

Abstract
Total synthesis of glycosylated seco-iridoid stereoisomers allows the identification and bypassing of the stereoselectivity of early steps in monoterpene indole alkaloid biosynthesis.
Stereochemistry often determines how a natural product interacts with biological systems. For example, different stereosisomers can have different pharmacological profiles.1 The biosynthetic enzymes that form natural products typically allow the production of only one stereoisomer, so fermentation of products with alternate stereochemistry must therefore bypass the stereochemical restrictions of biosynthetic pathway enzymes. Using synthetic seco-iridoid and strictosidine starting materials we show that the heteroyohimbine branch of monoterpene indole alkaloid (MIA) biosynthesis in the medicinal plant Catharanthus roseus2,3 has a surprisingly broad tolerance for stereochemical perturbations in vitro.
Strictosidine synthase catalyzes the first step in monoterpene indole alkaloid biosynthesis: an asymmetric Pictet–Spengler reaction between tryptamine 1 and secologanin 2 to yield strictosidine 3a (Scheme 1).4,5 In the second step of this alkaloid pathway, 3a is deglucosylated by strictosidine-β-D-glucosidase.6,7 This results in the formation of a hemiacetal that rearranges into a mixture of products that is channeled into one of several pathway branches. The enzymes responsible for these branching reactions are unknown at the genetic and biochemical levels. However, entry into the heteroyohimbine class of alkaloids (5a–c) is likely controlled by one or several NADPH-dependent reductases (Scheme 1).8,9 Different heteroyohimbine alkaloid stereoisomers have different pharmacological activities. The heteroyohimbine alkaloid ajmalicine (raubasine) 5a acts as a smooth muscle relaxant and as an α1 anti-adrenergic,10–12 while tetrahydroalstonine 5b acts as an α2 anti-adrenergic.12
Scheme 1.

Heteroyohimbine biosynthesis. STS (strictosidine synthase) and SGD (strictosidine glucosidase) catalyze reactions that lead to a hemiacetal, which rearranges into a mixture of isomers; cathenamine 4 is shown. Reduction leads to heteroyohimbines 5a–c.
The stereogenic centers of strictosidine 3a, the central intermediate for all monoterpene indole alkaloids, are either derived from the densely functionalized secologanin dihydropyran (C-15, C-20, and C-21) or the prochiral aldehyde carbon that is converted to the C-3 stereogenic center in strictosidine (Scheme 1). We recently reported that strictosidine glucosidase promotes not only deglucosylation of strictosidine 3a, but also deglucosylation of vincoside 3b (the 3-(R) diastereomer of 3a).13 We report here that strictosidine glucosidase has an 80-fold higher specificity constant (kcat/KM) for 3a compared to 3b (Table 1). The kinetics show that the change in kcat/KM is due to a change in turnover number (kcat) while the Michaelis constant (KM) remains the same for the two diastereomers (Table 1). In the crystal structure of SGD from the closely related Rauvolfia serpentina homolog (PDB: 2FJ6),14 the C-3 carbon atom of strictosidine 3a is located near the surface of the protein. We speculate that the orientation of the glucose moiety, which is in the interior of the glucosidase, remains unchanged during turnover of vincoside 3b, while the binding pocket for the more distal regions of the substrate, including the C-3 carbon, can accommodate 3b.
Table 1.
Steady-state kinetic constants
| Strictosidine glucosidase kineticsa | kcat [s−1] | KM [mM] | kcat/KM [M−1 s−1] | Reductase kineticsb | Vmax [U mg−1]b | KM [mM] | Vmax/KM [U M−1mg−1]b |
|---|---|---|---|---|---|---|---|
| 3a | 15 ± 0.8 | 0.15 ± 0.03 | 1.0 × 102 ± 0.2 × 102 | Deglycosylated 3a | 0.027 ± 0.001 | 0.10 ± 0.02 | 0.26 ± 0.04 |
| 3b | 0.19 ± 0.01 | 0.15 ± 0.04 | 1.3 ± 0.3 | Deglycosylated 3b | 0.012 ± 0.0003 | 0.66 ± 0.04 | 1.9 × 10−2 ± 0.1 × 10−2 |
Activity quantified by monitoring the disappearance of starting material at pH 6.0, 30 °C.
