

We investigated the poorly understood mechanism of plasma membrane targeting of formin mDia2 and found that its N terminus plays important roles in this process by binding acidic phospholipids through its N-terminal basic domain and by binding small GTPase Rif through direct interaction with the GTPase binding region and the diaphanous inhibitory domain.
Abstract
The formin mDia2 mediates the formation of lamellipodia and filopodia during cell locomotion. The subcellular localization of activated mDia2 depends on interactions with actin filaments and the plasma membrane. We investigated the poorly understood mechanism of plasma membrane targeting of mDia2 and found that the entire N-terminal region of mDia2 preceding the actin-polymerizing formin homology domains 1 and 2 (FH1–FH2) module was potently targeted to the membrane. This localization was enhanced by Rif, but not by other tested small GTPases, and depended on a positively charged N-terminal basic domain (BD). The BD bound acidic phospholipids in vitro, suggesting that in vivo it may associate with the plasma membrane through electrostatic interactions. Unexpectedly, a fragment consisting of the GTPase-binding region and the diaphanous inhibitory domain (G-DID), thought to mediate the interaction with GTPases, was not targeted to the plasma membrane even in the presence of constitutively active Rif. Addition of the BD or dimerization/coiled coil domains to G-DID rescued plasma membrane targeting in cells. Direct binding of Rif to mDia2 N terminus required the presence of both G and DID. These results suggest that the entire N terminus of mDia2 serves as a coincidence detection module, directing mDia2 to the plasma membrane through interactions with phospholipids and activated Rif.
INTRODUCTION
The ability of cells to move is a requisite for organismal development and survival. A common mode of cell motility entails shape changes, such as protrusion of the front and retraction of the rear of the cell. These processes are powered, in large part, through forces generated by the actin cytoskeleton. To protrude the leading edge, cells exert force on the plasma membrane through elongating actin filaments, with their barbed ends oriented toward the membrane (reviewed in Chhabra and Higgs, 2007). This process is regulated by a large number of structural and regulatory proteins.
Formin family proteins are key regulators of actin polymerization. They promote actin assembly by nucleating actin filaments de novo and by enhancing their elongation at the barbed end (Pruyne et al., 2002; Higgs, 2005; Paul and Pollard, 2009; Chesarone et al., 2010). Both of these functions are mediated by the evolutionarily conserved formin homology domain 2 (FH2). FH2 dimers associate processively with the barbed ends of elongating actin filaments, simultaneously allowing for the incorporation of actin monomers and protecting the barbed end against capping proteins. In the presence of profilin–actin complexes, the elongation-promoting activity of formins is greatly enhanced by the upstream proline-rich formin homology domain 1 (FH1), which is thought to mediate the transfer of profilin–actin complexes onto the barbed ends of growing filaments (Kovar et al., 2006).
The formin mDia2 controls actin polymerization in lamellipodia and filopodia (Yang et al., 2007), although its role in filopodia is better known (Peng et al., 2003; Pellegrin and Mellor, 2005; Schirenbeck et al., 2005). In cells, mDia2 is tightly regulated to allow actin polymerization at the right place and time. The regulation of mDia2, and the related formins mDia1 and mDia3, depends on domains that surround the actin-polymerizing module FH1–FH2. The region N-terminal to FH1–FH2 consists of a series of discrete domains, including the GTPase binding region (G), the diaphanous inhibitory domain (DID), the dimerization domain (DD), and the coiled coil (CC). The region C-terminal to the FH1–FH2 contains the diaphanous autoinhibitory domain (DAD) (Alberts et al., 1998; Alberts, 2001; Li and Higgs, 2005; Otomo et al., 2005; Rose et al., 2005). In addition, a nuclear localization signal was found within the first 41 amino acids of mDia2 (Miki et al., 2009).
In the resting state, full-length mDia proteins are autoinhibited through an intramolecular interaction between DID and DAD (Watanabe et al., 1999; Alberts, 2001; Li and Higgs, 2003). To reverse this inhibition, cells use small GTPases of the Rho family (Watanabe et al., 1997; Li and Higgs, 2005). Several small GTPases have been reported to control activation of mDia2: RhoA-C (Alberts et al., 1998; Wallar et al., 2007), Cdc42 (Peng et al., 2003), Rac1 and Rac2 (Ji et al., 2008; Lammers et al., 2008), and Rif (Pellegrin and Mellor, 2005), whereas RhoA-C are considered the primary regulators of mDia1 (Watanabe et al., 1999). Although the G region is believed to be the main site for the binding of GTPases, crystal structures show that both the G region and DID of mDia1 mediate interactions with Rho (Otomo et al., 2005; Rose et al., 2005; Lammers et al., 2008). The overlap between the binding sites of the DAD and Rho is only partial, so that Rho is thought to reverse autoinhibitory interactions by binding in two steps, first weakly to the exposed binding interface of the G and then strongly to the entire interface on the G-DID after displacement of the DAD (Lammers et al., 2005; Nezami et al., 2006). In vitro experiments with mDia1, however, have shown that RhoA alone is insufficient to fully activate mDia1 (Li and Higgs, 2003, 2005), suggesting that other factors are needed (Seth et al., 2006; Brandt et al., 2007). This notion is consistent with an emerging concept that multiple inputs are required to activate proteins to ensure their tight regulation and prevent spurious activity.
Subcellular targeting is another important aspect of protein regulation. Whereas formins use their FH2 to associate with actin filament barbed ends, they use other domains to find their precise locations in cells. Early results suggested a role of the N-terminal region, upstream of FH1–FH2, in targeting the fission yeast formin Fus1 to the membrane (Petersen et al., 1998). The N-terminal region is also involved in the subcellular localization of the mammalian formins mDia1 and FRLα (Seth et al., 2006; Copeland et al., 2007). Among the N-terminal domains, the G region is thought to play a key role in targeting formins to the plasma membrane by binding membrane-associated activated GTPases (Rose et al., 2005). Regions other than the G region, however, also have been shown to contribute to the localization of various formins (reviewed in Aspenstrom, 2010; Chesarone et al., 2010). Thus, a region within the DID of mDia1, but not of mDia2 or mDia3, was found to interact with Ras GTPase-activating-like protein (IQGAP1) and to be important for mDia1 localization (Brandt et al., 2007). In addition, sequences containing the DD and/or CC were reported to weakly target mDia1 to the mitotic spindle (Kato et al., 2001) or the plasma membrane (Copeland et al., 2007), and to interact with Abi1 (Yang et al., 2007; Ryu et al., 2009), a protein that localizes to the leading edge of lamellipodia (Stradal et al., 2001). The CC domain targets formin-1 to adherens junctions via interaction with α-catenin (Kobielak et al., 2004). Additionally, protein–protein interaction roles have been ascribed to the C-terminal region downstream to the FH1–FH2 module in the formins DAAM (Liu et al., 2008), FHOD1 (Gill et al., 2004), and Fmn-2 (Pechlivanis et al., 2009). FH1 could also participate in targeting, because in mDia1, mDia2, or DAAM this region has been shown to bind numerous SH3 domain-containing proteins having membrane-binding properties (Fujiwara et al., 2000; Hudson et al., 2008; Aspenstrom, 2010). Other mechanisms of formin targeting may involve covalent lipid modification, as shown for FMNL1γ (Han et al., 2009) and INF2 (Chhabra et al., 2009).
Thus, multiple mechanisms converge to fine-tune the subcellular localization of formins, involving virtually any formin domain. Among formins, subcellular localization has been most extensively studied for mDia1, but even for this formin the mechanism of targeting is not fully understood. Even less is known about the targeting mechanism of mDia2. Despite the close similarity and some functional overlap between these two formins, their cellular functions are different. Whereas mDia1 is mostly involved in the generation of contractile actin bundles and adhesions (Nakano et al., 1999; Watanabe et al., 1999; Yamana et al., 2006; Carramusa et al., 2007; Ryu et al., 2009), mDia2 is mostly implicated in the formation of membrane protrusions (Peng et al., 2003; Pellegrin and Mellor, 2005; Schirenbeck et al., 2005; Yang et al., 2007), although it also plays a role in cytokinesis (Watanabe et al., 2010). Accordingly, activated mDia2 functions at the interface between actin filament barbed ends and the plasma membrane, and the interactions with both surfaces control its subcellular localization (Yang et al., 2007). Our previous data suggested that the N-terminal region of mDia2 is required for stable association with the leading edge of the cell (Yang et al., 2007). Recent analysis of mDia2 targeting to the cytokinetic ring revealed that it involves interaction of the G region with RhoA and of the DID-DD-CC region with the scaffolding protein anillin (Watanabe et al., 2010). The mechanism of mDia2 interaction with the membrane during cell motility remains unknown, however.
