 3350. After transfer to Paratone oil (Hampton Research) and flash-freezing, diffraction data were collected at Beamline 8.3.1 (Advanced Light Source, Lawrence Berkeley National Laboratories) and processed using HKL2000 (ref. 47). A three-wavelength MAD experiment was performed on crystals of SeMet-substituted protein (Table 2). Twenty-five initial selenium positions were located using ShelxD48. Subsequent refinement, substructure completion and phasing were performed using SHARP49 (Supplementary Fig. 1). After density modification the figure of merit was 0.852. An initial model was built using RESOLVE50 and ARP/wARP51, manually extended with XtalView52 and refined against 2.00-Å native data using REFMAC5 (ref. 53). TLS parameters were used throughout the refinement. Side chains and full residues with missing electron densities were not modeled. The final model consists of three Ca2+/CaM–IQ domain complexes in the asymmetric unit with 95.2% of the residues in the core region of the Ramachandran plot and none in disallowed regions. A Ni2+ ion is observed in all three complexes and most likely was introduced into the sample from the Ni2+ affinity column (Poros20MC). Its presence in the crystal was confirmed by a fluorescence wavelength scan. The Ni2+ binds at the C lobe outer surface distal from the IQ domain and is not expected to interfere with Ca2+/CaM–IQ domain binding. Refinement data and statistics are shown in Table 2.
Table 2. Data collection, phasing and refinement statistics.
| Native | SeMet | |||
|---|---|---|---|---|
| Data collection | ||||
| Space group | P21 | P21 | ||
| Cell dimensions | ||||
| a, b, c (Å) | 84.73, 37.24, 86.86 | 82.55, 37.01, 87.55 | ||
| α, β, γ (°) | 90, 97.77, 90 | 90, 98.10, 90 | ||
| Peak | Inflection | Remote | ||
| Wavelength | 1.116 | 0.97957 | 0.97972 | 1.01986 |
| Resolution (Å) | 30.0–2.0 (2.07–2.0) | 30–2.5 (2.59–2.50) | 30–2.8 (2.90–2.80) | 30–2.4 (2.49–2.40) |
| Rsym | 6.6 (33.1) | 9.5 (42.8) | 7.4 (40.1) | 6.6 (34.3) |
| I / σI | 19.8 (2.2) | 19.0 (2.5) | 14.2 (2.1) | 15.0 (1.7) |
| Completeness (%) | 99.2 (98.3) | 97.1 (99.4) | 99.9 (100) | 87.1 (89.6) |
| Redundancy | 3.5 | 7.8 | 3.8 | 2.6 |
| Refinement | ||||
| Resolution (Å) | 30.0–2.0 | |||
| No. reflections | 36,436 | |||
| Rwork / Rfree | 20.23 / 25.47 | |||
| No. atoms | ||||
| Protein | 3,845 | |||
| Ligand/ion | 15 | |||
| Water | 260 | |||
| B-factors | ||||
| Protein | 31.50 | |||
| Ligand/ion | 38.16 | |||
| Water | 47.05 | |||
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.016 | |||
| Bond angles (°) | 1.363 | |||
One native and one SeMet crystal were used for structure solution. Highest-resolution shell is shown in parentheses.
Isothermal titration calorimetry
Samples were concentrated and dialyzed twice against 5 mM KCl, 10 mM HEPES (pH 7.4) and 1mM CaCl2. Higher salt concentrations reduced the solubility of the IQ domain. Samples were degassed for 5 min and titrations were performed on a VP-ITC calorimeter (MicroCal) at 15 °C. Data were processed with MicroCal Origin 7.0. For fitting of the F1628A N lobe data, the stoichiometry for the second binding site was set equal to 1. Because of the high titrant concentration required to measure weak interactions, and owing to limited solubility of the IQ domain, N lobe was titrated into peptide and not vice versa.
Protein concentrations were determined by absorbance54, except for N lobe CaM (which has no aromatic residues), where the BCA method was used, using known concentrations of full-length CaM and CaM C lobe as references. Each ITC experiment was repeated with different batches of purified protein, yielding similar thermodynamic parameters and stoichiometry values. Control injections, consisting of titrating one component into buffer, were used to adjust the baseline of each experiment.