1 U = 1 μmol product formed per minute at pH 7.0, 30°C. Apparent kinetic constants obtained from partially purified enzyme.
We asked whether strictosidine glucosidase can catalyze the conversion of alternate dihydropyran stereoisomers of 3a, derived from the C-2 and C-4 positions of secologanin 2. This has not been tested previously, as alternate stereoisomers of 2 are not readily available. To access alternate configurations, we applied and expanded the previously reported synthesis of acetal-protected 9,10-nor-secologanin aglycones.15,16 The key step is Tietze’s tandem Knoevenagel–hetero-Diels–Alder (KHDA) reaction, which assembles the dihydropyran from 7, 8, and an electron rich dienophile (e.g. vinyl ether). We adopted this strategy to, for the first time, obtain O-glucosylated 9,10-nor-strictosidine 12a and its stereoisomers 12 (Scheme 2). The KHDA substrate, 2,3,4,6-tetrabenzyl vinyl-glucose 6, was synthesized by Ir-catalyzed vinyl transfer from vinyl acetate to the corresponding alcohol.17 After the KHDA reaction of 6, 7, and 8, in the presence of potassium fluoride, the cycloadduct was subjected to methanolysis to generate methyl ester 9. Pd-catalyzed debenzylation of 9 afforded acetal-protected secologanin 10 as a mixture of inseparable stereoisomers. After acetal hydrolysis, 9,10-nor-secologanin 11 was carried into either an enzymatic or a non-enzymatic Pictet–Spengler reaction to generate the corresponding tetrahydro-β-carboline products 12a and 12 (Scheme 2).
Scheme 2.

Synthesis of 12 and 12a, 13a, and 13. a) KF, toluene; b) DBU, CH3OH, 38% over 2 steps; c) Pd/C, CH3OH, 77%; d) PPTS (2 eq., 0.2 M), acetone/H2O (2:1), ~10%; e) 1, C. roseus STS, NaPi (0.05 M, pH 7.0), 89%; f) 1, maleic acid buffer (0.01 M, pH 2.0); g) C. roseus SGD, citrate-phosphate buffer (0.15 M, pH 6.0); h) B. stearothermophilus α-glucosidase, almond β-glucosidase, strictosidine glucosidase, citrate phosphate buffer (0.15 M, pH 6.0); (i) reductase activity from C. roseus cell suspension culture, NADPH, NaPi (0.05 M, pH 7.0).
Isomers 12 were synthesized in acidic buffer from 1 and 11 (Scheme 2), and purified by preparative HPLC. Each separable peak was characterized by 1H NMR (ESI†). Analysis of the mixture by UPLC-MS resolved six peaks, each with a mass consistent with 18,19-nor-strictosidine 12 (m/z = 505 [M + H]+) (Fig. 1A(i): pk 1–6). In contrast, the strictosidine synthase-catalyzed reaction between 1 and 11 resulted in the appearance of a single product, 12a, which was isolated and characterized by NMR (ESI†). The doublet of a doublet at 5.8 ppm suggested a dihydropyran 2,4-trans configuration (Scheme 1),15,16 and the doublet at 4.8 ppm with a J-coupling constant of 7.8 Hz suggested a β-anomeric configuration for the glycoside linkage.18 Compound 12a therefore contains the same relative stereochemistry found in natural strictosidine. Compound 12a co-eluted with the third peak observed in the LC chromatogram of 12, indicating the presence and location of the natural stereoisomer among the mixture of the chemical reaction products. Therefore, strictosidine synthase, when challenged with an array of stereoisomers, displays a stringent preference for the diastereomer displaying the 2,4-trans configuration and β-glycoside bond found in the natural substrate. As an alternative to enzymatic synthesis, chemical synthesis of 12 from 1 and 11 allowed us to bypass the stringent selectivity of strictosidine synthase and assess stereochemical restrictions of subsequent biosynthetic steps. Analysis of the reaction of unnatural strictosidine stereoisomers with downstream enzymes therefore utilized chemically synthesized 12.