In this study, we performed a detailed structure–function analysis of mDia2 to define which domains are involved in its localization to the plasma membrane in an actin-independent manner. Because sequences N- and C-terminal to the FH1–FH2 module appear to play a targeting role in other formins, we considered these sequences to be candidates to target mDia2 to the membrane. In addition to the traditionally recognized N-terminal domains of mDia2, we also evaluated the role of the poorly characterized 90-aa region at the extreme N terminus preceding the G region. We refer to this region as the basic domain (BD), because of its positive charge, with a predicted isoelectric point of ∼10. We hypothesized that the BD could potentially play a role in targeting mDia2 to the membrane, because other proteins have been shown to bind charged plasma membrane phospholipids through their BDs (McLaughlin and Murray, 2005).
RESULTS
mDia2 is targeted to the plasma membrane through its N-terminal domains
The formin mDia2 is a modular protein consisting of a series of semi-independent functional domains (Goode and Eck, 2007). To test which of the domains of mDia2 localize to the plasma membrane in an actin-independent manner, we expressed green fluorescent protein (GFP) fusion of mDia2 domains, or their combinations, together with the cytoplasmic marker mRFP1 in HeLa cells (Figure 1, A and B). We excluded the region encompassing the FH1–FH2 domains (aa 529–1025) from this analysis because it can be targeted to the plasma membrane by binding to actin filament barbed ends. We analyzed the plasma membrane localization of GFP fusion proteins by confocal microscopy, using midplane optical sections of expressing cells (Figure 1B).
FIGURE 1:

Plasma membrane targeting capability of mDia2 resides in the N-terminal domains and depends on the BD. (A) Domain architecture of mDia2 and its truncation mutants used in this figure. Numbers indicate amino acid boundaries. (B) Confocal microscopy of HeLa cells coexpressing indicated GFP-tagged mDia2 proteins (top row) and mRFP1 (middle row). Merged images are shown in the bottom row. Cells expressing GFP-Nt were also treated with 2 μM latrunculin B for 30 min (+LatB). Midplane optical sections are shown for all cells. GFP-Nt is strongly targeted to the plasma membrane, in both the presence and the absence of latrunculin B, whereas GFP-BD and GFP-NtΔBD are weakly targeted. (C) Quantification of plasma membrane localization of mDia2 proteins. PM indices are shown as box-and-whisker plots, with boxes encompassing 75th/25th percentile and whiskers encompassing 95th/5th percentile. Dashed line is average, and solid line is median. Numbers of quantified cells are shown in parenthesis next to the name of the construct. (D) Line scan analysis of GFP-BD localization. Plot shows average fluorescence intensity of the GFP (green) and mRFP1 (red) signals within a 10-pixel-wide line drawn across the cell expressing GFP-BD and mRFP1 (inset). Two green peripheral peaks correspond to a plasma membrane pool of GFP-BD, and the broad central plateau corresponds to the nuclear pool of GFP-BD. Scale bars, 10 μm.
We first evaluated the localization of constructs flanking the FH1–FH2 module on both sides, the N-terminus (Nt, 1–528) or the C-terminus (Ct, 1026–1171), using GFP and full-length GFP-mDia2 as controls (Figure 1, A and B). As expected, GFP and mRFP1 had similar diffuse localizations in the cytoplasm and the nucleus (Figure 1B). Full-length GFP-mDia2 was also diffusely distributed in the cytoplasm, but was excluded from the nucleus, consistent with previous observations (Miki et al., 2009). Such a distribution is believed to reflect the autoinhibited conformation of mDia2. The C terminus of mDia2 was also cytoplasmic, suggesting that it is unlikely to contribute to membrane targeting of mDia2. In contrast, GFP-Nt was strongly localized to the plasma membrane and largely depleted from the cytoplasm, analogous to the reported plasma membrane localization of the N termini of mDia1 and FRLα (Seth et al., 2006). Treatment with the actin-depolymerizing drug latrunculin B (2 μM for 30 min) did not alter the membrane localization of GFP-Nt, consistent with its localization being actin-independent (Figure 1B).
To quantify the degree of plasma membrane localization of various GFP-tagged constructs, we devised a parameter termed the plasma membrane localization index (PM index; see Materials and Methods). The PM index equals zero for nonmembrane-targeted constructs, whereas enrichment at the membrane results in positive values of the PM index. PM indices ≤ 0.3 were close to the detection limit of our method, and constructs having PM indexes in this range were not considered to be targeted to the membrane. An increment of one unit in the PM index corresponds to a 100% increase in the average fluorescence intensity of a protein at the membrane as compared to that of the GFP control. The values of the PM index for different constructs (Figure 1C) confirmed the conclusions drawn from the visual inspection of confocal images. Specifically, the PM index for GFP, GFP-mDia2, and GFP-Ct were close to zero. In contrast, the PM index for GFP-Nt was 3.5 ± 1.9, indicating strong plasma membrane localization. This value did not change significantly after latrunculin B treatment (3.7 ± 1.5, Figure 1C), suggesting that plasma membrane targeting of GFP-Nt is independent of actin filaments.
The BD of mDia2 is important for plasma membrane targeting
The N terminus of mDia2 consists of the following characterized domains: G (91–148), DID (149–397), DD (398–468), and CC (469–528). The exact boundaries of these domains have been determined based on their crystal structures of mDia1 (Otomo et al., 2005) and have been deduced for mDia2 based on sequence alignment (Supplemental Figure 1). The first 90 amino acids of mDia2 are poorly characterized. We named this region the BD, because it is rich in positively charged residues (Supplemental Figure 1) and has a predicted isoelectric point of ∼10.
To determine which of the domains of the mDia2 Nt contribute to plasma membrane targeting, we generated GFP fusion constructs of BD, G, DID, and DD-CC (Figure 1A). We expressed these constructs in HeLa cells and tested their plasma membrane localization by confocal microscopy (Figure 1B). To our surprise, only GFP-BD showed significant localization to the plasma membrane (Figure 1, B and D) with a PM index of 1.0 ± 0.5 (Figure 1C), lower than that of GFP-Nt, but significantly higher than that of GFP alone. GFP-BD was also enriched in the nucleus, consistent with a previous finding that this region harbors a nuclear localization signal (NLS) (Miki et al., 2009). The lack of membrane localization of GFP-G and GFP-DID was not due to degradation as they were expressed at a correct molecular weight (Supplemental Figure 2).
FIGURE 2:

The BD of mDia2 is important for plasma membrane targeting and binds acidic phospholipids in vitro. (A) Subcellular fractionation of HeLa cells expressing GFP fusion proteins. Cytoplasmic (Cyt) and membrane (Mem) fractions of cells expressing indicated constructs (top row) were loaded on the gel in volume equivalents shown by numbers above the corresponding lanes. Western blotting with GFP antibody was used to detect the expressed proteins. Tubulin and IRSp53 were used as cytoplasmic and plasma membrane markers, respectively, to confirm successful fractionation. Calculated percentage of GFP fusion proteins in the membrane fraction (%) is shown at bottom. (B) Purified GST-BD (arrow) shown by Coomassie staining of SDS–PAGE gel. (C) Protein-lipid overlay assay showing GST-BD binding to acidic, but not neutral phospholipids (left panel). No binding is detected for GST alone (right panel). Abbreviations: LPA, lysophosphatidic acid; LPC, lysophosphocholine; PI, phosphatidylinositol; PI(3)P, phosphatidylinositol-3-phosphate; PI(4)P, phosphatidylinositol-4-phosphate; PI(5)P, phoshphatidylinositol-5-phosphate; PE, phosphatidylethanolamine; PC, phosphatidylcholine; S1P, sphingosine-1-phosphate; PI(3,4)P2, phosphatidylinositol-3,4-bisphosphate; PI(3,5)P2, phosphatidylinositol-3,5-bisphosphate; PI(4,5)P2, phosphatidylinositol-4,5-bisphosphate, PI(3,4,5)P3, phosphatidylinositol-3,4,5-trisphosphate; PA, phosphatidic acid; PS, phosphatidylserine. (D) Secondary structure predictions for the basic domain of mDia2. Top: Hydrophobic cluster analysis (HCA) reveals abundance of positively charged residues (blue) in the 1–20 region and two putative hydrophobic clusters in the 40–60 and 70–80 regions (outlined). In the HCA, stars are prolines, black diamonds are glycines, and empty and filled squares are threonines and serines, respectively. Middle: Helical wheel presentation of a putative amphipathic helix for amino acids 43–60 corresponding to the first hydrophobic cluster in the HCA plot. Nonpolar residues are clustered on the upper left side of the wheel. Bottom: Diagrams of mDia2 constructs used in panel E. (E) Confocal microscopy images of HeLa cells coexpressing indicated GFP-mDia2 fusion constructs and mRFP1 as a cytoplasmic marker. Average PM indices with standard deviations and numbers of quantified cells in parenthesis are shown to the right of the merged images. Scale bar, 10 μm.