Electrophysiology
Constructs for electrophysiology consisted of human CaV1.2 (splice variant α1c77) in pcDNA3.1(+)/hygro (Invitrogen), CaVβ2a in pGEM (Promega) and rabbit CaVα2δ in pcDNA3 (Invitrogen). Mutants of the α1c77 subunit were made using the QuikChange protocol (Stratagene). RNA transcripts were prepared using a T7 mMessage mMachine kit (Ambion). 50 nl of a complementary RNA mixture containing 30–100 nM CaV1.2 α1c77, 33 nM CaVβ2a and 33 nM CaVα2δ was microinjected into Xenopus oocytes, which were then kept at 18 °C in ND96 medium supplemented with penicillin and streptomycin. Recordings were performed 3–7 d after injection. Before recording, oocytes were injected with 25–50 nl 100 mM BAPTA to minimize contaminating Ca2+-activated Cl− current. During recordings, the oocytes were superfused using a Valvelink 16 (Automate Scientific) controller with either a Ba2+-containing solution (40 mM Ba(OH)2, 50 mM NaOH, 1 mM KOH, 0.4% (w/v) niflumic acid, 10 mM HEPES) or a Ca2+-containing solution (Ba(OH)2 replaced with Ca(NO3)2). Both solutions were adjusted to pH 7.4 using HNO3. Two-electrode voltage-clamp experiments were performed using a GeneClamp 500B amplifier (Axon Instruments) controlled by a computer with a 1,200 MHz processor (Celeron, Gateway) using CLAMPEX 8.2.0.224 (Axon Instruments) and digitized at 1 kHz with a Digidata1332A (Axon Instruments). Electrodes were filled with 3 M KCl and had resistances of 0.1–1.5 MΩ. Leak currents were subtracted using a P/4 protocol. Ionic currents were analyzed with Clampfit 8.2 (Axon Instruments).
Supplementary Material
Supplementary Fig. 1 a, Experimentally phased and density modified electron density map, contoured at 1.2 σ, shown for the IQ domain in complex A. b, Residual difference electron density map contoured at 2 σ for the IQ domain in complex B. N-lobe and C-lobe are displayed in green and blue, respectively. IQ domain residues are shown as red sticks.
Supplementary Fig. 2 a-g, Details of interactions of CaV1.2 IQ domain residues with Ca2+/CaM. IQ domain residues are displayed in red, Ca2+/N-lobe residues in green, and Ca2+/C-lobe residues in blue. Dark colours correspond to complex A and light colours correspond to complex C. For the Ca2+/N-lobe interaction figures, the Ca2+/N-lobes of complex A and complex C have been superposed, outlining the differences in positions of IQ domain residues. For the Ca2+/C-lobe interactions, only chain A is shown, as the interactions in complex A and C are very similar. h, Superposition of complex A Ca2+/C-lobe (blue) with the Ca2+/C-lobe of CaM complexed to a CaMKII peptide (PDB entry 1CDM) (yellow), showing the different conformation of Met109 to produce a pocket for the deep anchor F1628. The CamKII peptide is not shown for clarity.
Supplementary Fig. 3 Overview of mainchain and sidechain interactions a, complex A and b, complex C. CaM residues ≤ 4 Å to the IQ domain are shown. IQ domain residues (red) are in three rows: contacts to Ca2+/N-lobe only (upper), none or both (middle) or Ca2+/C-lobe only (lower). Because contacts that are just below 4Å in one complex but just above 4 Å in the other would appear only in one figure and hinder appreciation of the main differences, contacts ≤ 4.2 Å are included, provided they are ≤ 4.0 Å in the other complex. Water-mediated hydrogen bonds are indicated (o). Residues N-terminal or C-terminal to the interaction site with are not shown.
Supplementary Fig. 4 Sequence alignment of the IQ domains of CaV1, CaV2, and NaV1 channels. CaV residues are coloured according to their corresponding contacts in complex A: green, Ca2+/N-lobe; cyan, Ca2+/C-lobe; purple, Ca2+/N-lobe and Ca2+/C-lobe. NaV residues are coloured according to their degree of conservation: grey, identical; orange, conserved. Asterisks indicate principal aromatic anchor positions of the CaV1.2 IQ domain. The consensus IQ motif is indicated.
Acknowledgments
We thank K. Brejc and D. Fass for comments on the manuscript; J. Holton at Beamline 8.3.1 at the Advanced Light Source for help with data collection; C.B. Klee (US National Institutes of Health, Bethesda, Maryland, USA) for the calmodulin clone; R.W. Tsien (Stanford University School of Medicine, Stanford, California, USA) and D.T. Yue (Johns Hopkins University School of Medicine, Baltimore, USA) for calcium channel clones; and members of the Minor laboratory for support at all stages of this work. This work was supported by awards to D.L.M. from the McKnight Foundation for Neuroscience, the Rita Allen Foundation, the Alfred P. Sloan Foundation and the US National Institutes of Health and to F.V.P. from the American Heart Association Western States Affiliate. D.L.M. is a McKnight Scholar in Neurosciences, an Alfred P. Sloan Research Fellow and a Rita Allen Foundation Scholar.
Footnotes
Accession codes. Protein Data Bank: Coordinates have been deposited with accession code 2BE6.
