

Podosomes are actin-based matrix contacts in a variety of cell types. This study identifies the kinesin KIF9 and reggie/flotillin proteins as novel regulators of macrophage podosomes and shows that their interaction through the unique C-terminal domain of KIF9 is critical for the matrix-degrading ability of these structures.
Abstract
Podosomes are actin-based matrix contacts in a variety of cell types, most notably monocytic cells, and are characterized by their ability to lyse extracellular matrix material. Besides their dependence on actin regulation, podosomes are also influenced by microtubules and microtubule-dependent transport processes. Here we describe a novel role for KIF9, a previously little-characterized member of the kinesin motor family, in the regulation of podosomes in primary human macrophages. We find that small interfering RNA (siRNA)/short-hairpin RNA–induced knockdown of KIF9 significantly affects both numbers and matrix degradation of podosomes. Overexpression and microinjection experiments reveal that the unique C-terminal region of KIF9 is crucial for these effects, presumably through binding of specific interactors. Indeed, we further identify reggie-1/flotillin-2, a signaling mediator between intracellular vesicles and the cell periphery, as an interactor of the KIF9 C-terminus. Reggie-1 dynamically colocalizes with KIF9 in living cells, and, consistent with KIF9-mediated effects, siRNA-induced knockdown of reggies/flotillins significantly impairs matrix degradation by podosomes. In sum, we identify the kinesin KIF9 and reggie/flotillin proteins as novel regulators of macrophage podosomes and show that their interaction is critical for the matrix-degrading ability of these structures.
INTRODUCTION
Cells use two strategies for invasion: amoeboid, nonlytic migration through gaps in the extracellular matrix meshwork and mesenchymal, protease-dependent migration involving the cleavage of matrix components (reviewed in Friedl and Wolf, 2003). Podosomes and invadopodia, collectively called “invadosomes,” are matrix contacts with the ability to lyse matrix components and are thus considered as potential key structures of proteolytic cell invasion (reviewed in Gimona et al., 2008; Buccione et al., 2009; Linder, 2009).
Podosomes are constitutively formed in cells that have to cross tissue boundaries, most notably monocytic cells such as monocytes, macrophages, dendritic cells, and osteoclasts (reviewed in Linder and Aepfelbacher, 2003). Comparable to focal adhesions (reviewed in Zamir and Geiger, 2001), podosomes have emerged as highly complex organelles that comprise a large variety of components ranging from matrix contact proteins such as integrins (reviewed in Gimona et al., 2008) or CD44 (Chabadel et al., 2007), to adhesion plaque proteins such as talin (Zambonin-Zallone et al., 1989) and paxillin (Pfaff and Jurdic, 2001), to actin regulators such as Arp2/3 complex (Linder et al., 2000a) or cortactin (Webb et al., 2006). Podosomes are highly dynamic structures with lifetimes of 2–12 min and show an even higher internal actin turnover (Destaing et al., 2003).
Besides their dependence on actin regulation, podosomes are also influenced by microtubules and microtubule-dependent transport processes (Linder et al., 2000b; Cougoule et al., 2005). In this context, microtubules function as regulators of podosome dynamics (Kopp et al., 2006; reviewed in Linder, 2009) and their subcellular positioning (Destaing et al., 2005; Ory et al., 2008; McMichael et al., 2010). Moreover, microtubules also influence the matrix lytic ability of both podosomes and invadopodia (reviewed in Linder, 2007; Poincloux et al., 2009), which is probably based on the transport of signaling molecules and proteases to sites of degradation (Sakurai-Yageta et al., 2008; Steffen et al., 2008; Wiesner et al., 2010).
As podosomes are contacted by the plus ends of microtubules (Kopp et al., 2006), transport of material to podosomes is likely to involve plus end–directed motors of the kinesin family. Previously, we could show that kinesin-like family 1C (KIF1C), a kinesin-3 member, is involved in the regulation of podosome dynamics in primary human macrophages (Kopp et al., 2006). We have now expanded our screen for kinesins involved in podosome regulation and identified the little-characterized kinesin KIF9, a member of the kinesin-9 family (Lawrence et al., 2004).
KIF9 was discovered through interaction with the GTP-binding protein Gem (Piddini et al., 2001) and reported to associate with microtubules in primary glial cells (Piddini et al., 2001). So far KIF9 has not been characterized further, and no cellular function has been described for this motor. Here we demonstrate a function for KIF9 as a regulator of podosomes and of podosomal matrix degradation in primary human macrophages. Furthermore, we show that its C-terminal region interacts with reggie-1/flotillin-2.
Reggie/flotillin proteins were discovered as neuronal proteins involved in axon regeneration (Schulte et al., 1997; reviewed in Stuermer, 2010) and are now thought to act as ubiquitous signaling mediators between intracellular vesicles and the plasma membrane (reviewed in Langhorst et al., 2008; Stuermer, 2010). To date, two isoforms have been described, reggie-1 and ‑2 (flotillin-2 and ‑1), which can form hetero-oligomers through interaction of their C-terminal regions (reviewed in Glebov et al., 2006, Babuke and Tikkanen, 2007). Reggie proteins have been found both at the plasma membrane and at intracellular pools ranging from Golgi-derived vesicles and late endosomes/lysosomes to multivesicular bodies and lipid bodies (reviewed in Stuermer, 2010).
We now show that reggie-1 interacts with the C-terminal region of KIF9, that both proteins cross-precipitate from macrophage lysates, and that they dynamically colocalize in living cells. Consistent with the observed effects of KIF9 knockdown, knockdown of reggie impairs matrix degradation by podosomes. These experiments reveal both KIF9 and reggie proteins as novel regulators of matrix degradation by podosomes. (Note: The term “regulation” is used here in the broad sense of having a direct or indirect influence on the formation and/or functionality of podosomes.)
RESULTS
KIF9 regulates podosome numbers and matrix degradation in macrophages
To identify novel kinesin isoforms involved in podosome regulation, we used a small interfering RNA (siRNA)–based knockdown approach in primary human macrophages. These cells constitutively form numerous podosomes and are thus amenable to statistical evaluation of podosome formation. Among others, an siRNA specific for KIF9 was validated in HeLa cells and found to be highly effective (>85% knockdown; see Materials and Methods). Primary macrophages were transfected with the respective siRNA, and the number of cells containing podosomes (i.e., ≥10 podosomes/cell) was evaluated 24 and 48 h after transfection. Compared with control cells transfected with plasmid enhanced green fluorescent protein (pEGFP)–N1 or an siRNA specific for firefly luciferase, the number of podosome-forming cells in KIF9 siRNA-transfected macrophages was reduced to approximately 40% of controls (Figure 1A; see also Table 1). To get detailed confirmation of this effect, we next generated a KIF9-specific short-hairpin RNA (shRNA) construct, which allows bicistronic expression of EGFP. Primary macrophages transfected with this construct showed a clear reduction of podosome levels (Figure 1, B and C). A detailed evaluation of podosome numbers in KIF9-shRNA–expressing cells at 24, 48, and 72 h posttransfection showed that the number of cells containing numerous (>50) podosomes was reduced, while the number of cells containing few (0–10) podosomes was increased, compared with cells expressing a scrambled control sequence (Figure 1, D and E; Table 1). Combined, these findings indicate that knockdown of KIF9 leads to decreased podosome numbers in primary macrophages.
FIGURE 1:

KIF9 influences podosome numbers. (A) Evaluation of podosome formation in macrophages transfected with EGFP-C1, luciferase-specific siRNA, and KIF9-specific siRNA. Influence of siRNA was analyzed 24 h (left) and 48 h (right) after transfection. For each value, 3 × 30 cells were evaluated. Cells containing less than 10 podosomes at a given time point were scored as “containing no podosomes.” Values are given as mean percentage ± SD of total counts in Table 1. For differences between control values and values gained with KIF9 siRNA, a P value < 0.05 was considered significant (indicated by asterisk). (B, C) Fluorescence micrographs of primary human macrophages expressing scrambled shRNA (B) or KIF9-specific shRNA (C) 72 h after transfection. F-actin stained with rhodamine-labeled phalloidin. Inserts show respective GFP signals. White bar indicates 10 μm. (D, E) Evaluation of podosome formation in primary human macrophages transfected with psiSTRIKE vector bicistronically end encoding EGFP and scrambled shRNA, (D) or KIF9-specific shRNA (E). Influence of each shRNA was evaluated 24, 48, and 72 h after transfection. For each value, 3 × 30 cells were evaluated. Values are given as mean percentage ± SD of total counts in Table 1. For differences between control values and values gained with kinesin shRNAs, a P value < 0.05 was considered significant (indicated by asterisk).