Fig. 1.
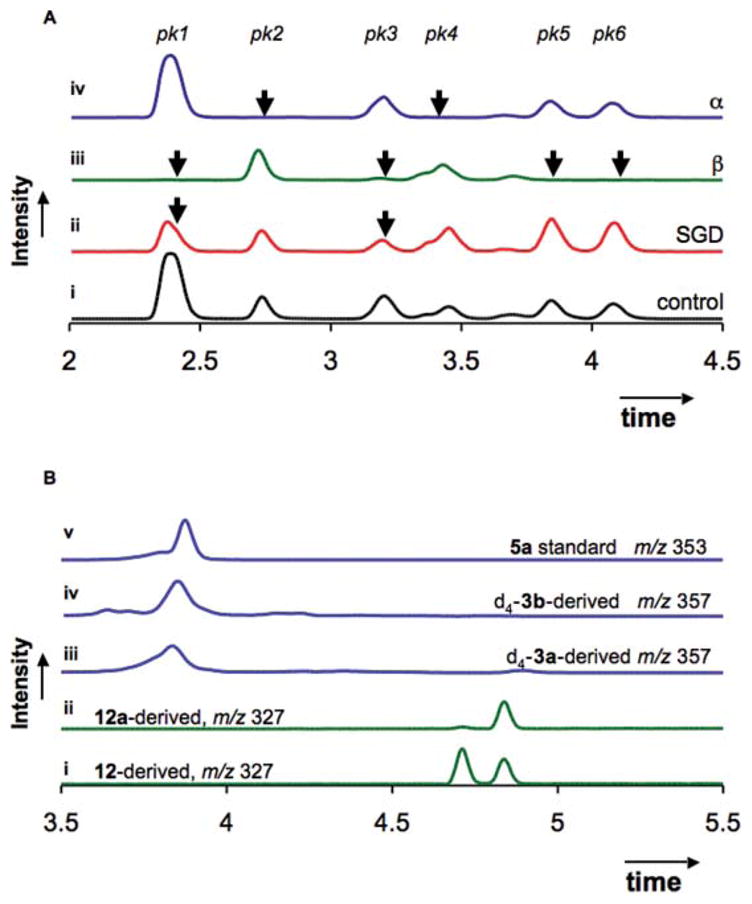
A Remaining starting material after deglucosylation of 12 (m/z 505.1 ± 0.5) by C. roseus strictosidine glucosidase (ii), almond β-glucosidase (β, iii), and B. stearothermophilus α-glucosidase (α, iv) monitored by UPLC-MS, (i) no-enzyme control; B. Reductase assay. (i) reduction of 12 yields two separable stereoisomers 13; (ii) reduction of deglucosylated 12a yields a single product 13a; (iii) reduction of deglucosylated d4-strictosidine 3a (m/z 357); (iv) reduction of deglucosylated d4-vincoside 3b (m/z 357); (v) ajmalicine 5a standard (m/z 353).
Strictosidine glucosidase could efficiently deglucosylate tetrahydro-β-carboline 12a, the isomer with the natural 2,4-trans configuration. When strictosidine glucosidase was incubated with all isomers of 12, only two of the six separable peaks decreased in area (Fig. 1A(ii): pk 1 and pk 3). One peak (pk 3) co-eluted with 12a (not shown), suggesting that the compound with natural trans stereochemistry is turned over. Pk 1 was isolated by preparative HPLC and subjected to NMR spectroscopy. The 1H NMR spectrum showed the presence of two stereoisomers, both with H15/H21-cis configuration, as evidenced by the triplets at 5.9 and 5.7 ppm, assigned to H-21 (ESI†).15,16 The anomeric configuration of the glucose moiety is likely β as indicated by the J-coupling constant of 8.0 Hz.18 We conclude that strictosidine glucosidase accommodates stereochemical perturbation in the secologanin dihydropyran moiety, but the anomeric configuration must be β for turnover to occur.