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
Competing Interests Statement: The authors declare that they have no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Hille B. Ion Channels of Excitable Membranes. Sinauer Associates, Inc.; Sunderland, Massachusetts, USA: 2001. [Google Scholar]
- 2.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 3.Saimi Y, Kung C. Calmodulin as an ion channel subunit. Annu Rev Physiol. 2002;64:289–311. doi: 10.1146/annurev.physiol.64.100301.111649. [DOI] [PubMed] [Google Scholar]
- 4.Kang MG, Campbell KP. Gamma subunit of voltage-activated calcium channels. J Biol Chem. 2003;278:21315–21318. doi: 10.1074/jbc.R300004200. [DOI] [PubMed] [Google Scholar]
- 5.Sheng ZH, Westenbroek RE, Catterall WA. Physical link and functional coupling of presynaptic calcium channels and the synaptic vesicle docking/fusion machinery. J Bioenerg Biomembr. 1998;30:335–345. doi: 10.1023/a:1021985521748. [DOI] [PubMed] [Google Scholar]
- 6.Walker D, De Waard M. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends Neurosci. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- 7.Kandel ER, Schwartz JH, Jessell TM. Principles of Neural Science. McGraw-Hill; New York, USA: 2000. [Google Scholar]
- 8.Budde T, Meuth S, Pape HC. Calcium-dependent inactivation of neuronal calcium channels. Nat Rev Neurosci. 2002;3:873–883. doi: 10.1038/nrn959. [DOI] [PubMed] [Google Scholar]
- 9.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 10.Lee A, et al. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 11.Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca2+/calmodulin-dependent regulation of Cav2.1 channels. Proc Natl Acad Sci USA. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 13.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 15.Zühlke RD, Pitt GS, Tsien RW, Reuter H. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the (alpha)1C subunit. J Biol Chem. 2000;275:21121–21129. doi: 10.1074/jbc.M002986200. [DOI] [PubMed] [Google Scholar]
- 16.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci USA. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pitt GS, et al. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- 18.Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- 19.Romanin C, et al. Ca(2+) sensors of L-type Ca(2+) channel. FEBS Lett. 2000;487:301–306. doi: 10.1016/s0014-5793(00)02361-9. [DOI] [PubMed] [Google Scholar]
- 20.Tang W, et al. Apocalmodulin and Ca2+ calmodulin-binding sites on the CaV1.2 channel. Biophys J. 2003;85:1538–1547. doi: 10.1016/s0006-3495(03)74586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pate P, et al. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J Biol Chem. 2000;275:39786–39792. doi: 10.1074/jbc.M007158200. [DOI] [PubMed] [Google Scholar]
- 22.Black DJ, et al. Calmodulin interactions with IQ peptides from voltage-dependent calcium channels. Am J Physiol Cell Physiol. 2005;288:C669–C676. doi: 10.1152/ajpcell.00191.2004. [DOI] [PubMed] [Google Scholar]
- 23.Liang H, et al. Unified mechanisms of Ca(2+) regulation across the Ca(2+) channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- 24.Osawa M, et al. A novel target recognition revealed by calmodulin in complex with Ca2+-calmodulin-dependent kinase kinase. Nat Struct Biol. 1999;6:819–824. doi: 10.1038/12271. [DOI] [PubMed] [Google Scholar]
- 25.Kurokawa H, et al. Target-induced conformational adaptation of calmodulin revealed by the crystal structure of a complex with nematode Ca(2+)/calmodulin-dependent kinase kinase peptide. J Mol Biol. 2001;312:59–68. doi: 10.1006/jmbi.2001.4822. [DOI] [PubMed] [Google Scholar]
- 26.Bahler M, Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002;513:107–113. doi: 10.1016/s0014-5793(01)03239-2. [DOI] [PubMed] [Google Scholar]
- 27.Jurado LA, Chockalingam PS, Jarrett HW. Apocalmodulin. Physiol Rev. 1999;79:661–682. doi: 10.1152/physrev.1999.79.3.661. [DOI] [PubMed] [Google Scholar]
- 28.Dunitz JD. Win some, lose some: enthalpy-entropy compensation in weak intermolecular interactions. Chem Biol. 1995;2:709–712. doi: 10.1016/1074-5521(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 29.Gao T, Bunemann M, Gerhardstein BL, Ma H, Hosey MM. Role of the C terminus of the alpha 1C (CaV1.2) subunit in membrane targeting of cardiac L-type calcium channels. J Biol Chem. 2000;275:25436–25444. doi: 10.1074/jbc.M003465200. [DOI] [PubMed] [Google Scholar]
- 30.Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zühlke RD, Reuter H. Ca2+-sensitive inactivation of L-type Ca2+ channels depends on multiple cytoplasmic amino acid sequences of the alpha1C subunit. Proc Natl Acad Sci USA. 1998;95:3287–3294. doi: 10.1073/pnas.95.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 33.Deschenes I, et al. Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ Res. 2002;90:E49–E57. doi: 10.1161/01.res.0000012502.92751.e6. [DOI] [PubMed] [Google Scholar]