Table 1.
Values for podosome formation or matrix degradation following various treatments. Statistical evaluation of podosome numbers or matrix degradation in macrophages transfected with siRNA or shRNA or microinjected with proteins. For each value, each time at least 30 randomly chosen cells from three independent experiments were evaluated. Values are given as mean percentage ± SD of total counts.
| Effect on podosome numbers | |||
| Expression of | 24 h | 48 h | |
| GFP-N1 | 78.0% ± 7.0% | 80.0% ± 7.0% | |
| Luciferase | 74.0% ± 4.0% | 68.0% ± 17.0% | |
| KIF9-specific siRNA | 61.1% ± 18.4% | 32.2% ± 1.9% | |
| Effect of KIF9-specific shRNA on podosome numbers | |||
| Podosomes/cell | 24 h | 48 h | 72 h |
| 0–10 | 7.4% ± 4.9% | 40.0% ± 3.3% | 48.2% ± 18.3% |
| 11–50 | 42.6% ± 6.7% | 36.7% ± 11.5% | 21.8% ± 6.9% |
| >50 | 50.0% ± 4.9% | 22.2% ± 1.9% | 30.0% ± 21.8% |
| Effect of scrambled shRNA on podosome numbers | |||
| Podosomes/cell | 24 h | 48 h | 72 h |
| 0–10 | 0.0% ± 0.0% | 1.1% ± 1.9% | 1.3% ± 2.2% |
| 11–50 | 7.1% ± 3.5% | 8.1% ± 5.3% | 0.0% ± 0.0% |
| >50 | 92.9% ± 8.9% | 90.8% ± 1.9% | 98.8% ± 20.7% |
| Degradation of gelatin matrix after siRNA treatment | |||
| Target of siRNA | 0–40% | 41–100% | |
| Luciferase | 31.1% ± 10.7% | 68.9% ± 17.1% | |
| KIF9 | 88.9% ± 5.1% | 11.1% ± 5.1% | |
| Microinjection of GST-KIF9-CT81 | |||
| Microinjection of | 0 h | 1 h | |
| GST | 6.7% ± 3.3% | 72.2% ± 8.4% | |
| GST-KIF9-CT81 | 7.8% ± 1.9% | 32.2% ± 1.9% | |
| Effect on podosome numbers | |||
| Target of siRNA | Podosomes/cell | 72 h | |
| Luciferase | 0–10 | 0.0% ± 0.0% | |
| 11–50 | 1.1% ± 1.9% | ||
| 50 | 95.6% ± 5.1% | ||
| Reggie-1 | 0–10 | 1.1% ± 1.9% | |
| 11–50 | 3.3% ± 3.3% | ||
| 50 | 95.6% ± 1.9% | ||
| Reggie-2 | 0–10 | 0.0% ± 0.0% | |
| 11–50 | 1.1% ± 1.9% | ||
| 50 | 98.9% ± 1.9% | ||
| Reggie-1 + reggie-2 | 0–10 | 0.0% ± 0.0% | |
| 11–50 | 0.0% ± 0.0% | ||
| 50 | 100.0% ± 0.0% | ||
| Degradation of gelatin matrix after siRNA treatment | |||
| Target of siRNA | 0–40% | 41–100% | |
| Luciferase | 14.6% ± 7.6% | 85.3% ± 7.6% | |
| Reggie-1 | 37.3% ± 16.6% | 62.7% ± 16.6% | |
| Reggie-2 | 50.6% ± 26.6% | 49.4% ± 26.6% | |
| Reggie-1 + reggie-2 | 60.0% ± 15.0% | 40.0% ± 15.0% | |
| Degradation of gelatin matrix after overexpression | |||
| Construct | 0–40% | 41–100% | |
| KIF9-CT402-GFP | 24.3% ± 7.0% | 75.7% ± 7.0% | |
| KIF9-NT709-GFP | 11.0% ± 5.0% | 89.0% ± 5.0% | |
To further investigate potential additional roles of KIF9, 7-d-old macrophages were transfected with siRNA specific for KIF9 or for firefly luciferase as a control. After 3 d, cells were reseeded on fluorescently labeled gelatin matrix, and their matrix-degrading ability was assessed after 5 h. Matrix degradation was evaluated only in cells containing numerous (>50) podosomes, to distinguish the potential effects of KIF9 in matrix degradation from its role in regulation of the podosome structure itself. Matrix degradation was analyzed by measuring the rhodamine-based fluorescence of the podosome-covered area, and cells were scored into groups according to the degree of matrix degradation (0–40% and 41–100%; Figure 2, A–C). Importantly, the number of cells showing low (0–40%) matrix degradation was significantly enhanced in cells transfected with siRNA specific for KIF9, compared with the control (Figure 2C; Table 1). Combined, these results indicate that KIF9 regulates not only the podosome structure itself but also the ability of podosomes to degrade matrix material.
FIGURE 2:

Knockdown of KIF9 influences matrix degradation. Confocal laser scanning micrographs of primary human macrophages transfected with siRNA-luciferase (A) or siRNA-KIF9 (B), seeded on rhodamine-labeled gelatin matrix (red). Matrix degradation is visible as dark areas; insets show relevant F-actin staining by Cy5-labeled phalloidin (white). White bar, 10 μm. (C) Evaluation of matrix degradation in cells treated with siRNAs. The degree of matrix degradation was analyzed by fluorescence measurements of 3 × 30 cells each time. Complete absence of labeled matrix beneath cells was set as 100% degradation. Cells were scored into groups according to matrix degradation (0–40% and 41–100%). For differences between control values and values gained with KIF9 siRNA, a P value < 0.01 was considered highly significant (indicated by asterisks).
KIF9-GFP vesicles associate with microtubules and contact podosomes
So that KIF9 localization and dynamics in macrophages could be visualized, both GFP- and mCherry-fusion constructs (KIF9-GFP and KIF9-mCherry) were generated and transfected into macrophages. KIF9-GFP was found to be present in vesicle-like accumulations, which were often closely associated with microtubules (Figure 3A). Live cell imaging of cells coexpressing KIF9-mCherry and GFP-α-tubulin revealed that KIF9-GFP vesicles move along microtubules (Figure 3B and Supplemental Video 1), comparable to other kinesins such as KIF1C (Kopp et al., 2006).
FIGURE 3:

KIF9-GFP contacts microtubules and podosomes. (A) Confocal laser scanning micrographs of primary human macrophage expressing KIF9-GFP (green), labeled for α-tubulin (red). White boxes in overview image are enlarged on the right. White bar, 10 μm. (B) Image from confocal time-lapse video of a primary human macrophage expressing KIF9-GFP (green) and α-tubulin-mCherry, labeling microtubules (see Supplemental Video 1). White frame indicates detail images on the right (left panel: overlay; middle panel: α-tubulin-mCherry signal, right panel: KIF9-GFP signal). White bar, 5 μm. Elapsed time since start of the experiment is given in seconds on the right. (C) Image from confocal time-lapse video of a primary human macrophage expressing KIF9-GFP (green) and mRFP-β-actin (red), labeling podosomes (see Supplemental Video 2). Cell circumference is depicted by the dashed white line. (C) White frame indicates area of detail images on the right, elapsed time since start of the experiment is given in seconds in lower left corners. Note dynamic contact (white arrows) of KIF9-GFP particles with podosomes in the central area but not in the cell periphery.
To analyze potential interactions between KIF9-positive vesicles and podosomes, macrophages were cotransfected with KIF9-GFP and mRFP-β-actin constructs, the latter for labeling actin-rich podosome cores, and analyzed by time-lapse confocal video microscopy. KIF9-GFP–positive vesicles were found to localize at the podosome-containing ventral cell side (Figure 3C), where they dynamically and repeatedly contacted podosomes (Figure 3C and Supplemental Video 2). Interestingly, KIF9-GFP vesicles contacted mostly podosomes at the inner region of the ventral cell side, and not the larger podosome precursors at the cell periphery, which are contacted preferentially by the KIF1C kinesin (Kopp et al., 2006).