To fully explore the stereochemical promiscuity of the subsequent reductase catalyzed step, complete deglucosylation of 12 is required. However, strictosidine glucosidase only deglycosylated a few of the stereoisomers of 12. Since it has been previously shown that strictosidine 3a can be deglycosylated by bacterial glycosidases,19 we examined whether two commercially available glucosyl hydrolases, Bacillus stearothermophilus α-glucosidase and almond β-glucosidase, display different deglucosylation patterns compared to strictosidine glucosidase. Almond β-glucosidase was considerably more permissive than strictosidine glucosidase, consuming four out of the six peaks in the chromatogram (Fig. 1A(iii): pk 1, 3, 5, and 6), B. stearothermophilus α-glucosidase facilitated the consumption of two peaks that were not converted by either β-glucosidase (Fig. 1A(iv): pk 2 and 4). Strictosidine glucosidase therefore appears to be more specific for its substrate, 12a, while the two commercially available glucosidases likely have active sites that allow a greater diversity of substrates to be accepted. By using glucosidases from different metabolic pathways, it is possible to bypass the native biosynthetic pathway to fully deglucosylate 12.
In the heteroyohimbine biosynthetic pathway, one or more reductases catalyze the NADPH-dependent reduction of deglycosylated strictosidine 3a to form monoterpene indole alkaloids such as 5a–c (Scheme 1). A cell-free extract from C. roseus cell suspension culture was used to reconstitute the reductase activity. Control experiments revealed that 3a, strictosidine glucosidase, NADPH, and the reductase activity were each a necessary component for formation of a reduced product that eluted near an authentic standard of ajmalicine 5a (Fig. 1B). We determined the steady-state kinetics for reduction of the natural substrate, deglucosylated strictosidine 3a, and the unnatural substrate, deglucosylated vincoside 3b (Table 1). The enzyme showed a 15-fold catalytic preference (Vmax/KM) for the 3-(S) stereochemistry of strictosidine 3a (0.26 U M−1 mg−1) over the 3-(R) stereochemistry of vincoside 3b (0.019 U M−1 mg−1). This was mainly due to a 7-fold difference in KM (0.1 mM compared to 0.7 mM with 3a and 3b, respectively). The catalytic differentiation between 3a and 3b suggests that the reductase activity derives from the monoterpene indole alkaloid pathway. However, since the enzyme was not purified to homogeneity, despite extensive efforts, we cannot rigorously exclude the involvement of additional reductases in the turnover of deglucosylated 3a or 3b.
Deglucosylated 18,19-nor-strictosidine 12a was converted by the reductase into a compound with a 1H NMR spectrum and mass consistent with 13a (Scheme 2, and ESI†). When reductase activity and NADPH was added to deglycosylated 12, which contained all stereoisomers, two separable reduced products formed; one product coeluted with 13a (Fig. 1B). The reduced product contains two stereogenic centers, C-3 and C-15 (Scheme 2), and only two sets of enantiomers are expected to separate under the chromatographic conditions. Since 12 was completely deglucosylated, and because all deglucosylated 12 was completely consumed upon addition of the reductase activity (ESI†), we conclude that the reductase turns over all secologanin dihydropyran configurations. The reductase(s) that generate heteroyohimbine alkaloids appear to be capable of acting upon a wide variety of substrates. This suggests that this reductase(s) will have broad applications in chemoenzymatic synthesis after efforts to clone the enzyme are successful. Synthetic installation of the vinyl group on 11 will allow access to an even broader range of alkaloid structures.
The first step of the pathway, catalyzed by strictosidine synthase, may have evolved stringent substrate specificity to ensure the integrity of the first committed intermediate strictosidine 3a. Strictosidine glucosidase also shows strict stereocontrol, but accepts at least one unnatural dihydropyran stereoisomer with relative cis stereochemistry. Recruitment of glucosidases from other metabolic pathways highlights the potential to bypass stereochemical restrictions and to diversify alkaloid biosynthesis. Assay of 3a, 3b, 12a, and 12 with heteroyohimbine reductase activity suggests that C. roseus harbors at least one enzyme that converts these stereoisomers to reduced alkaloids in vitro. Regardless of whether the observed activity is functional in vivo, this enzyme can be used in heterologous expression systems to yield novel variants of the heteroyohimbine framework. This approach requires the gene encoding this enzyme, and efforts to identify the NADPH dependent reductases of C. roseus are ongoing.