- 34.Kim J, et al. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279:45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- 35.Mori M, et al. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-sensitivity? Biochemistry. 2000;39:1316–1323. doi: 10.1021/bi9912600. [DOI] [PubMed] [Google Scholar]
- 36.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 37.Leavitt S, Freire E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr Opin Struct Biol. 2001;11:560–566. doi: 10.1016/s0959-440x(00)00248-7. [DOI] [PubMed] [Google Scholar]
- 38.Jencks WP. On the attribution of additivity of binding energies. Proc Natl Acad Sci USA. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mouton J, Feltz A, Maulet Y. Interactions of calmodulin with two peptides derived from the c-terminal cytoplasmic domain of the Ca(v)1.2 Ca2+ channel provide evidence for a molecular switch involved in Ca2+-induced inactivation. J Biol Chem. 2001;276:22359–22367. doi: 10.1074/jbc.M100755200. [DOI] [PubMed] [Google Scholar]
- 40.Xiong L, Kleerekoper QK, He R, Putkey JA, Hamilton SL. Sites on calmodulin that interact with the C-terminal tail of Cav1.2 channel. J Biol Chem. 2005;280:7070–7079. doi: 10.1074/jbc.M410558200. [DOI] [PubMed] [Google Scholar]
- 41.Hoeflich KP, Ikura M. Calmodulin in action: diversity in target recognition and activation mechanisms. Cell. 2002;108:739–742. doi: 10.1016/s0092-8674(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 42.Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kholod N, Mustelin T. Novel vectors for co-expression of two proteins in E coli. Biotechniques. 2001;31:322–323. 326–328. doi: 10.2144/01312st03. [DOI] [PubMed] [Google Scholar]
- 44.Kapust RB, et al. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 45.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 46.McPherson A. Crystallization of Biological Macromolecules. Cold Spring Harbor Press; Cold Spring Harbor, New York, USA: 1999. [Google Scholar]
- 47.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 48.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 49.d Fortelle El, Bricogne G. Maximum-likelihood heavy atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 50.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 52.McRee DE. XtalView/Xfit–A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 53.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 54.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 a, Experimentally phased and density modified electron density map, contoured at 1.2 σ, shown for the IQ domain in complex A. b, Residual difference electron density map contoured at 2 σ for the IQ domain in complex B. N-lobe and C-lobe are displayed in green and blue, respectively. IQ domain residues are shown as red sticks.
Supplementary Fig. 2 a-g, Details of interactions of CaV1.2 IQ domain residues with Ca2+/CaM. IQ domain residues are displayed in red, Ca2+/N-lobe residues in green, and Ca2+/C-lobe residues in blue. Dark colours correspond to complex A and light colours correspond to complex C. For the Ca2+/N-lobe interaction figures, the Ca2+/N-lobes of complex A and complex C have been superposed, outlining the differences in positions of IQ domain residues. For the Ca2+/C-lobe interactions, only chain A is shown, as the interactions in complex A and C are very similar. h, Superposition of complex A Ca2+/C-lobe (blue) with the Ca2+/C-lobe of CaM complexed to a CaMKII peptide (PDB entry 1CDM) (yellow), showing the different conformation of Met109 to produce a pocket for the deep anchor F1628. The CamKII peptide is not shown for clarity.
Supplementary Fig. 3 Overview of mainchain and sidechain interactions a, complex A and b, complex C. CaM residues ≤ 4 Å to the IQ domain are shown. IQ domain residues (red) are in three rows: contacts to Ca2+/N-lobe only (upper), none or both (middle) or Ca2+/C-lobe only (lower). Because contacts that are just below 4Å in one complex but just above 4 Å in the other would appear only in one figure and hinder appreciation of the main differences, contacts ≤ 4.2 Å are included, provided they are ≤ 4.0 Å in the other complex. Water-mediated hydrogen bonds are indicated (o). Residues N-terminal or C-terminal to the interaction site with are not shown.
Supplementary Fig. 4 Sequence alignment of the IQ domains of CaV1, CaV2, and NaV1 channels. CaV residues are coloured according to their corresponding contacts in complex A: green, Ca2+/N-lobe; cyan, Ca2+/C-lobe; purple, Ca2+/N-lobe and Ca2+/C-lobe. NaV residues are coloured according to their degree of conservation: grey, identical; orange, conserved. Asterisks indicate principal aromatic anchor positions of the CaV1.2 IQ domain. The consensus IQ motif is indicated.