KIF9 is expressed in primary macrophages and forms oligomers
To get direct proof that KIF9 is expressed in primary macrophages, reverse transcriptase PCR was performed using mRNA prepared from 7-d cultured primary human macrophages and primers specific for the KIF9 sequence comprising nucleotides 1125–1579. A band of the respective size (454 base pairs) was detected on agarose gels (Figure 4A), indicating the presence of the KIF9 transcript. To detect the KIF9 protein, a polyclonal antibody was raised against the unique C-terminal region of KIF9 comprising amino acid (aa) residues 710–790 (“CT81”; see Materials and Methods) as part of a GST fusion protein (GST-KIF9-CT81; see also Figure 5A) and subsequently affinity purified using the CT81 polypeptide of thrombin-cleaved GST-KIF9-CT81. On Western blots, this antibody recognized the respective KIF9 sequence both as part of the GST-KIF9-CT81 fusion protein (Figure 4B, left lane) and as the thrombin-cleaved polypeptide of the appropriate size (9 kDa; Figure 4B, right lane). Using this antibody on Western blots of macrophage lysates, we could not detect a band corresponding to endogenous KIF9 (∼87 kDa), possibly owing to the relatively low abundance of kinesins in whole cell lysates, comparable to KIF1C (Kopp et al., 2006).
FIGURE 4:
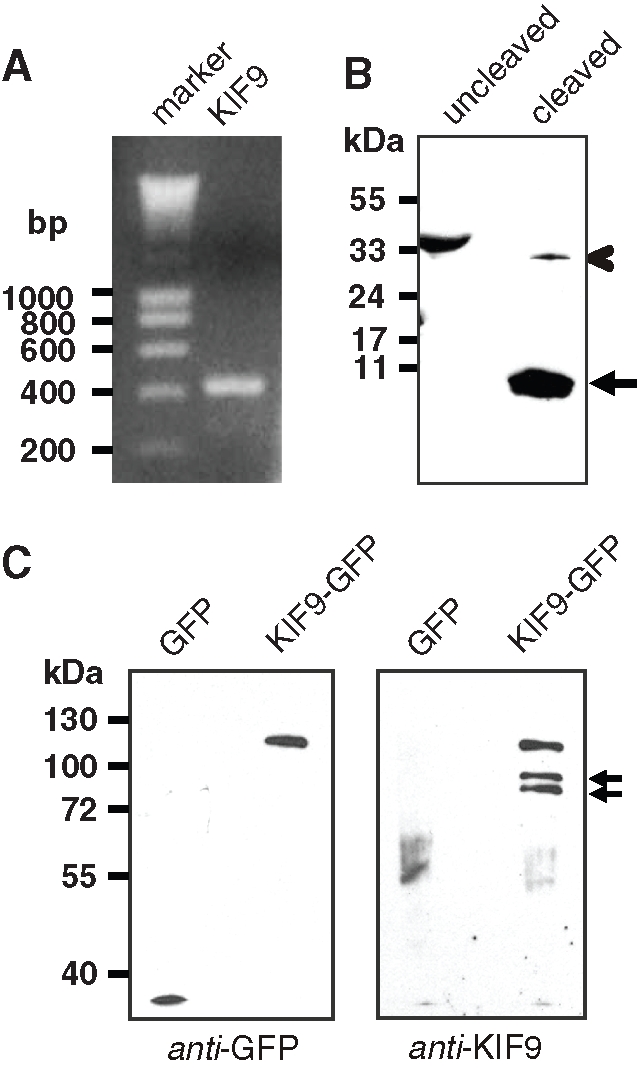
KIF9-GFP is expressed in primary human macrophages and exists as a dimer. (A) KIF9 is expressed in primary human macrophages. Reverse transcriptase-PCR using KIF9-specific primers. A band of the expected size (435 base pairs) is detected. Agarose gel stained with ethidium bromide; size in base pairs on left. (B) Western blot developed with polyclonal rabbit antibody against C-terminal tail of KIF9. GST-KIF9-CT81 fusion construct before (lane 1) and after proteolytic cleavage (lane 2). Note reactivity of anti-KIF9 antibody with the fusion construct (indicated by chevron) and the cleaved CT81 peptide (10 kDa; arrowhead), but not with the GST tag (26 kDa). (C) GFP immunoprecipitation of macrophage lysates expressing GFP (left lane) or GFP-fused KIF9 (right lane); Western blot probed with anti-GFP antibody (left blot) or anti–KIF9-CT81 (right blot). Note coprecipitated bands that react with the anti-KIF9 antibody (indicated by arrows). Molecular mass in kilodaltons is indicated on the left.
FIGURE 5:

KIF9 expression constructs and their localization in cells. (A) Domain organization of KIF9 and expression constructs used in this study: P-loop sequence (aa 93–100), motor domain signature (aa 1–300), stalk (aa 301–709), and unique tail (aa 710–790). Numbers indicate first and last amino acid residues of constructs. (B–J) Confocal laser scanning micrographs of primary human macrophages expressing different GFP-fused KIF9 constructs. (B, C) Primary human macrophage expressing GFP-fused KIF9 (C), stained for F-actin (B), with overlay shown in (D). (E, F) Primary human macrophage expressing GFP-fused KIF9-CT402 (F), stained for F-actin (E), with overlay shown in (G). (H, I). Primary human macrophage expressing GFP-fused KIF9-NT709 (H), stained for F-actin (I), with overlay shown in (J). (K, L) Primary human macrophage expressing GFP-fused KIF9-CT81 (K), stained for F-actin (L), with overlay shown in (M). White bar, 10 μm.
As kinesins are present mostly as dimers (reviewed in Woehlke and Schliwa, 2000), and the KIF9 sequence contains coiled-coil sequences predicted to be involved in dimerization (Piddini et al., 2001), we next tried to concentrate endogenous KIF9 protein by coprecipitation with overexpressed KIF9-GFP. Immunoprecipitations from lysates of macrophages expressing KIF9-GFP or GFP alone were performed with anti-GFP specific antibody coupled to magnetic beads. Western blots of respective fractions probed with anti-GFP antibody showed successful precipitation of both KIF9-GFP and the GFP control (Figure 4C, left blot). Comparable Western blots developed with anti-KIF9 antibody showed a band corresponding to the KIF9-GFP fusion construct, and in addition two bands in the 85–90-kDa range. Both of these bands are likely to correspond to endogenous KIF9 and may represent either splice or phosphorylation variants. These results indicate that endogenous KIF9 can be coprecipitated together with overexpressed KIF9-GFP, which is probably based on the formation of heterodimers between overexpressed and endogenous forms.
The KIF9 C-terminus induces podosome disruption and Golgi dispersal
We next set out to identify domains responsible for the observed effects of KIF9 on podosomes. KIF9 shows an N-terminal motor domain with a P-loop sequence, which is involved in ATP hydrolysis of kinesins (Sack et al., 1999); a stalk domain containing predicted coiled-coil sequences; and a unique tail comprising the C-terminal 81 aa residues (Figure 5A; Piddini et al., 2001). We thus generated deletion constructs comprising the stalk and tail regions (KIF9-CT402-GFP; aa residues 389–790; see also Supplemental Figure 1) or the tail region (GFP-KIF9-CT81; aa residues 710–790; Figure 5A) and, vice versa, a construct lacking the tail region (KIF9-NT709-GFP). Upon expression in macrophages, KIF9-CT402-GFP localized to vesicular structures (Figure 5, E–G), comparable to full-length (fl) KIF9-GFP (Figure 5, B–D). Time-lapse videos of cells expressing KIF9-CT402-GFP together with fl KIF9-mCherry showed that this construct localizes to the same vesicle population as the full-length construct (unpublished data). Accordingly, extensive vesicle movement and contact of podosomes were observed in cells showing moderate overexpression of KIF9-CT402-GFP (Supplemental Figure 1 and Supplemental Videos 3 and 4). Upon higher overexpression, less KIF9-CT402-GFP–positive vesicles appeared to be motile, which probably reflects competitive binding of this motorless, nonprocessive construct with the endogenous and processive fl KIF9. Consistently, cells overexpressing KIF9-CT402-GFP showed reduced matrix-degrading ability, compared with controls (Supplemental Figure 2).
The construct lacking the C-terminal region (KIF9-NT709-GFP) also localized in a vesicular pattern (Figure 5, H–J). Coexpression with KIF9-mCherry confirmed that this construct colocalizes at the same vesicle population with fl KIF9 (Supplemental Figure 3, A–C). Live cell imaging further showed that these vesicles repeatedly contact podosomes (Supplemental Figure 3, D and D′; Supplemental Video 5), comparable to fl KIF9. In contrast to GFP-KIF9-CT402, however, overexpression of KIF9-NT709-GFP, also at high levels, did not influence gelatin matrix degradation (Supplemental Figure 2).
The vesicular localization of both deletion constructs may be based on the ability for dimerization with endogenous KIF9 (discussed previously), which is probably conferred by the stalk domain (Figure 5A; Piddini et al., 2001). We also observed no detrimental effects on cell viability or podosome physiology upon expression of these constructs, comparable to fl KIF9-GFP (unpublished data). By contrast, overexpressed GFP-KIF9-CT81 did not localize to vesicles and was found mostly in a perinuclear accumulation (Figure 5, K–M). Moreover, cells expressing this construct detached from coverslips 6–8 h after transfection, indicating a profound effect of GFP-KIF9-CT81 on macrophage viability.