If the BD plays an important role in plasma membrane targeting, its deletion should compromise plasma membrane localization of the other N-terminal domains. Consistent with this idea, plasma membrane targeting of GFP-NtΔBD (91–528) was severely compromised, as reflected by a decrease of the PM index to 0.7 ± 0.4 (Figure 1, A–C). These results strongly suggest that the BD is a key determinant for plasma membrane localization of mDia2 Nt, and most likely also contributes to the targeting of activated full-length mDia2.
To verify the data obtained by confocal microscopy, we performed subcellular fractionation of HeLa cells expressing GFP, GFP-BD, GFP-NtΔBD, and GFP-Nt (Figure 2A) to determine the extent to which these constructs partition with cellular membranes. The results showed no recovery of GFP in the membrane fraction, whereas 28 ± 8% of GFP-BD, 15 ± 3% of GFP-NtΔBD, and 41 ± 8% of GFP-Nt were present in the membrane fraction, suggesting that the ability of these proteins to bind the membrane decreases in the following order: Nt > BD > NtΔBD > GFP. These results are consistent with the data obtained by confocal microscopy (Figure 1C), and confirm that the BD of mDia2 associates with the plasma membrane and contributes to plasma membrane targeting of mDia2 Nt.
We hypothesized that the positively charged BD could be targeted to the plasma membrane by direct interaction with negatively charged phospholipids. To test this idea, we performed a lipid–protein overlay assay with purified GST-BD and found that it binds acidic lipids but not neutral lipids (Figure 2, B and C). The interaction with acidic phospholipids seemed to be nonspecific, suggesting an electrostatic mode of interaction.
The N-terminal portion of the BD (1–37) contains most of the positively charged amino acids, but lacks clusters of hydrophobic amino acids and is, therefore, predicted to be intrinsically disordered. In contrast, hydrophobic cluster analysis (Gaboriaud et al., 1987) reveals the presence of two clusters of hydrophobic amino acids within the C-terminal portion of the BD (38–90), displaying a characteristic helical pattern (Figure 2D). Using a helical wheel representation of this region, we found that hydrophobic amino acids and charged (or polar) amino acids are mostly clustered on two opposite sides of the helical wheel, indicative of amphipathic helices (Figure 2D). To test the role of the two predicted amphipathic helices of the BD in membrane localization, we prepared constructs GFP-BD1–64 and GFP-BD1–37 lacking one or both of the hydrophobic clusters, respectively (Figure 2, D and E). Confocal microscopy of HeLa cells revealed that GFP-BD1–64 was somewhat enriched at the plasma membrane (PM index = 0.6 ± 0.3), whereas GFP-B1–37 was mostly cytoplasmic (PM index = 0.2 ± 0.2). These data suggest that the C-terminal helical portion of the BD is important for its localization to the plasma membrane.
To further explore the roles of the basic stretch and hydrophobic clusters of the BD in plasma membrane targeting, we generated construct GFP-NtΔ1–37 (38–528), which lacks the N-terminal basic residues stretch, and evaluated its membrane localization in cells (Figure 2, D and E). The PM index of GFP-NtΔ1–37 was 2.1 ± 1.4, which is significantly higher than that of GFP-NtΔBD (0.7 ± 0.4) and points to an important role of two predicted amphipathic helices of the BD in plasma membrane targeting. The plasma membrane localization of GFP-NtΔ1–37 was significantly lower than that of GFP-Nt (PM index = 3.5 ± 1.9), however, suggesting that the N-terminal basic stretch of BD also contributes in a significant way to plasma membrane binding. The isolated construct GFP-B1–37 did not significantly localize to the membrane, possibly due to degradation of this short unstructured peptide. Together, these data indicate that the BD is an important membrane-targeting region of mDia2 and that its association with the plasma membrane depends on electrostatic interactions and the presence of two predicted amphipathic helices.
G region, DID, and DD-CC collectively contribute to plasma membrane localization
Although the plasma membrane localization of GFP-NtΔBD was drastically impaired by the absence of the BD, GFP-NtΔBD was not completely cytoplasmic based on confocal microscopy and cell fractionation results. In other words, even though individual G, DID, and DD-CC fragments did not detectably localize to the plasma membrane, collectively they did (Figure 1C). Therefore, we assessed the contributions of various combinations of these domains toward targeting of GFP-NtΔBD to the plasma membrane (Figure 3).
FIGURE 3:

G, DID, and DD-CC collectively localize to the plasma membrane. (A) Domain architecture of mDia2 constructs used in this figure (left) and average PM indices of HeLa cells expressing corresponding GFP fusion proteins. Asterisk represents a point mutation. The numeric values of PM indices and numbers of quantified cells in parenthesis are shown to the right of each bar. Error bars, SEM. (B) Confocal microscopy images of HeLa cells coexpressing indicated GFP-mDia2 constructs and mRFP1 as a cytoplasmic marker. Merged images are shown in the bottom row. Scale bar, 10 μm.
We observed negligible plasma membrane enrichment of the isolated G region (PM index = 0.2 ± 0.1; Figure 1B) even though this region, in conjunction with a part of DID, is thought to target formins to the membrane through GTPase binding (Rose et al., 2005). We then asked whether the two predicted amphipathic helices of the BD that lie immediately N-terminal to the G (Figure 2D) form a common GTPase-binding unit with the G region in mDia2. To answer this question, we extended the G construct to include the two predicted helices of the BD (Figure 3A). The resulting construct GFP-BD38–91-G (38–148) remained cytosolic, however (PM index = 0.3 ± 0.2; Figure 3B). Only when the entire BD was added to the G did the resulting fragment GFP-BD-G (1–148) show appreciable plasma membrane localization (Figure 3), most of which can be accounted for by the BD, because the contribution of the G region was still undetectable.
The crystal structure of a complex of mDia1 and RhoC demonstrated that both the G region and adjacent DID mediate the interaction with the GTPase (Rose et al., 2005), providing another potential explanation for the lack of plasma membrane localization of GFP-G. Therefore, we tested whether adding the DID to the G or to BD-G would improve plasma membrane localization. GFP-G-DID, however, was still largely cytosolic (PM index = 0.3 ± 0.1), similar to GFP-G (Figure 3). Likewise, the localization of GFP-BD-G-DID was comparable to that of GFP-BD-G (Figure 3).
Because GFP-G-DID did not significantly bind the membrane, but GFP-NtΔBD did, we considered a role of DD-CC in membrane localization. Neither GFP-DD-CC (PM index = 0.1 ± 0.1; Figure 1B) nor GFP-DID-DD-CC (PM index = 0.3 ± 0.2) localized to the plasma membrane (Figure 3), however, suggesting that the membrane targeting of GFP-NtΔBD was not caused by direct binding of DD-CC to the membrane or by dimerization with endogenous mDia2. These results further suggested that the G region is necessary for membrane targeting of GFP-NtΔBD, and DD-CC appeared to potentiate the membrane-binding ability of G-DID.
Of interest, the addition of DD-CC to GFP-BD-G-DID enhanced membrane targeting as evident from an increase of the PM index from ~0.6 to ~3.5 in the resulting Nt construct (Figure 3). Yet, removal of the CC domain from the mDia2 N terminus dramatically reduced membrane targeting of the resulting construct GFP-NtΔCC (PM index = 0.6 ± 0.7). Thus, the CC domain plays a critical role in targeting the N terminus of mDia2 to the membrane.
The above data raised the possibility that the G-DID had membrane-targeting capabilities that were uncovered only in the presence of the surrounding domains. To test more directly whether the G-DID participates in membrane targeting through interaction with GTPases, we substituted residue Ser-184 of the DID by glutamic acid (S184E) within construct GFP-Nt (Figure 3). Indeed, structural and biochemical studies of mDia1 (Rose et al., 2005; Lammers et al., 2008) have shown that Ser-184 occupies a central position at the binding interface between the G-DID and the GTPase, such that the S184E mutation would be expected to abrogate GTPase binding. Consistent with this expectation, the localization of the mutant GFP-Nt-S184E was severely impaired (PM index = 0.7 ± 0.3). Any residual membrane-binding activity of this mutant might be ascribed to the BD.
Collectively, these results confirm the importance of G-DID in binding to the plasma membrane and suggest that DD-CC potentiates membrane binding of G-DID by increasing its avidity through dimerization and/or by presenting it in a conformation that is more optimal for membrane binding.