Supplementary Material
Acknowledgments
We gratefully acknowledge funding from GM074820.
Footnotes
Electronic supplementary information (ESI) available: Chemical synthesis, supplementary figures, methods. See DOI: 10.1039/b916027m
Notes and references
- 1.De Camp WH. Chirality. 1989;1:2–6. doi: 10.1002/chir.530010103. [DOI] [PubMed] [Google Scholar]
- 2.Van der Heijden R, Jacobs DI, Snoeijer W, Hallard DVR. Curr Med Chem. 2004;11:607–628. doi: 10.2174/0929867043455846. [DOI] [PubMed] [Google Scholar]
- 3.O’Connor SE, Maresh J. Nat Prod Rep. 2006;23:532–547. doi: 10.1039/b512615k. [DOI] [PubMed] [Google Scholar]
- 4.Ma X, Panjikar S, Koepke J, Loris E, Stöckigt J. Plant Cell. 2006;18:907–920. doi: 10.1105/tpc.105.038018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maresh JJ, Giddings LA, Friedrich A, Loris EA, Panjikar S, Trout BL, Stöckigt J, Peters B, O’Connor SE. J Am Chem Soc. 2008;130:710–723. doi: 10.1021/ja077190z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerasimenko I, Sheludko Y, Ma X, Stöckigt J. Eur J Biochem. 2002;269:2204–2213. doi: 10.1046/j.1432-1033.2002.02878.x. [DOI] [PubMed] [Google Scholar]
- 7.Geerlings A, Ibanez MML, Memelink J, Van der Heijden R, Verpoorte R. J Biol Chem. 2000;275:3051–3056. doi: 10.1074/jbc.275.5.3051. [DOI] [PubMed] [Google Scholar]
- 8.Hemscheidt T, Zenk MH. Plant Cell Rep. 1985;4:216–219. doi: 10.1007/BF00269293. [DOI] [PubMed] [Google Scholar]
- 9.Stöckigt J, Hemscheidt T, Hofle G, Heinstein P, Formacek V. Biochemistry. 1983;22:3448–3452. [Google Scholar]
- 10.Brevetti G, Chiariello M, Verrienti S, Spena M, Desiderati M, Condorelli M. Angiology. 1983;34:517–526. doi: 10.1177/000331978303400803. [DOI] [PubMed] [Google Scholar]
- 11.Li S, Long J, Ma Z, Xu Z, Li J, Zhang Z. Curr Med Res Opin. 2004;20:409–415. doi: 10.1185/030079904125003080. [DOI] [PubMed] [Google Scholar]
- 12.Roquebert J, Demichel P. Eur J Pharmacol. 1984;106:203–205. doi: 10.1016/0014-2999(84)90698-8. [DOI] [PubMed] [Google Scholar]
- 13.Yerkes N, Wu JX, McCoy E, Galan MC, Chen S, O’Connor SE. Bioorg Med Chem Lett. 2008;18:3095–3098. doi: 10.1016/j.bmcl.2007.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barleben L, Panjikar S, Ruppert M, Koepke J, Stöckigt J. Plant Cell. 2007;19:2886–2897. doi: 10.1105/tpc.106.045682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tietze LF, Meier H, Nutt H. Liebigs Ann Chem. 1990:253–260. [Google Scholar]
- 16.Tietze LF. Angew Chem. 1983;95:840–853. [Google Scholar]
- 17.Okimoto Y, Sakaguchi S, Ishii Y. J Am Chem Soc. 2002;124:1590–1591. doi: 10.1021/ja0173932. [DOI] [PubMed] [Google Scholar]
- 18.Sinnott ML. Carbohydrate Chemistry and Biochemistry: Structure and Mechanism. Royal Society of Chemistry; 2007. [Google Scholar]
- 19.Zhengwu S, Eisenreich W, Kutchan TM. Phytochemistry. 1998;48:293–296. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.