Cells overexpressing GFP-KIF9-CT81 often showed a loss of podosomes; however, this phenomenon was inconsistent and could also have been due to a general detachment of cells. To assess potentially specific effects of the KIF9 C-terminus on podosomes, we thus generated a GST-fused version of KIF9-CT81 (GST-KIF9-CT81; Figure 5A) and used it in a microinjection-based podosome reformation assay (Linder et al., 1999; Hufner et al., 2001). This procedure takes advantage of a microinjection artifact, as injection of macrophages leads to initial loss of podosomes, with subsequent reformation within 1 h (Linder et al., 2000b). Assessing podosome reformation after microinjection, we found that control cells injected with GST initially lost most of their podosomes, but mostly reformed them within 1 h, whereas cells injected with GST-KIF9-CT81 showed a clear reduction in podosome reformation (Figure 6, A and B; Table 1). Within the observed time period, microinjected cells showed normal spreading and no obvious loss of viability, arguing for a specific effect of GST-KIF9-CT81 on podosomes. We conclude from these experiments that the C-terminal region of KIF9 is important for the de novo formation of macrophage podosomes.
FIGURE 6:

Microinjection of the KIF9 C-terminal region inhibits podosome reformation. (A) Evaluation of podosome numbers in macrophages microinjected with GST-KIF9-CT81 (0 h) and podosome reformation (1 h). For each bar, 3 × 30 cells were evaluated. Cells containing less than 10 podosomes at a given time point were scored as “containing no podosomes.” Values for podosome formation are given as mean percentage ± SD of total counts in Table 1. (B) Primary human macrophage 1 h after injection of 2 µg/µl GST-KIF9-CT81, labeled for F-actin (red), confocal laser scanning micrograph of substrate-attached part of cell. Injected cells were identified by labeling coinjected rat IgG (5 mg/ml) with FITC-labeled goat anti–rat IgG antibody (green). Note absence of podosomes in cell injected with GST-KIF9-CT81. White bar, 10 μm.
To characterize the localization and effects of the KIF9 C-terminus in more detail, we immunostained cells expressing GFP-KIF9-CT81 for a variety of vesicular and cytoskeletal markers. We observed partial colocalization of GFP-KIF9-CT81 with Golgi proteins, such as the trans-Golgi marker TGN46 (Figure 7, A–F). Strikingly, cells expressing GFP-KIF9-CT81 also showed a dispersed localization of the Golgi (Figure 7, A–F), compared with control cells (93.3% ± 2.6% for cells expressing GFP-KIF9-CT81; 13.3% ± 8.8% for cells expressing GFP; Figure 7G). These results indicate that the KIF9 C-terminus partially colocalizes with Golgi proteins and that overexpression of GFP-KIF9-CT81 leads to Golgi dispersal, which may contribute to the observed loss of cell viability. Interestingly, siRNA-induced knockdown of KIF9 did not lead to significant alterations in Golgi architecture (Supplemental Figure 4), indicating that KIF9 per se is not involved in regulating Golgi integrity.
FIGURE 7:

Golgi integrity is influenced by GFP-KIF9-CT81. (A–F) Confocal laser scanning micrographs of primary human macrophages expressing GPF-fused KIF9-CT81 (green, A and D), stained with specific primary antibody for TGN 46 (red, B and E) 7 h after transfection, with overlay shown in (C and F). Note compact Golgi appearance in untransfected cells, as opposed to dispersed Golgi morphology in cells expressing GFP-KIF9-CT81. White boxes in (D–F) indicate areas shown in enlarged insets. Cell circumferences are depicted by dashed white lines. White bars, 10 μm. (G) Evaluation of Golgi integrity 7 h after transfection of GFP-KIF9-CT81 or GFP as control. For each value, 3 × 30 cells were evaluated. Values are given as mean percentage ± SD of total counts in Table 1.
KIF9 interacts and colocalizes with reggie-1/flotillin-2
We next investigated potential interaction partners of the KIF9 C-terminus by overexpressing GFP-KIF9-CT81 in macrophages with subsequent anti-GFP immunoprecipitation. Silver staining of respective polyacrylamide (PAA) gels showed that additional bands coprecipitated with GFP-KIF9-CT81, compared with the GFP control (Figure 8A). Subsequent mass spectrometry (MS) and Western blot analyses revealed that a prominent band at ∼45 kDa (Figure 8A) corresponded to reggie-1 and -2 (also named flotillin-2 and -1; reviewed in Stuermer, 2010).
FIGURE 8:

KIF9 interacts with reggie-1/flotillin-2. (A) Lysates of primary human macrophages immunoprecipitated with anti-GFP antibody coupled to magnetic beads. Silver-stained PAA gel, left lane: cells transfected with pEGFP-N1 as control; right lane: cells transfected with GFP-KIF9-CT81 construct. Arrow indicates band subsequently identified by mass spectrometry as reggie-1/2. Arrowheads indicate bands corresponding to GFP (left lane) and GFP-KIF9-CT81 (right lane), as judged by their mobility on PAA gels. Molecular mass in kilodaltons is indicated on the left. (For a comparison of immunoprecipitations performed with GFP-KIF9-CT81 and fl KIF9-GFP, see Supplemental Figure 7.) (B) GFP immunoprecipitation of KIF9-GFP–transfected macrophages, using GFP-N1 as control. Western blot developed with anti–reggie-1 antibody (left side) or with anti-GFP antibody (right side). (C) GFP immunoprecipitation of reggie-1-GFP–transfected macrophages, using GFP-N1 as control. Western blot developed with anti-KIF9 antibody (left side) or with anti-GFP antibody (right side). (D) GFP immunoprecipitation of KIF9-NT709-GFP–transfected macrophages, using GFP-N1 as control. Western blot developed with anti–reggie-1 antibody (left side) or with anti-GFP antibody (right side). Dashed lines indicate that lanes were not directly adjacent on original blots.
Reggies/flotillins are known for their strong tendency toward hetero-oligomerization (Langhorst et al., 2008; reviewed in Stuermer, 2010). We thus focused initially on reggie-1/flotillin-2 to verify a potential interaction of KIF9 with reggie proteins. As available antibodies against reggie-1 and KIF9 are not suitable for immunoprecipitation, we expressed GFP-tagged versions of these proteins and performed anti-GFP immunoprecipitations from macrophage lysates. Indeed, endogenous reggie-1 coprecipitated with KIF9-GFP (Figure 8B), and, vice versa, endogenous KIF9 coprecipitated with reggie-1-GFP (Figure 8C), indicating a close interaction between both proteins. Still, the construct lacking the C-terminal domain of KIF9 (KIF9-NT709-GFP) showed residual binding of reggie-1, indicating that the C-terminal 81 aa residues constitute not the only or not the complete binding site for reggie-1 in KIF9 (Figure 8C). Alternatively, this may reflect binding of reggie-1 by endogenous KIF9, which can form homodimers with GFP-fused forms of KIF9 containing the coiled-coil region (discussed previously).
To confirm the biochemical data, we next investigated whether reggie-1 and KIF9 also colocalize in fixed and living cells. Overexpression of GFP-KIF9-CT81 showed that endogenous reggie-1 partially colocalized with this construct (Figure 9, A–C) in a perinuclear accumulation, comparable to our previous observations (Figures 5 and 7). As the antibody generated against KIF9 is not suitable for immunofluorescence, KIF9-mCherry was expressed together with reggie-1-GFP in macrophages. Both proteins showed extensive colocalization in vesicle-like structures (Figure 9, D–F). However, confocal time-lapse video microscopy revealed that colocalization of reggie-1 and KIF9 is not permanent, but is a dynamic process, with two vesicle populations constantly interacting, either in a “kiss-and-run” scenario or in a more prolonged contact involving combined movement in the same direction (Figure 9, G and H; Supplemental Videos 5–7).
FIGURE 9:

KIF9 and reggie-1 colocalize in fixed and living cells. (A–F) Confocal laser scanning micrographs of primary human macrophages expressing GFP-KIF9-CT81 (green, A), stained for reggie-1 (red, B) with specific primary antibody, with overlay shown in (C). Cell circumference is depicted by the dashed white line. White boxes indicate areas shown in detail images. (D–F) Confocal laser scanning micrographs of primary human macrophages coexpressing KIF9-mCherry (red, D) and reggie-1-GFP (green, E) with overlay shown in (F). (G, H) Images from confocal time-lapse videos of primary human macrophages expressing reggie-1-GFP (green) and KIF9-mCherry (red). White frames indicate areas of detail images. White bar, 10 μm. (G′, H′, H″) Time-lapse sequences from Supplemental Videos 6–8, taken from respective detail regions indicated in (G, H). Note close and repeated nonrandom contact between reggie-1-GFP (arrowheads) and KIF9-mCherry vesicles (arrows). Time since start of the experiments is indicated in seconds in upper right corners.