Effects of small GTPases on membrane targeting of mDia2
The inability of G-DID to target the plasma membrane might be explained by insufficient amounts of activated GTPases at the plasma membrane. To address this possibility, we investigated whether coexpression of constitutively active small GTPases would enhance plasma membrane targeting of G-containing mDia2 constructs. Several small GTPases of the Rho family, including RhoA, Cdc42, Rif, Rac1, and Rac2, have been proposed to regulate mDia2 functions in cells (Alberts et al., 1998; Peng et al., 2003; Pellegrin and Mellor, 2005; Ji et al., 2008; Lammers et al., 2008). We first tested which of these GTPases in their constitutively active form is the most effective in enhancing plasma membrane localization of coexpressed mDia2 Nt.
We found that only Rif significantly increased the PM index of GFP-Nt, whereas active RhoA, Cdc42, and Rac1 did not (Figure 4, A and B). Moreover, both active and inactive forms of Cdc42 lowered the plasma membrane localization of GFP-Nt, although the statistical significance of these differences was low (p = 0.01 and 0.03, respectively). These results are consistent with efficient targeting of Rif, but not RhoA, Cdc42, and Rac1, to the plasma membrane (Figure 4C) and do not contradict the previous reports that all these GTPases interact with mDia2 (Alberts et al., 1998; Peng et al., 2003; Pellegrin and Mellor, 2005; Ji et al., 2008; Lammers et al., 2008). We did not detect strong effects of dominant negative Rif on GFP-Nt localization (Figure 4B). Although from visual inspection it sometimes appeared that Cdc42 and Rac1 also enhanced plasma membrane targeting of GFP-Nt, this effect might be due to changes in cell morphology caused by the overexpression of these GTPases, as monomeric red fluorescent protein (mRFP1) also showed equivalent membrane enrichment (Figure 4A). Because we normalized the GFP membrane/cytoplasm intensity ratio against the mRFP1 membrane/cytoplasm ratio during calculation of the PM index, we took these changes into account to determine the actual degree of plasma membrane localization.
FIGURE 4:

Constitutively active Rif specifically enhances plasma membrane localization of mDia2 N-terminus. (A) Confocal microscopy of HeLa cells coexpressing GFP-Nt (top row), mRFP1 (second row), and indicated Myc or HA-tagged constitutively active GTPases immunostained with respective tag antibodies (bottom row). Merged GFP-Nt/mRFP1 images are shown in the third row. Scale bar, 10 μm. (B) Quantification of plasma membrane localization of GFP-Nt in control HeLa cells (green) and in cells coexpressing indicated constitutively active (red) or dominant negative (blue) GTPases. Statistically significant difference with p < 0.01 as compared to control cells is marked by two asterisks. Values of p in the 0.01–0.05 range are marked with one asterisk. Among tested GTPases, only Rif expression significantly increases plasma membrane targeting of GFP-Nt. (C) Average PM index of indicated GTPases. Error bars, SEM; N is shown in parentheses.
In contrast to the entire N terminus of mDia2, the plasma membrane localization of a shorter construct GFP-BD-G-DID was enhanced not only by constitutively active Rif, but also by Cdc42 and RhoA, although Rif still had the greatest effect (Supplemental Figure 3). These data show that Rif is the most relevant GTPase targeting the mDia2 N terminus to the membrane and suggest that the presence of DD-CC confers added specificity to mDia2 for GTPase binding.
Role of N-terminal domains of mDia2 in Rif-dependent membrane targeting
Having established that Rif causes the greatest increase in plasma membrane targeting of mDia2 Nt, we decided to further characterize the interactions of Rif with N-terminal domains of mDia2. Coexpression of constitutively active Rif with various N-terminal mDia2 constructs produced several unexpected and quite striking results, along with some expected findings (Figure 5). In contrast to increased targeting of GFP-Nt and GFP-BD-G-DID (Figure 4), Rif unexpectedly had no effect on the localization of GFP-G-DID, which remained mostly cytoplasmic (Figure 5). These findings supported our earlier conclusion that the G-DID alone is insufficient to mediate membrane binding.
FIGURE 5:

Small GTPase Rif enhances plasma membrane localization of mDia2 N-terminal constructs in a BD- and G-dependent manner. (A) Confocal microscopy images of HeLa cells coexpressing indicated GFP-mDia2 constructs (first row) and mRFP1 (second row) with (+Rif) or without (–Rif) Myc-Rif Q75L. Merged GFP/mRFP1 images are shown in the third row. Rif stained with Myc antibody is shown in white in the bottom row. Scale bar, 10 μm. GFP-G-DID localization is not changed by coexpression of active Rif, whereas localization of GFP-BD and, especially GFP-BD-G, is enhanced by Rif. (B) Average PM indices of indicated mDia2 constructs in the absence (blue) or presence (orange) of active Rif are shown with SEM. Statistically significant difference between PM indices of nonexpressing and Rif Q75L-expressing cells for a given GFP-mDia2 construct is marked by two asterisks for values of p < 0.01 and by one asterisk for values of p in the 0.01–0.05 range.
The potentiating effect of DD-CC on membrane targeting detected in the absence of ectopically expressed Rif was even more striking in the presence of active Rif (Figure 5B). Thus, the PM index of GFP-Nt in the presence of Rif was significantly higher than that of GFP-BD-G-DID (Figure 5B). Similarly, in the absence of BD, the membrane enrichment of GFP-NtΔBD was substantially higher as compared to GFP-G-DID in active Rif-expressing cells. This effect was likely dependent on G-region binding to the GTPase, because membrane localization of GFP-DID-DD-CC, which lacks the G region, was not significantly increased by active Rif (Figure 5B). These findings are consistent with the idea that the addition of DD-CC enhances the ability of the G-DID to respond to plasma membrane–bound Rif.
Surprisingly, the plasma membrane localization of BD-containing constructs was significantly increased by coexpression with active Rif (Figure 5). This observation is illustrated, for example, by the lack of response of G-DID to Rif (Figure 5) in contrast to a prominent response of BD-G-DID (Figure 4). Even isolated GFP-BD responded to the presence of active Rif with a slight but significant increase in membrane localization (p = 0.009). Additionally, Rif greatly enhanced membrane localization of the GFP-BD-G construct. Interestingly, active Rif also decreased the nuclear localization of GFP-BD and, even more, that of GFP-BD-G (Figure 5A), further supporting the idea that the response of these constructs to Rif was specific. The addition of the DID to BD-G, however, did not result in a further increase of membrane targeting in the presence of Rif (Figure 5B), suggesting that Rif enhances membrane localization of these constructs through an indirect mechanism. Active Rif also increased the plasma membrane localization of the GTPase-binding mutant GFP-Nt-S184E to an extent similar to that of GFP-BD, implying that Nt-S184E is likely targeted to the membrane by the BD. Together these results suggest that in cells the BD can respond to the presence of active Rif at the membrane and even confer this sensitivity to the G region.
Direct interaction of Rif with mDia2 requires both G and DID, but not BD
One possibility of how Rif may enhance membrane localization of BD-containing mDia2 constructs is through direct binding to the BD. We tested this possibility by performing protein–protein binding assays with purified GST-Rif and various MBP-mDia2 constructs (Figure 6). To date, this interaction has been tested only by yeast two-hybrid screen and coimmunoprecipitation from mammalian cell lysates, which may reflect both direct and indirect binding patterns (Pellegrin and Mellor, 2005). Furthermore, multidomain fragments of the mDia2 (1–297 and 47–800) that were used for these binding assays do not allow one to separate the contributions of individual mDia2 domains to this interaction.
FIGURE 6:

Small GTPase Rif binds mDia2 through domains G-DID. (A) Coomassie staining of MBP or MBP-tagged mDia2 proteins (Nt and NtΔBD) that were used in a GTPase-binding assay with GST-Rif Q75L or GST as control. The presence of BD does not increase binding to active Rif in vitro. (B) An immunoblot of MBP-mDia2 proteins Nt, NtΔBD, BD-G-DID, and BD-G that were used in a GTPase binding assay with GST-Rif Q75L or GST control. Constructs Nt, NtΔBD, and BD-G-DID bound Rif, whereas construct BD-G could not be detected unless high exposure times were used. Quantification of band intensities relative to respective controls shows a 2.4-fold (A) and 3.8-fold (B) increase for MBP-Nt and a 6.0-fold (A) and 9.2-fold (B) increase for MBP-NtΔBD. There is no difference in the MBP band intensity between GST and GST-Rif (A). Panels A and B represent separate experiments.