We conclude from these experiments that reggie-1 interacts with the KIF9 C-terminus in macrophage lysates and colocalizes with it in cells and that fl reggie-1 and KIF9 dynamically colocalize in living cells. Collectively, these data argue for a close interaction of both proteins in living cells, mainly through the KIF9 C-terminal region.
Reggie knockdown leads to reduced matrix degradation
We next investigated whether reggies/flotillins are involved in the KIF9-mediated regulation of podosomes. To assess a potential effect of reggie proteins on podosome numbers, macrophages were transfected with siRNAs specific for reggie-1 or ‑2 (Solis et al., 2007), or a combination of both, as well as for firefly luciferase as a control, and assessed for their ability to form podosomes. However, neither knockdown of single isoforms nor a combined knockdown of reggie-1 and ‑2 showed effects on podosome morphology, subcellular distribution, or numbers (Supplemental Figure 5 and unpublished data), compared with controls.
To assess the potential involvement of reggie proteins in podosomal matrix degradation, siRNA-transfected cells were reseeded on fluorescently labeled gelatin matrix after 3 d and incubated for an additional 5 h. Matrix degradation was evaluated only in cells that contained numerous (>50) podosomes, by measuring the rhodamine-based fluorescence of the podosome-covered area. Each time, 3 × 30 cells were evaluated and scored into groups according to the degree of matrix degradation (0–40% or 41–100%; Figure 10). Importantly, the percentage of cells showing low matrix degradation was significantly enhanced in cells treated with siRNA specific for either reggie-1 or reggie-2 (Figure 10E; Table 1). Combined knockdown of both reggie isoforms showed a more pronounced effect, although this was not statistically significant compared with knockdown of single isoforms (Figure 10E; Table 1). We conclude from these experiments that, comparable to KIF9, reggie proteins are also important for the matrix-degrading ability of macrophage podosomes.
FIGURE 10:

Knockdown of reggie proteins influences matrix degradation. Confocal laser scanning micrographs of primary human macrophages transfected with siRNA-luciferase (A), siRNA-reggie-1 (B), siRNA-reggie-2 (C), or a combination of both reggie siRNAs (D), seeded on rhodamine-labeled gelatin matrix (red). Matrix degradation is visible as dark areas; insets show relevant F-actin staining by Cy5-labeled phalloidin (white). White bar, 10 μm. (E) Evaluation of matrix degradation in cells treated with siRNAs. The degree of matrix degradation was analyzed by fluorescence measurements of 3 × 30 cells each time. Complete absence of labeled matrix beneath cells was set as 100% degradation. Cells were scored into groups according to matrix degradation (0–40% and 41–100%). For differences between control values and values gained with reggie siRNAs, a P value < 0.05 was considered significant, and a P value < 0.01 was considered highly significant (indicated by asterisks). Values are given as mean percentage ± SD of total counts in Table 1.
DISCUSSION
In this report, we focus on the microtubule-dependent regulation of podosomes in primary human macrophages. In addition to their actin-based architecture, podosomes also depend on an intact microtubule cytoskeleton and on microtubule-based transport processes (Linder et al., 2000b; Cougoule et al., 2005). This is most probably based on the repeated and dynamic contact between microtubule plus ends and podosomes (Kopp et al., 2006) and involves the regulation of both podosome positioning (Destaing et al., 2005; Ory et al., 2008; McMichael et al., 2010) and turnover (Kopp et al., 2006).
Microtubule-based regulation, in consequence, indicates the necessity for transport processes involving plus end–directed motor proteins of the kinesin family. Indeed, KIF1C, a kinesin-3 family member, has previously been identified as an important player in the regulation of podosome dynamics (Kopp et al., 2006). We have now expanded our screen for podosome-associated kinesins and report a critical role for KIF9 in the regulation of podosomal matrix degradation in macrophages.
KIF9 is a little-characterized member of the kinesin-9 family. So far, expression of KIF9 mRNA has been reported only for brain and kidney (Piddini et al., 2001). We now add to these findings and show, on both mRNA and protein levels, that KIF9 is also expressed in primary human macrophages. Importantly, anti-GFP immunoprecipitation of GFP-fused KIF9 resulted in coprecipitation of endogenous KIF9. This indicates an ability of KIF9 for oligomerization, presumably as a dimer, as is typical for kinesins (reviewed in Woehlke and Schliwa, 2000). Dimer formation is likely to involve the coiled-coil regions of KIF9 (Piddini et al., 2001), comparable to the general mode of kinesin dimerization (Wade and Kozielski, 2000). This is also supported by the observations that a construct lacking the motor domain (KIF9-CT402) or a construct lacking the tail region (KIF9-NT709), but in both cases containing the coiled-coil regions, localizes to vesicles, presumably through interaction with the endogenous motor. By contrast, a shorter construct lacking the coiled-coil regions (KIF9-CT81) localizes more diffusely.
Interestingly, GFP-KIF9 coprecipitates two proteins that react with the newly developed anti-KIF9 antibody and that migrate within the expected size range for KIF9 (85–90 kDa) on PAA gels. Apart from potential unspecific cross-reaction of the antibody, this 1) may be based on degradation of KIF9 in the cell lysate or 2) could indicate that KIF9 is present in two forms in macrophages, which may represent either splice or phosphorylation variants.
Knockdown of KIF9, using two independent siRNA/shRNAs, resulted in a pronounced loss of podosome numbers, indicating a role for KIF9 in the formation and/or turnover of these structures, comparable to the effect previously described for KIF1C (Kopp et al., 2006). Moreover, in KIF9 siRNA-treated cells that still form numerous podosomes, gelatin matrix degradation was almost completely blocked, pointing to an additional role of KIF9 in the regulation of podosomal matrix degradation. These effects may be independent but could also be linked through previously reported feedback loops connecting podosome formation and podosomal matrix degradation (reviewed in Linder, 2007).
Comparable to other kinesin motors, KIF9 contains an N-terminal motor domain (Piddini et al., 2001), which is expected to bind to the microtubule lattice. Indeed, KIF9 has been reported to associate with the microtubule cytoskeleton in primary glial cells and to cosediment with taxol-stabilized microtubules (Piddini et al., 2001). Here we show that KIF9-GFP–positive vesicles associate with microtubules and repeatedly contact podosomes, revealing a direct connection between KIF9-mediated, microtubule-based transport and actin-rich podosomes, which are regulated by KIF9.
Interestingly, KIF1C regulates the dynamics of podosome precursors in the cell periphery (Kopp et al., 2006) and is not involved in the regulation of matrix degradation by podosomes (Wiesner et al., 2010). By contrast, KIF9 contacts podosomes mostly in the inner region of the ventral cell surface, which are more efficient in matrix degradation (Wiesner et al., unpublished observations), and has a profound effect on the matrix-degrading ability of these structures. These findings indicate that specific kinesins exert differential effects on podosomes. The molecular/structural basis of this fine-tuned regulation will be an important point to address in future studies. It may be mediated by trafficking of specific kinesins on differentially modified microtubule subsets, as shown previously for movement of kinesin-1 along acetylated (Reed et al., 2006), or of KIF5c along detyrosinated, microtubules (Dunn et al., 2008; reviewed in Verhey and Hammond, 2009). Furthermore, it is likely to involve differential transport of specific cargo molecules of various kinesin isoforms to podosomes.
In this context, the C-terminal regions of kinesins are of particular interest, as they are thought to function as cargo binding sites of adaptors or regulators (reviewed in Woehlke and Schliwa, 2000). Indeed, the C-terminal 81 aa residues of KIF9 comprise a unique sequence (Piddini et al., 2001) that could potentially act as a hub for specific interaction partners. In a first step to investigate this, we generated a respective GFP-fused construct (GFP-KIF9-CT81). However, overexpression of GFP-KIF9-CT81 for more than 6 h resulted in cell detachment. To circumvent this problem, we microinjected a GST-fused version into macrophages, which allows for manifestation of potential effects within 1 h. This resulted in a pronounced defect of cells to reform podosomes, indicating that the C-terminal region of KIF9 is involved in podosome regulation, presumably by binding to specific interaction partners.
Immunoprecipitation of GFP-KIF9-CT81 from macrophage lysates and subsequent MS analysis revealed the presence of reggie-1 and ‑2 in the precipitates, indicating a potential interaction of reggie proteins with the KIF9 C-terminus. Indeed, GFP-fused full-length constructs of either KIF9 or reggie-1 were able to cross-precipitate the respective endogenous proteins, arguing for a close interaction of both proteins within cells. Interestingly, only one band of endogenous KIF9 was coprecipitated by GFP–reggie-1, indicating that reggie may interact with only one (splice or phospho) variant of the motor. Moreover, a construct lacking the unique C-terminal region of KIF9 (KIF9-NT709-GFP) still showed binding to endogenous reggie-1, although at a diminished degree. 1) This could indicate that both proteins interact through the KIF9 C-terminal region, but this region may constitute only a part or not the single reggie-1 binding site in KIF9, or 2) it may reflect recruitment of reggie-1 by endogenous KIF9, which can form dimers with KIF9-NT709-GFP. Both scenarios would be consistent with the observation that overexpression of KIF9-NT709-GFP did not reduce gelatin degradation in the matrix-degradation assay.
Consistent with the biochemical analysis, we observed partial colocalization of GFP-KIF9-CT81 with reggie-1 at the above-mentioned dispersed Golgi localization and colocalization of fl KIF9-mCherry with GFP-fused reggie-1 at vesicular structures in fixed cells. Both localizations are in line with earlier observations showing the presence of reggie proteins at the Golgi or at cytoplasmic vesicles (Langhorst et al., 2008; Stuermer, 2010). Interestingly, live cell imaging of macrophages coexpressing reggie-1-GFP and KIF9-mCherry revealed that, although a clear overlap between reggie-1– or KIF9-positive vesicles was observed, contact between both populations is mostly transient and more reminiscent of a kiss-and-run scenario. The extensive colocalization observed in fixed specimens thus probably represents a fixation artifact.
The dispersed Golgi morphology upon overexpression of GFP-KIF9-CT81 may be due to the KIF9 polypeptide interacting with reggie and other Golgi-localized proteins. As siRNA-induced knockdown of reggie-1 did not result in Golgi dispersal, it is unlikely that KIF9 per se is involved in the regulation of Golgi architecture. In a physiological scenario, fl KIF9, containing the motor domain, would be able to transport this cargo along microtubules. The isolated C-terminus, however, is unable to bind to and move processively along microtubules, and its overexpression may thus lead to sequestering of Golgi-derived proteins. This may result in the aberrant Golgi morphology and, ultimately, in cell detachment.
A central issue concerns the question of how the KIF9/reggie interaction affects podosomes and their function. Interestingly, siRNA-induced knockdown of reggie proteins did not influence podosome numbers but significantly impaired podosomal matrix degradation. This is in partial contrast to the results gained with knockdown of KIF9, which resulted in both reduction of podosome numbers and matrix degradation, and indicates that the KIF9 C-terminus probably binds to other regulatory factors, apart from reggie-1.
Furthermore, as reggie-1 and ‑2 have a pronounced tendency to form hetero-oligomers (Langhorst et al., 2008), both reggie-1 and ‑2 are likely to form a complex together with KIF9. Consistently, knockdown of either reggie-1 or ‑2 had comparable effects on matrix degradation, while a combined knockdown did not show a significant enhancement over single isoform down-regulation. This indicates that 1) reggie-1 and ‑2 cannot compensate for each other and 2) only the reggie hetero-oligomer is functional in this regard. Interestingly, reggie proteins are not found in purified podosome fractions (our own unpublished observations), which may indicate that reggie proteins are only in transient contact with podosomes and that they are not actual components of these structures.
Reggie proteins function in the targeted delivery of membrane and membrane proteins from internal vesicle pools to privileged sites at the plasma membrane, including cell–cell contacts or growth cones (Stuermer, 2010). In this context, it is tempting to speculate that reggie proteins may be involved in the delivery of membrane material to podosomes. Involvement of membrane delivery to the related invadopodia has been demonstrated (reviewed in Caldieri and Buccione, 2010), and this mechanism may be involved in the protrusive growth of these structures. However, it is currently unclear whether podosomes are also protrusive (Gimona et al., 2008; McMichael et al., 2010). Alternatively, a potential reggie-dependent membrane influx at podosomes may be involved in the delivery or recycling of lytic enzymes, for example, through vesicles containing matrix metalloproteinases (MMPs), which are important for the lytic ability of podosomes (Linder, 2007). However, colocalization studies performed by staining several important MMP isoforms such as MMP-2, MMP-7, MMP-8, MMP-9, MMP-12, MT1-MMP, and MT4-MMP in reggie-1-GFP–expressing macrophages revealed no significant degree of colocalization (Supplemental Figure 6), which makes this scenario unlikely.
Reggie proteins cluster in complexes that also comprise Src family kinases and Rho GTPases (Kawase et al., 2006). Interestingly, Src family members such as Src (Sounni et al., 2004), Fyn (Redondo-Muñoz et al., 2010), or Lyn (Zhao et al., 2006) and Rho GTPase signaling (Deroanne et al., 2005; Guegan et al., 2008) have been shown to be involved in MMP-dependent signaling in various cell systems. The potential involvement of reggie proteins in the indirect regulation of MMPs is thus an important direction for future studies.
In sum, we show here a novel role for the kinesin KIF9 in the regulation of both numbers and matrix-degradation ability of human macrophage podosomes and demonstrate that the unique C-terminal region of KIF9 is central for these effects. KIF9 is only the second kinesin to be identified as a regulator of podosomes, while, in turn, podosome regulation is the first reported cellular function for this motor. We further show a specific interaction of the KIF9 C-terminus with reggie-1/flotillin-2. Consistently, reggie proteins also have a significant role in the regulation of podosomal matrix degradation. KIF9 thus appears to mediate several activities through interaction with distinct cargo molecules. A future challenge is thus to identify further interactors of KIF9 and to elucidate their effects on specific aspects of podosome regulation.
MATERIALS AND METHODS
Cell isolation and cell culture
Human peripheral blood monocytes were isolated from buffy coats (kindly provided by Frank Bentzien, University Medical Center Hamburg-Eppendorf, Germany) and differentiated into macrophages as described previously (Linder et al., 1999).
Microinjection of proteins
Cells for microinjection experiments were cultured for 5–8 d. Proteins were expressed in Escherichia coli as described in Linder et al. (2000). For microinjection, proteins were dialyzed against microinjection buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2), concentrated in Vivaspin filters (Sartorius, Göttingen, Germany), shock frozen, and stored at −80°C. Microinjection was performed using the Eppendorf Transjector 5246 and a Compic Inject micromanipulator (Cell Biology Trading, Hamburg, Germany). GST-KIF9-CT81 was injected into the cytoplasm at 2 µm/µl. Control experiments were performed with comparable concentrations of GST. Injected cells were identified by labeling coinjected rat immunoglobulin G (IgG) (5 mg/ml; Dianova, Hamburg, Germany) with fluorescein isothiocyanate transferrin (FITC)–labeled goat anti–rat IgG antibody (Dianova). Cells containing less than 10 podosomes at a given time point were scored as “containing no podosomes.”
Transfection of cells
Cells were transiently transfected using the Microporator (Peqlab, Erlangen, Germany). For transfection of primary human macrophages, the following parameters were used: 1000 V, 40 ms, 2 pulses, and 0.5 μg DNA per 1 × 105 cells.