Our binding assays showed that MBP-Nt and MBP-NtΔBD bound GST-Rif Q75L to a similar extent (Figure 6, A and B), suggesting that the BD does not enhance Rif-binding in vitro. Thus stronger plasma membrane localization of GFP-BD-G-DID than of GFP-G-DID in Rif-expressing cells was likely mediated by another mechanism, distinct from direct binding of the BD to the GTPase. We also found a clear interaction between GST-RifQ75L and MBP-BD-G-DID (Figure 6B) but weak interaction with MBP-BD-G detectable only at higher exposure times (unpublished data). These results suggest that the DID is necessary for strong binding of mDia2 to Rif, as previously shown for the mDia1–RhoC interaction. In addition, the fact that both NtΔBD and BD-G-DID bind Rif implies that domains common to these constructs, namely G-DID, mediate the interaction with Rif GTPase.
DISCUSSION
Accumulating evidence in various systems converges on the idea that multiple inputs regulate protein activity and subcellular localization, a concept referred to as coincidence detection. The regulation of actin filament nucleation may also follow this scheme. For example, N-WASP, an autoinhibited regulator of the Arp2/3 complex, can be cooperatively activated and recruited to the membrane by interactions with Cdc42 through its GBD, phosphoinositides through its adjacent BD (Prehoda et al., 2000), and SH3 domain-containing proteins through its proline-rich region (Takenawa and Suetsugu, 2007; Derivery and Gautreau, 2010).
The regulation of mDia formins resembles that of N-WASP, as they are also autoinhibited proteins regulated by small GTPases cooperating with other coactivators (Seth et al., 2006; Dominguez, 2010). Protein interaction with phospholipids at the membrane frequently plays a role in coincidence detection, but its importance is poorly understood for mDia formins, and in particular for mDia2. Here, we found that interactions of mDia2 with GTPases and phospholipids contribute to its localization at the plasma membrane in an actin-independent manner and that this activity is mediated by the coordinated actions of several N-terminal domains in a previously unappreciated manner.
Our main finding is that the BD plays an essential role in plasma membrane targeting of the Nt of mDia2. Its effect appears to be specific, as the C-terminal basic stretch adjacent to DAD (Wallar et al., 2006) is insufficient to recruit the Ct of mDia2 to the plasma membrane. Interestingly, a computer algorithm that searches for potential unstructured membrane-binding sites in protein sequences (Brzeska et al., 2010) also identifies the BD of mDia2 as a potential membrane-binding site.
Protein–lipid interactions commonly rely on electrostatic forces and hydrophobic interactions, consistent with the amphipathic nature of membrane phospholipids. Electrostatic interactions likely contribute to the association of the BD with the plasma membrane in vivo, first, because the deletion of the highly basic stretch within the first 37 amino acids of the BD significantly impairs membrane targeting of mDia2 fragments and, second, because the BD binds to acidic phospholipids in vitro. The apparent broad specificity of the BD for acidic phospholipids is reminiscent of the regulation of the WAVE complex, an activator of the Arp2/3 complex, by a range of charged acidic phospholipids (Lebensohn and Kirschner, 2009). The remainder of the BD contains two clusters of hydrophobic amino acids, predicted to form amphipathic helices. Deletion of these clusters decreases membrane targeting, suggesting that they also contribute to plasma membrane binding. These putative helices could promote membrane binding by several nonexclusive mechanisms: formation of a single folding unit with the GBD for GTPase binding; insertion into the membrane bilayer, as has been suggested for the BD of mDia1 (Ramalingam et al., 2010); clustering of basic amino acids into a common membrane-binding interface; or interaction with some membrane-associated proteins.
Although the BD is important for membrane targeting, it is not solely responsible for the strong binding of the mDia2 Nt. Generally, proteins activated by small GTPases are thought to be also recruited to the membrane by these GTPases. Indeed, RhoA-dependent recruitment of mDia2 to the cytokinetic ring has been recently demonstrated (Watanabe et al., 2010). The identity of the small GTPase targeting mDia2 to the plasma membrane in interphase remained unclear, however. Among several GTPases reported to interact with mDia2 (Alberts et al., 1998; Peng et al., 2003; Pellegrin and Mellor, 2005; Wallar et al., 2007; Ji et al., 2008; Lammers et al., 2008), only Rif potently and specifically enhanced the membrane localization of mDia2 constructs, suggesting that RhoA, Cdc42, and Rac1 may target mDia2 to other subcellular locations. Indeed, we have observed that overexpressed Rif is much more enriched at the membrane than are other GTPases, all of which are believed to interact with the membrane through prenylation of their C-terminal CAAX motifs. Thus, additional mechanisms may be involved in enhancing the membrane localization of Rif.
The structural basis for the interaction of mDia formins with GTPases is best known for the mDia1–RhoC complex, the crystal structure of which has been determined (Otomo et al., 2005; Rose et al., 2005). It showed that both the G region and the DID of mDia1 make specific contacts with the GTPase. Similar contacts were observed for a complex of an mDia2-mimicking mutant of mDia1 and Cdc42 or Rac1 (Lammers et al., 2008), although the structure of the actual mDia2 with any GTPase has not yet been determined. The biochemical analysis of the mDia2-Rif interaction (Pellegrin and Mellor, 2005) did not focus on whether both the G region and DID were required for binding. Here, we used purified proteins to demonstrate a direct interaction between active Rif and G-DID–containing constructs of mDia2, whereas the interaction of the fragment BD-G with was much weaker. Thus, both G and DID of mDia2 are needed for optimal binding to Rif, which is analogous to the interactions of mDia1 with Rho family GTPases. Despite the ability of the BD to enhance the plasma membrane localization of the N-terminal mDia2 constructs in a Rif-dependent manner in cells, however, the BD is not involved in direct interaction between the N terminus of mDia2 and Rif. These findings suggest that Rif indirectly enhances the recruitment of the BD to the plasma membrane in cells, for example, by changing the plasma membrane composition through other effectors or signaling pathways.
Although both G and DID participate in GTPase-dependent targeting of mDia2 to the plasma membrane, surprisingly, they are not sufficient, as the construct G-DID fails to localize appreciably to the plasma membrane even in Rif-expressing cells. The addition of DD-CC to G-DID, however, rescues plasma membrane targeting, possibly through dimerization, which allows for multivalent binding (increased avidity) of the BD-G-DID module. If this idea is correct, the dimerization may require both the DD and CC domains, as the removal of the CC from Nt severely decreases plasma membrane targeting. In mDia1, however, the DD is sufficient to mediate dimerization in vitro (Otomo et al., 2005) suggesting that the NtΔCC of mDia2 may also be a dimer. Another possibility is that the DD-CC–containing region has membrane-targeting capabilities of its own, as proposed for mDia1 (Copeland et al., 2007). Consistent with this idea, DID-DD-CC of mDia2 localizes to the cytokinetic ring by interacting with anillin (Watanabe et al., 2010). The N-terminal region of mDia2 containing partial DID, DD, and CC is also involved in the Abi1-dependent stabilization of mDia2 at filopodial tips (Yang et al., 2007). Inability of DD-CC or DID-DD-CC to accumulate at the plasma membrane, however, is not consistent with this possibility or with a scenario in which DD-CC–containing constructs dimerize with endogenous mDia2. Therefore, we currently favor an idea that the DD-CC module, in addition to dimerization, may cause a conformational change that allows for better binding of the GTPase by G-DID or improves binding of another target (for instance Abi1) by N-terminal domains. Consistent with this idea, it has been found recently that the N terminus of mDia1 correctly interacts with its C terminus only when the N terminus contains the CC domain (Nezami et al., 2010; Otomo et al., 2010).
Together, our results suggest a model for a mechanism of mDia2 targeting with implications for its activation (Figure 7). We propose that the BD, which is expected to be accessible in the autoinhibited conformation of mDia2, mediates initial binding to the membrane through electrostatic, and possibly also hydrophobic, interactions. This initial binding allows mDia2 to linger at the plasma membrane until it encounters active Rif. Next, weak binding of Rif to the G region causes the displacement of the DAD from the DID, as proposed previously (Lammers et al., 2005), which would allow the GTPase to engage the DID to form a more stable complex at the membrane. The role of the DD-CC module in mDia2 is to dimerize and optimally arrange the N-terminal mDia2 domains for efficient Rif binding and/or for engagement of additional targeting molecules.
FIGURE 7:

Model for plasma membrane targeting of mDia2. Step 1: Phospholipid binding. The initial targeting event occurs while mDia2 is still autoinhibited, yet its BD is accessible to bind acidic phospholipids of the plasma membrane through electrostatic interactions. This transient binding allows mDia2 to linger at the plasma membrane until it encounters active GTPase Rif there. Step 2: Weak Rif binding. Active Rif binds to G region of mDia2. Now, the mDia2 dimer is additionally attached to the plasma membrane via weak interaction with the membrane-associated active Rif. The BD-G–bound Rif begins to displace the DAD peptide of the C terminus from DID (black arrows). Step3: Strong Rif binding and activation. Rif, possibly in concert with additional coactivator(s), causes disruption of the DID/DAD bond. This event allows DID to bind Rif, resulting in a more stable association of mDia2 with the membrane and also relieves autoinhibition of mDia2 to allow the FH1-FH2 domains to nucleate and elongate actin filaments.