Expression vectors
For cloning of GST-KIF9-CT81, part of the coding sequence of KIF9 was amplified using the following primers: (CT81 forward) 5′-CTGGTACAATAGATCTTTTGTCATCCCTG-3′ and (CT81 reverse) 3′-ATCTGGTACGGATCCTCCTTTGTC-5′, generating 5′ BamHI and 3′ BglII restriction sites, and cloned into pGEX-4T2, resulting in a construct coding for aa residues 712–793. For cloning and expression of KIF9-CT81-GFP, the same coding sequence of KIF9 was amplified using primer (CT81 forward) and primer 3′-GGCACATAGAGGATCCAACCTCATCG-5′, generating 5′ BamHI and 3′ BglII restriction sites, and cloned into pEGFP-C1 (Clontech, Saint-Germain-en-Laye, France). For cloning and expression of wild-type GFP-KIF9, the KIF9 coding sequence was amplified from pBluescript-KIF9 using the primers (KIF9-GFP forward) 5′-TCGCTGCCTGGTAAGCTTAGAATGGGACTAG-3′ and (KIF9-GFP reverse) 3′-GTACTGGCGATGACCGGTTTTTTTCTATGTGC-5′, generating 5′ HindIII and 3′ AgeI restriction sites. The PCR product was cleaved by using the internal KpnI restriction site, and the resulting two regions were cloned successively into vector pEGFP-N1 (Clontech, Palo Alto, CA). For cloning and expression of KIF9-CT402-GFP, the fl KIF9-GFP construct was digested by AgeI and KpnI, and the region coding for aa 388–790 was cloned into pEGFP-N1. For cloning of the KIF9-NT709 tailless construct, part of the coding sequence of KIF9 was amplified using the following primers: (KIF9-GFP NT1 forward) 5′-ATATAGTATTTAACTCGAGATGGGTACTAGGA-3′ and (KIF9-GFP NT709 reverse) 3′-CTGGTACAATGAGTCCCGGGCCCATAAAATT-5′, generating 5′ XhoI and 3′ ApaI restriction sites, and cloned into pEGFP-N1, resulting in a construct coding for aa residues 1–709. For cloning of KIF9-mCherry, GFP and mCherry sequences of KIF9-GFP and mCherry-N1 (Clontech, Mountain View, CA) were exchanged by digestion with AgeI and NotI, with subsequent ligation of the mCherry sequence into the KIF9 vector backbone. For cloning of KIF9-pTagRFP, GFP and pTagRFP sequences of KIF9-GFP and pTagRFP-N (Evrogen, Moscow, Russia) were exchanged by digestion with AgeI and NotI, with subsequent ligation of the pTagRFP sequence into the KIF9 vector backbone. Generation of mRFP-β-actin has been described in Osiak et al. (2005). Vector-based shRNAs for KIF9 and scrambled control sequences were generated using siSTRIKE U6 Hairpin Cloning System (Human)-hMGFP (Promega, Madison, WI) according to the manufacturer’s instructions. The sequence for KIF9shRNA was 5′-GAGAGGAGTTGTCAATAA-3′, targeting nucleotides 126 to 143. The sequence for scrambled control was 5′-GTACCTAAATCCAAAGAA-3′. pEGFP-N1 was purchased from Clontech, and reggie-1-GFP was a kind gift of R. Tikkanen (Universitätsklinik Frankfurt, Institute for Biochemistry, Germany).
Immunofluorescence and microscopy
Cells were fixed for 10 min in 3.7% formaldehyde solution and permeabilized for 5 min in ice-cold acetone. Actin was stained with Alexa 568–labeled phalloidin (Molecular Probes, Leiden, Netherlands), TGN 46 was stained with specific primary polyclonal antibody (AbD Serotec, Oxford, UK), and α-tubulin was stained with specific primary monoclonal antibody (Sigma, St. Louis, MO). MMPs were stained with specific primary monoclonal antibodies: MMP-2 (Calbiochem, Darmstadt, Germany), MMP-7 (Acris, Herford, Germany), MMP-8 (Millipore, Billerica, MA), MMP-9 (Calbiochem), MMP-12 (Sigma), MT1-MMP (Millipore), or MT4-MMP (Epitomics, Burlingame, CA).
Secondary antibodies were Alexa 568–labeled goat anti-mouse or goat anti-sheep (Molecular Probes). Coverslips were mounted in Mowiol (Calbiochem) containing p-phenylenediamine (Sigma-Aldrich, St. Louis, MO) as an antifading reagent and sealed with nail polish.
Microscopy was performed as described previously (Kopp et al., 2006). Images of fixed samples were acquired with a confocal laser-scanning microscope (Leica DM IRE2 with a Leica TCS SP2 AOBS confocal point scanner) equipped with an oil-immersion plan Apo 63× NA 1.4 objective. Acquisition and processing of images was performed with Leica Confocal Software (Leica, Wetzlar, Germany).
Live cell imaging
Images were acquired with a spinning disk confocal system (Spinning disk CSU22, Yokogawa, Japan) fitted on a Zeiss Axiovert 200M microscope with a temperature- and CO2- controllable environmental chamber (Solent Scientific, Regensworth, UK), an oil immersion HCX PL Apo 63× NA 1.4–0.6 lambda blue objective, and a CCD camera (EM-CCD C-9100-2, Hamamatsu, Japan). Acquisition and processing of images were performed with Volocity Software (Improvision, Coventry, UK). Cells were seeded on 35-mm μ-dishes (ibidi, Martinsried, Germany) at a density of 2 × 105 and incubated 5 or 72 h, as indicated, before the start of the experiment.
Podosomes were counted by taking confocal images of the substrate-attached plane of macrophages and by counting separate F-actin–containing dots with a diameter of 0.5–1.5 μm. In the vast majority of cases, this allows a clear scoring of cells into the groups described (e.g., Figure 1D: containing 0–10, 11–50, and >50 podosomes). To be scored as a contact between podosomes (labeled with mRFP-actin) and KIF9-GFP vesicles, the respective mRFP and GFP signals had to be directly adjacent or overlapping for at least two pixels, without intermediate black pixels.
Immunoblotting
Immunolabeling was performed by standard procedure, using the following primary antibodies: mouse monoclonal reggie-1 was from BD Biosciences (Franklin Lakes, NJ), mouse monoclonal HA was from Cell Signaling (Danvers, MA), and mouse polyclonal GFP was a kind gift of J. Faix (Medical University Hannover, Hannover, Germany). For generating the KIF9 antibody, GST-KIF9-CT81 was thrombin cleaved, and the KIF9-CT81 peptide was separated by PAA gel electrophoresis, purified from gels, and injected into New Zealand White rabbits. Rabbit serum was affinity purified with GST-KIF9-CT81 spotted on nitrocellulose membrane. Secondary antibodies were horseradish peroxidase–coupled anti–mouse or anti–rabbit IgG (Dianova). Protein bands were visualized by using a SuperSignal kit (Pierce, Rockford, IL) and X-Omat AR film (Kodak, Stuttgart, Germany).
Quantitative real-time PCR
SiRNAs were validated by quantitative real-time PCR (qPCR) using lysates of transfected HeLa cells, as previously described (Machuy et al., 2005). Briefly, 0.1–0.25 μg siRNA (final concentration 80–200 nM) directed against KIF9 or luciferase as control and 2 μl TransMessenger reagent (Qiagen, Hilden, Germany) were added to 10 × 104 cells seeded in 96-well plates. RNA was isolated 48 h later using the RNeasy 96 BioRobot 8000 system (Qiagen). The relative amount of target mRNA was determined by qPCR using QuantiTect SYBR Green RT-PCR Kit following the manufacturer’s instructions (Qiagen). The sequence of the KIF9-specific siRNA was 5′-CAGGACTTGGTTTATGAGACA-3′, targeting nucleotides 196 to 216. SiRNA for firefly luciferase, used as a control, was generated as described in Kopp et al. (2006). siRNAs for reggie-1 and reggie-2 were generated (MWG, Ebersberg, Germany) according to Solis et al. (2007). Primary human macrophages were transfected with siRNA (650 ng) twice at 0 and 72 h and evaluated after a further incubation period of 5 h.
Reverse transcriptase reaction
A total of 6 × 106 cells were cultured for 7 d, and mRNA was isolated using 1 ml Trizol Reagent (Invitrogen, Carlsbad, CA). DNA was removed by DNAse digestion (Novagen, Madison, WI). For cDNA-synthesis, 1 μg random primer (Promega) was annealed to 2 μg RNA for 5 min at 70°C, and first-strand synthesis was performed using Moloney murine leukemia virus reverse transcriptase (Promega). Second-strand synthesis was performed using an oligonucleotide primer pair corresponding to nucleotides 1125–1150 and 1531–1579 of the KIF9 coding sequence (accession number BC030657), respectively. As a control for quantitative removal of residual DNA, oligonucleotide primers specific for an exon in the human ß-actin gene were used, corresponding to nucleotides 1161–1142 and 716–735, respectively.
Immunoprecipitation
Immunoprecipitations of GFP-fused proteins were performed using the μMACS GFP Tagged Protein Isolation Kit (Miltenyi Biotec, Bergisch-Gladbach, Germany) according to the manufacturer’s instructions. For lysis, preparation of columns, and washing, the following buffers were used: lysis buffer (150 mM NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 8.0) with Complete Mini Protease Inhibitor (Roche Diagnostics, Penzberg, Germany), wash buffer 1 (150 mM NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 8.0), and wash buffer 2 (20 mM Tris-HCl, pH 7.5).