Thus, mDia2 targeting, and possibly activation as well, occurs through extensive cooperation of all N-terminal domains, which together serve as a coincidence detection module recognizing at least two inputs: membrane phospholipids and a small GTPase. A similar mechanism may also be used to some extent by other mDia formins. Thus, mDia1 has a similarly charged, albeit slightly shorter, BD at the N terminus (Ramalingam et al., 2010), whereas the corresponding region of mDia3 has a slightly lower predicted pI of ∼8, because it lacks the first cluster of basic amino acids. In contrast, other formins containing an N-terminal GBD (Schonichen and Geyer, 2010) are not associated with an upstream basic sequence, suggesting that the BD-G module is specific for Diaphanous-related formins.
MATERIALS AND METHODS
Constructs
Truncation mutants of mDia2 were generated by PCR amplification from the full-length mDia2 template (Yang et al., 2007) and subcloned into pEGFP-C1 or -C2 vectors using either Sac1/Sal1 restriction sites or EcoRI/Sal1 sites to produce GFP-tagged proteins. The BD of mDia2 (aa 1–91) was also subcloned into the pGEX-5x-3 vector (GE Healthcare, Piscataway, NJ) using BamHI/Sal1 restriction sites to produce GST-tagged protein. Point mutation was introduced using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Myc-tagged Rif Q75L and T33N in pcDNA3 vector was a gift from Harry Mellor (University of Bristol); Myc-tagged RhoA A14V in pEXV vector, Myc-tagged Rac1 Q61L in pRK5 vector, and HA-tagged Cdc42 G12V in pcDNA vector were gifts from Margaret Chou (University of Pennsylvania); HA-Cdc 42 T17N was a gift from Wei Guo (University of Pennsylvania); and mRFP1-N1 was a gift from Roger Tsien (University of California at San Diego) (Campbell et al., 2002).
Cell culture, transfection, and reagents
HeLa cells were maintained in culture medium containing 45% DMEM, 45% F-10, 10% fetal bovine serum (ThermoScientific, Waltham, MA), penicillin, and streptomycin. Cells were transfected overnight using Lipofectamine LTX or Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions, replated onto laminin-coated coverslips (20 μg/ml; Sigma, St. Louis, MO), and fixed ∼3 or 24 h after replating with 4% paraformaldehyde in PBS. No significant difference in plasma membrane localization of several mDia2 constructs was found between the two conditions. Latrunculin B (Calbiochem, EMD Chemicals, Gibbstown, NJ) was added to the culture medium at 2 μM for 30 min. Immunostaining of GTPases was performed using mouse monoclonal Myc (clone 9E10; Abcam, Cambridge, MA) or HA antibody (Covance, Princeton, NJ), followed by Cy5-conjugated anti–mouse antibody (Jackson ImmunoResearch, West Grove, PA). For the Western blot analysis shown in Supplemental Figure S2, lysates were prepared from transfected HeLa cells by addition of 1% Triton X-100 in PBS. After low-speed centrifugation to remove nuclei, supernatants were mixed with 6X SDS buffer and boiled for 3 min. SDS–PAGE was performed in the NuPAGE system (Invitrogen), using Bis-Tris gels. Immobilon-P transfer membranes (Millipore, Billerica, MA) were blocked with a 5% solution of nonfat dry milk in TBS-T (Tris-buffered saline, Tween-20) and probed with rabbit GFP antibody (Abcam).
Confocal microscopy and image analysis
Confocal microscopy of HeLa cells coexpressing mRFP1 and GFP-tagged mDia2 proteins was performed using a Leica (Richmond, IL) LCS laser-scanning confocal microscope with a 63× oil immersion objective. Images were acquired using 122-μm pinhole and were averaged over four frames in the 1024:1024 pixel format. Midplane optical sections were used for presentation and quantification. Image brightness was linearly adjusted in Adobe Photoshop for optimal presentation.
The PM index was calculated using the following equation:
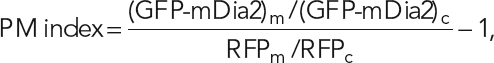 |
where (GFP-mDia2)m and (GFP-mDia2)c are average GFP fluorescence intensities at the plasma membrane and in the cytoplasm, respectively, and RFPm and RFPc are average mRFP1 fluorescence intensities at the plasma membrane and in the cytoplasm, respectively. Intensities were calculated using MetaMorph 7.5 imaging software (Molecular Devices, Sunnyvale, CA). To determine the average fluorescence intensity of GFP and RFP at the plasma membrane, (GFP-mDia2)m and RFPm, respectively, a three-pixel-wide line was drawn along the entire margin of the cell, and the intensity profile was obtained along this line using the Metamorph line scan tool. To determine the average fluorescence intensity in the cytoplasm, (GFP-mDia2)c, and RFPc, we used the MetaMorph multiline region tool to draw an irregularly shaped region between the nucleus and the membrane that includes only the cytoplasmic compartment. The obtained values for (GFP-mDia2)m, (GFP-mDia2)c, RFPm, and RFPc were used to calculate the PM index as shown earlier in the text. The PM index equals zero for cytoplasmic proteins, whereas plasma membrane–targeted constructs have a positive PM index. At least ten cells per construct were analyzed, except for cotransfection of Rif with GFP-DID-DD-CC, for which seven cells were analyzed. To calculate the PM indices of expressed GTPases, the intensity of the immunofluorescence signal was used instead of GFP fluorescence. Statistical significance was determined using Student’s t test in Microsoft Excel with a two-tailed heteroscedastic comparison; box-and-whisker plots were generated in SigmaPlot.
Subcellular fractionation
Membrane and cytoplasmic cell fractions were separated as described (Chandra Roy et al., 2009) with minor modifications. Briefly, HeLa cells were washed with ice-cold PBS and scraped in hypotonic buffer (10 mM Tris-HCl, pH 7.5; 1 mM EGTA; 1 mM MgCl2) supplemented with 1 mM PMSF and protease inhibitor cocktail (one tablet per 10 ml; Roche, Indianapolis, IN). Cells were disrupted by 10 passages through a 22-gauge needle, and lysates were clarified by centrifugation at 4300 × g for 10 min. Supernatants were subsequently centrifuged at 50,000 × g for 1 h to produce cytosolic and membrane fractions. The membrane fraction was washed three times with the hypotonic buffer and dissolved in 1% SDS in buffer A (50 mM Tris-HCl, pH 7.5; 140 mM NaCl; 10% glycerol; 1% Triton X-100) supplemented with 1 mM PMSF and protease inhibitor cocktail. The resulting cytoplasmic and membrane fractions were loaded on a NuPAGE 10% Bis-Tris gel (Invitrogen) in increasing volumes for the membrane fraction. GFP-mDia2 proteins were detected by Western blotting using GFP antibody (Abcam), and efficiency of fractionation was confirmed by probing gels with α-tubulin antibody (Sigma) and IRSp53 monoclonal antibody (a gift from Giorgio Scita, IFOM-IEO [FIRC Institute of Molecular Oncology/European Institute of Oncology], Milan, Italy). Secondary HRP-tagged antibodies (GE Healthcare) were detected using the ECL Plus reagents (GE Healthcare). Protein band intensities were measured using Adobe Photoshop. The intensity of the band corresponding to the cytoplasmic fraction was compared to a linear fit for the intensities of graded membrane fractions to estimate the percentage of the GFP-fusion protein in the membrane fraction.
Protein expression and purification
Escherichia coli BL21 star (DE3; Invitrogen) transformed with GST-BD was inoculated into 200 ml of Luria-Bertani (LB) medium and grown overnight at 37°C; then 1.2 l of LB medium was added to the culture and grown to OD600nm = 1.0. After stimulation of protein expression by 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h at 28°C, bacteria were pelleted at 5000 × g, slowly resuspended by stirring at 4°C in 30 ml of buffer T (50 mM Tris-HCl, pH 7.7; 100 mM NaCl; 1 mM DTT; 45 mg of protease inhibitors [Sigma], and 1 mM PMSF), ultrasonicated, and clarified by centrifugation at 15,000 × g for 15 min. GSH-Sepharose beads (0.8 ml; Amersham Pharmacia, Piscataway, NJ) were incubated with GST-BD–containing supernatant and washed five times in 40 ml of buffer T. GST-BD was eluted from beads with 20 mM glutathione (pH 8.6) and dialyzed overnight against buffer T. Concentration was determined by the Bradford assay.
E. coli BL21 RP strain (a gift from Wei Guo) was used for expression of MBP-mDia2 proteins and GST-RifQ75L. Protein expression was stimulated by adding 0.5 mM IPTG to 0.2 l of bacterial culture at OD600nm = 0.8 followed by overnight incubation at 18°C. Bacterial lysates were prepared as described earlier in this article, except that DNAse I at 33 μg/ml (Sigma) was added to cell suspension prior to sonication. Following centrifugation at 10,000 × g for 20 min, 20-ml supernatants containing MBP-mDia2 proteins were incubated with approximately 1.0 ml of washed amylose resin (New England Biolabs, Ipswich, MA), and those with GST-RifQ75L were incubated with 1.0 ml of washed GSH-Sepharose (Amersham) overnight at 4°C with agitation. Beads with bound protein were extensively washed with 0.1 M NaCl, 50 mM Tris pH7.5, 1 mM DTT and then five times in high-salt buffer (0.5 M NaCl; 50 mM Tris, pH7.5; 1 mM DTT). GSH beads with bound proteins were also washed with buffer containing 1% Triton X-100. MBP and MBP-mDia2 proteins were eluted with 10 mM maltose solution, and concentration was measured using the Bradford reagent (BioRad, Hercules, CA).
Rif binding assay
GSH-Sepharose beads (60 μl wet volume, 20 μl dry volume) bearing 100 pmol of GST-RifQ75L were loaded with 1 mM GTP or GTP-γ-S in 25 mM Tris (pH 7.5), 100 mM NaCl, 20 mM EDTA for 30 min at 30°C and were stabilized by 70 mM MgCl2. Control beads with immobilized GST were mixed with empty GSH beads to equalize inputs. Twenty microliters (dry volume) of GST or GST-RifQ75L beads were incubated with 200 pmol of MBP-mDia2 or MBP control overnight at 4°C with agitation in buffer containing 50 mM Tris (pH7.5), 0.1 M NaCl, 5 mM MgCl2, 1 mM DTT. Subsequently, buffer with 1.0% Triton X-100 was used to wash beads five times at 4°C, 10 min each, to remove nonspecifically bound proteins. Proteins were eluted with SDS buffer, boiled for 3 min at 100°C, and analyzed by SDS–PAGE or Western blot. Two separate experiments are shown in Figure 6. For Western blot, bead samples were diluted in SDS buffer 100-fold before loading onto SDS–PAGE gel. A pipetting error inherent to large dilutions is the likely reason why band intensities for GST-Rif in the anti-GST blot differ between binding reaction samples. MBP rabbit antibody was a gift from Mecky Pohlschroder, University of Pennsylvania. GST antibody (Amersham) was used in conjunction with secondary anti–donkey HRP-conjugated antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Protein-lipid overlay assay
PIP microstrips (Echelon Biosciences, Salt Lake City, UT) were processed according to the manufacturer’s instructions with some modifications. Briefly, strips were blocked with 10% nonfat dry milk in TBS-T buffer (10 mM Tris-HCl, pH 7.4; 150 mM NaCl; 0.1% [vol/vol] Tween-20) and incubated with 50 nM GST-BD in 2% milk in TBS-T. After extensive washes with TBS-T, strips were probed with goat polyclonal GST antibody (GE Healthcare), washed, and probed with anti–goat HRP-conjugated secondary antibody (Santa Cruz Biotechnology), followed by final TBS-T washes. Secondary antibody was detected using ECL Plus reagents (GE Healthcare).
Supplementary Material
Acknowledgments
We are grateful to Harry Mellor for Rif constructs; Margaret Chou for RhoA, Rac1, and Cdc42 constructs; Giorgio Scita for IRSp53 antibody; Wei Guo for purified GST protein, BL21 RP bacteria, and HA antibody; and Mecky Pohlschroder for MBP antibody. We thank Suk Namgoong and Kelley Bethoney for their advice on protein purification and lipid binding studies, respectively, and Zachary Essel, Jonathan Chia, and Zoya Svitkina for technical assistance. This work was supported by National Institutes of Health Grants GM70898 to T.S., HD07516 (training grant) to R.G., and GM073791 to R.D.
Abbreviations used:
- BD
basic domain
- CC
coiled coil
- DAD
diaphanous autoinhibitory domain
- DD
dimerization domain
- DID
diaphanous inhibitory domain
- FH1
formin homology domain 1
- FH2
formin homology domain 2
- G
GTPase binding region.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-03-0256) on November 30, 2010.
REFERENCES
- Alberts AS. Identification of a carboxyl-terminal diaphanous-related formin homology protein autoregulatory domain. J Biol Chem. 2001;276:2824–2830. doi: 10.1074/jbc.M006205200. [DOI] [PubMed] [Google Scholar]
- Alberts AS, Bouquin N, Johnston LH, Treisman R. Analysis of RhoA-binding proteins reveals an interaction domain conserved in heterotrimeric G protein beta subunits and the yeast response regulator protein Skn7. J Biol Chem. 1998;273:8616–8622. doi: 10.1074/jbc.273.15.8616. [DOI] [PubMed] [Google Scholar]
- Aspenstrom P. Formin-binding proteins: Modulators of formin-dependent actin polymerization. Biochim Biophys Acta. 2010;1803:174–182. doi: 10.1016/j.bbamcr.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Brandt DT, Marion S, Griffiths G, Watanabe T, Kaibuchi K, Grosse R. Dia1 and IQGAP1 interact in cell migration and phagocytic cup formation. J Cell Biol. 2007;178:193–200. doi: 10.1083/jcb.200612071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzeska H, Guag J, Remmert K, Chacko S, Korn ED. An experimentally based computer search identifies unstructured membrane-binding sites in proteins: application to class I myosins, PAKs, and CARMIL. J Biol Chem. 2010;285:5738–5747. doi: 10.1074/jbc.M109.066910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carramusa L, Ballestrem C, Zilberman Y, Bershadsky AD. Mammalian diaphanous-related formin Dia1 controls the organization of E-cadherin-mediated cell-cell junctions. J Cell Sci. 2007;120:3870–3882. doi: 10.1242/jcs.014365. [DOI] [PubMed] [Google Scholar]
- Chandra Roy B, Kakinuma N, Kiyama R. Kank attenuates actin remodeling by preventing interaction between IRSp53 and Rac1. J Cell Biol. 2009;184:253–267. doi: 10.1083/jcb.200805147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11:62–74. doi: 10.1038/nrm2816. [DOI] [PubMed] [Google Scholar]
- Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- Chhabra ES, Ramabhadran V, Gerber SA, Higgs HN. INF2 is an endoplasmic reticulum-associated formin protein. J Cell Sci. 2009;122:1430–1440. doi: 10.1242/jcs.040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland SJ, Green BJ, Burchat S, Papalia GA, Banner D, Copeland JW. The diaphanous inhibitory domain/diaphanous autoregulatory domain interaction is able to mediate heterodimerization between mDia1 and mDia2. J Biol Chem. 2007;282:30120–30130. doi: 10.1074/jbc.M703834200. [DOI] [PubMed] [Google Scholar]
- Derivery E, Gautreau A. Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. Bioessays. 2010;32:119–131. doi: 10.1002/bies.200900123. [DOI] [PubMed] [Google Scholar]
- Dominguez R. Structural insights into de novo actin polymerization. Curr Opin Struct Biol. 2010;20:217–225. doi: 10.1016/j.sbi.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Mammoto A, Kim Y, Takai Y. Rho small G-protein-dependent binding of mDia to an Src homology 3 domain-containing IRSp53/BAIAP2. Biochem Biophys Res Commun. 2000;271:626–629. doi: 10.1006/bbrc.2000.2671. [DOI] [PubMed] [Google Scholar]
- Gaboriaud C, Bissery V, Benchetrit T, Mornon JP. Hydrophobic cluster analysis: an efficient new way to compare and analyse amino acid sequences. FEBS Lett. 1987;224:149–155. doi: 10.1016/0014-5793(87)80439-8. [DOI] [PubMed] [Google Scholar]
- Gill MB, Roecklein-Canfield J, Sage DR, Zambela-Soediono M, Longtine N, Uknis M, Fingeroth JD. EBV attachment stimulates FHOS/FHOD1 redistribution and co-aggregation with CD21: formin interactions with the cytoplasmic domain of human CD21. J Cell Sci. 2004;117:2709–2720. doi: 10.1242/jcs.01113. [DOI] [PubMed] [Google Scholar]
- Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- Han Y, Eppinger E, Schuster IG, Weigand LU, Liang X, Kremmer E, Peschel C, Krackhardt AM. Formin-like 1 (FMNL1) is regulated by N-terminal myristoylation and induces polarized membrane blebbing. J Biol Chem. 2009;284:33409–33417. doi: 10.1074/jbc.M109.060699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs HN. Formin proteins: a domain-based approach. Trends Biochem Sci. 2005;30:342–353. doi: 10.1016/j.tibs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM. Interaction of the RAGE cytoplasmic domain with Diaphanous-1 Is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem. 2008;283:34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji P, Jayapal SR, Lodish HF. Enucleation of cultured mouse fetal erythroblasts requires Rac GTPases and mDia2. Nat Cell Biol. 2008;10:314–321. doi: 10.1038/ncb1693. [DOI] [PubMed] [Google Scholar]
- Kato T, Watanabe N, Morishima Y, Fujita A, Ishizaki T, Narumiya S. Localization of a mammalian homolog of diaphanous, mDia1, to the mitotic spindle in HeLa cells. J Cell Sci. 2001;114:775–784. doi: 10.1242/jcs.114.4.775. [DOI] [PubMed] [Google Scholar]
- Kobielak A, Pasolli HA, Fuchs E. Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol. 2004;6:21–30. doi: 10.1038/ncb1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar DR, Harris ES, Mahaffy R, Higgs HN, Pollard TD. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell. 2006;124:423–435. doi: 10.1016/j.cell.2005.11.038. [DOI] [PubMed] [Google Scholar]
- Lammers M, Meyer S, Kuhlmann D, Wittinghofer A. Specificity of interactions between mDia isoforms and Rho proteins. J Biol Chem. 2008;283:35236–35246. doi: 10.1074/jbc.M805634200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers M, Rose R, Scrima A, Wittinghofer A. The regulation of mDia1 by autoinhibition and its release by Rho*GTP. EMBO J. 2005;24:4176–4187. doi: 10.1038/sj.emboj.7600879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Higgs HN. The mouse Formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol. 2003;13:1335–1340. doi: 10.1016/s0960-9822(03)00540-2. [DOI] [PubMed] [Google Scholar]
- Li F, Higgs HN. Dissecting requirements for auto-inhibition of actin nucleation by the formin, mDia1. J Biol Chem. 2005;280:6986–6992. doi: 10.1074/jbc.M411605200. [DOI] [PubMed] [Google Scholar]
- Liu W, Sato A, Khadka D, Bharti R, Diaz H, Runnels LW, Habas R. Mechanism of activation of the Formin protein Daam1. Proc Natl Acad Sci USA. 2008;105:210–215. doi: 10.1073/pnas.0707277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- Miki T, Okawa K, Sekimoto T, Yoneda Y, Watanabe S, Ishizaki T, Narumiya S. mDia2 shuttles between the nucleus and the cytoplasm through the importin-α/β- and CRM1-mediated nuclear transport mechanism. J Biol Chem. 2009;284:5753–5762. doi: 10.1074/jbc.M806191200. [DOI] [PubMed] [Google Scholar]
- Nakano K, Takaishi K, Kodama A, Mammoto A, Shiozaki H, Monden M, Takai Y. Distinct actions and cooperative roles of ROCK and mDia in Rho small G protein-induced reorganization of the actin cytoskeleton in Madin-Darby Canine Kidney cells. Mol Biol Cell. 1999;10:2481–2491. doi: 10.1091/mbc.10.8.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezami A, Poy F, Toms A, Zheng W, Eck MJ. Crystal structure of a complex between amino and carboxy terminal fragments of mDia1: insights into autoinhibition of diaphanous-related formins. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezami AG, Poy F, Eck MJ. Structure of the autoinhibitory switch in formin mDia1. Structure. 2006;14:257–263. doi: 10.1016/j.str.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Otomo T, Otomo C, Tomchick DR, Machius M, Rosen MK. Structural basis of Rho GTPase-mediated activation of the formin mDia1. Mol Cell. 2005;18:273–281. doi: 10.1016/j.molcel.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Otomo T, Tomchick DR, Otomo C, Machius M, Rosen MK. Crystal structure of the Formin mDia1 in autoinhibited conformation. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul AS, Pollard TD. Review of the mechanism of processive actin filament elongation by formins. Cell Motil Cytoskeleton. 2009;66:606–617. doi: 10.1002/cm.20379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechlivanis M, Samol A, Kerkhoff E. Identification of a short Spir interaction sequence at the C-terminal end of formin subgroup proteins. J Biol Chem. 2009;284:25324–25333. doi: 10.1074/jbc.M109.030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrin S, Mellor H. The Rho family GTPase Rif induces filopodia through mDia2. Curr Biol. 2005;15:129–133. doi: 10.1016/j.cub.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Peng J, Wallar BJ, Flanders A, Swiatek PJ, Alberts AS. Disruption of the Diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr Biol. 2003;13:534–545. doi: 10.1016/s0960-9822(03)00170-2. [DOI] [PubMed] [Google Scholar]
- Petersen J, Nielsen O, Egel R, Hagan IM. FH3, a domain found in formins, targets the fission yeast formin Fus1 to the projection tip during conjugation. J Cell Biol. 1998;141:1217–1228. doi: 10.1083/jcb.141.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehoda KE, Scott JA, Dyche Mullins R, Lim WA. Integration of multiple signals through cooperative regulation of the N- WASP-Arp2/3 complex. Science. 2000;290:801–806. doi: 10.1126/science.290.5492.801. [DOI] [PubMed] [Google Scholar]
- Pruyne D, Evangelista M, Yang C, Bi E, Zigmond S, Bretscher A, Boone C. Role of formins in actin assembly: nucleation and barbed-end association. Science. 2002;297:612–615. doi: 10.1126/science.1072309. [DOI] [PubMed] [Google Scholar]
- Ramalingam N, Zhao H, Breitsprecher D, Lappalainen P, Faix J, Schleicher M. Phospholipids regulate localization and activity of mDia1 formin. Eur J Cell Biol. 2010;89:723–732. doi: 10.1016/j.ejcb.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Rose R, Weyand M, Lammers M, Ishizaki T, Ahmadian MR, Wittinghofer A. Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature. 2005;435:513–518. doi: 10.1038/nature03604. [DOI] [PubMed] [Google Scholar]
- Ryu JR, Echarri A, Li R, Pendergast AM. Regulation of cell-cell adhesion by Abi/Diaphanous complexes. Mol Cell Biol. 2009;29:1735–1748. doi: 10.1128/MCB.01483-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirenbeck A, Bretschneider T, Arasada R, Schleicher M, Faix J. The Diaphanous-related formin dDia2 is required for the formation and maintenance of filopodia. Nat Cell Biol. 2005;7:619–625. doi: 10.1038/ncb1266. [DOI] [PubMed] [Google Scholar]
- Schonichen A, Geyer M. Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim Biophys Acta. 2010;1803:152–163. doi: 10.1016/j.bbamcr.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Seth A, Otomo C, Rosen MK. Autoinhibition regulates cellular localization and actin assembly activity of the diaphanous-related formins FRLalpha and mDia1. J Cell Biol. 2006;174:701–713. doi: 10.1083/jcb.200605006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradal T, Courtney KD, Rottner K, Hahne P, Small JV, Pendergast AM. The Abl interactor proteins localize to sites of actin polymerization at the tips of lamellipodia and filopodia. Curr Biol. 2001;11:891–895. doi: 10.1016/s0960-9822(01)00239-1. [DOI] [PubMed] [Google Scholar]
- Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- Wallar BJ, Deward AD, Resau JH, Alberts AS. RhoB and the mammalian Diaphanous-related formin mDia2 in endosome trafficking. Exp Cell Res. 2007;313:560–571. doi: 10.1016/j.yexcr.2006.10.033. [DOI] [PubMed] [Google Scholar]
- Wallar BJ, Stropich BN, Schoenherr JA, Holman HA, Kitchen SM, Alberts AS. The basic region of the diaphanous-autoregulatory domain (DAD) is required for autoregulatory interactions with the diaphanous-related formin inhibitory domain. J Biol Chem. 2006;281:4300–4307. doi: 10.1074/jbc.M510277200. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16:3044–3056. doi: 10.1093/emboj/16.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Okawa K, Miki T, Sakamoto S, Morinaga T, Segawa K, Arakawa T, Kinoshita M, Ishizaki T, Narumiya S. Rho and anillin-dependent control of mDia2 localization and function in cytokinesis. Mol Biol Cell. 2010;21:3193–3204. doi: 10.1091/mbc.E10-04-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamana N, et al. et al. The Rho-mDia1 pathway regulates cell polarity and focal adhesion turnover in migrating cells through mobilizing Apc and c-Src. Mol Cell Biol. 2006;26:6844–6858. doi: 10.1128/MCB.00283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Czech L, Gerboth S, Kojima S, Scita G, Svitkina T. Novel roles of formin mDia2 in lamellipodia and filopodia formation in motile cells. PLoS Biol. 2007;5:e317. doi: 10.1371/journal.pbio.0050317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.