Mass spectrometry
After SDS–PAGE and silver staining of proteins, gel bands were excised from the gel and subjected to in-gel digest as described in Shevchenko et al. (2006). Extracted peptides were desalted and concentrated with “STAGE” t. Reverse-phase liquid chromatography–tandem MS (LC-MS/MS) was done by using an Agilent 1200 Nanoflow LC system (Agilent Technologies, Böblingen, Germany). The LC system was online coupled to an LTQ-Orbitrap (Thermo Scientific, Waltham, MA) equipped with a nanoelectrospray source (Proxeon, Odense, Denmark). Chromatographic separation of peptides was performed with a custom-made capillary needle packed with reverse-phase ReproSil-Pur C18 resin (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany). The tryptic peptide mixtures were auto sampled at a flow rate of 0.5 μl/min and then eluted with a linear gradient at a flow rate of 0.25 μl/min. The mass spectrometers were operated in the data-dependent mode to automatically measure MS and MS/MS (Krüger et al., 2008). Full-scan MS spectra were acquired with a resolution r = 60,000 at m/z 400. Raw MS spectra were processed using the MaxQuant software, which performed peak list generation and false discovery rate calculation based on search engine results (Cox and Mann, 2008). The derived peak list was searched with the Mascot search engine (Matrix Science, Boston, MA) against a concatenated database (IPI 3.54 human) (Elias and Gygi, 2007).
Matrix labeling and degradation
Gelatin (from swine; Roth, Karlsruhe, Germany) was fluorescently labeled with normal human serum (NHS)–rhodamine (Thermo Scientific, Rockford, IL) according to Chen (1996). Coverslips were coated with labeled gelatin solution, fixed in 0.5% glutaraldehyde (Roth), and washed with 70% ethanol and medium. Cells were seeded on coated coverslips with a density of 8 × 105 cells/coverslip. Cells previously transfected with siRNA and incubated for the times indicated were seeded on coated coverslips with a density of 8 × 105 cells/coverslip and incubated for a further 5 h, followed by fixation and staining.
Statistics
ImageJ software was used to analyze Cy5-labeled F-actin fluorescence intensity. Values of matrix degradation were determined by loss of fluorescence intensity, with intensity of undegraded areas set to 100%. For comparability, laser intensity was not changed between measurements. For each value, 3 × 30 cells were evaluated. Statistical analysis was performed with Excel software. When indicated, differences between mean values were analyzed using the Student’s t test. P < 0.05 was considered as statistically significant and P < 0.01 as statistically highly significant.
For vesicle tracking, the ImageJ software (plug-in “manual tracking”) was used to analyze vesicle movement of KIF9-GFP–transfected cells. Each vesicle was tracked manually throughout the video.
In selected time-lapse movies, line intensity profiles were determined at time points indicated using Volocity 5.3 for Mac imaging software. The resulting intensity values (gray levels of the acquired 14-bit image) of EGFP and mRFP fluorescence were offset corrected and plotted over the length of the line drawn using GraphPad Prism 5.0c for Mac.
Supplementary Material
Acknowledgments
We thank Jan Faix for the anti-GFP antibody, Ritva Tikkanen for GFP-reggie-1, Jens Cornils and Barbara Böhlig for expert technical assistance, and Martin Aepfelbacher and Peter C. Weber for continuous support. This work is part of the doctoral thesis of SC. Work in the SL lab is supported by Deutsche Forschungsgemeinschaft (LI925/2-1, LI925/3-1), Wilhelm Sander-Stiftung (2007.020.02), and the European FP7 program (Marie Curie Actions, T3Net).
Abbreviations used:
- FITC
fluorescein isothiocyanate
- fl
full length
- Ig
immunoglobulin
- KIF
kinesin-like family
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MMP
matrix metalloproteinases
- MS
mass spectrometry
- NHS
normal human serum
- PAA
polyacrylamide
- pEGFP
plasmid enhanced green fluorescent protein
- qPCR
quantitative PCR
- shRNA
short-hairpin RNA
- siRNA
small interfering RNA
- TGN
trans-Golgi network
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-05-0394) on November 30, 2010.
REFERENCES
- Babuke T, Tikkanen R. Dissecting the molecular function of reggie/flotillin proteins. Eur J Cell Biol. 2007;86:525–532. doi: 10.1016/j.ejcb.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Buccione R, Caldieri G, Ayala I. Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 2009;28:137–149. doi: 10.1007/s10555-008-9176-1. [DOI] [PubMed] [Google Scholar]
- Caldieri G, Buccione R. Aiming for invadopodia: organizing polarized delivery at sites of invasion. Trends Cell Biol. 2010;20:64–70. doi: 10.1016/j.tcb.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Chabadel A, Banon-Rodriguez I, Cluet D, Rudkin BB, Wehrle-Haller B, Genot E, Jurdic P, Anton IM, Saltel F. CD44 and beta3 integrin organize two functionally distinct actin-based domains in osteoclasts. Mol Biol Cell. 2007;18:4899–4910. doi: 10.1091/mbc.E07-04-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Proteases associated with invadopodia, and their role in degradation of extracellular matrix. Enzyme Protein. 1996;49:59–71. doi: 10.1159/000468616. [DOI] [PubMed] [Google Scholar]
- Cougoule C, Carreno S, Castandet J, Labrousse A, Astarie-Dequeker C, Poincloux R, Le CV, Maridonneau-Parini I. Activation of the lysosome-associated p61Hck isoform triggers the biogenesis of podosomes. Traffic. 2005;6:682–694. doi: 10.1111/j.1600-0854.2005.00307.x. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Deroanne CF, Hamelryckx D, Ho TT, Lambert CA, Catroux P, Lapiere CM, Nusgens BV. Cdc42 downregulates MMP-1 expression by inhibiting the ERK1/2 pathway. J Cell Sci. 2005;118:1173–1183. doi: 10.1242/jcs.01707. [DOI] [PubMed] [Google Scholar]
- Destaing O, Saltel F, Geminard JC, Jurdic P, Bard F. Podosomes display actin turnover and dynamic self-organization in osteoclasts expressing actin-green fluorescent protein. Mol Biol Cell. 2003;14:407–416. doi: 10.1091/mbc.E02-07-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destaing O, Saltel F, Gilquin B, Chabadel A, Khochbin S, Ory S, Jurdic P. A novel Rho-mDia2-HDAC6 pathway controls podosome patterning through microtubule acetylation in osteoclasts. J Cell Sci. 2005;118:2901–2911. doi: 10.1242/jcs.02425. [DOI] [PubMed] [Google Scholar]
- Dunn S, Morrison EE, Liverpool TB, Molina-Paris C, Cross RA, Alonso MC, Peckham M. Differential trafficking of Kif5c on tyrosinated and detyrosinated microtubules in live cells. J Cell Sci. 2008;121:1085–1095. doi: 10.1242/jcs.026492. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Proteolytic and non-proteolytic migration of tumour cells and leucocytes. Biochem Soc Symp. 2003:277–285. doi: 10.1042/bss0700277. [DOI] [PubMed] [Google Scholar]
- Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol. 2008;20:235–241. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Glebov OO, Bright NA, Nichols BJ. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat Cell Biol. 2006;8:46–54. doi: 10.1038/ncb1342. [DOI] [PubMed] [Google Scholar]
- Guegan F, Tatin F, Leste-Lasserre T, Drutel G, Genot E, Moreau V. p190B RhoGAP regulates endothelial-cell-associated proteolysis through MT1-MMP and MMP2. J Cell Sci. 2008;121:2054–2061. doi: 10.1242/jcs.025817. [DOI] [PubMed] [Google Scholar]
- Hufner K, Higgs HN, Pollard TD, Jacobi C, Aepfelbacher M, Linder S. The verprolin-like central (vc) region of Wiskott-Aldrich syndrome protein induces Arp2/3 complex-dependent actin nucleation. J Biol Chem. 2001;276:35761–35767. doi: 10.1074/jbc.M106520200. [DOI] [PubMed] [Google Scholar]
- Kawase K, Nakamura T, Takaya A, Aoki K, Namikawa K, Kiyama H, Inagaki S, Takemoto H, Saltiel AR, Matsuda M. GTP hydrolysis by the Rho family GTPase TC10 promotes exocytic vesicle fusion. Dev Cell. 2006;11:411–421. doi: 10.1016/j.devcel.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Kopp P, Lammers R, Aepfelbacher M, Woehlke G, Rudel T, Machuy N, Steffen W, Linder S. The kinesin KIF1C and microtubule plus ends regulate podosome dynamics in macrophages. Mol Biol Cell. 2006;17:2811–2823. doi: 10.1091/mbc.E05-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger M, Moser M, Ussan S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fässler R, Mann M. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell. 2008;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- Langhorst MF, Reuter A, Jaeger FA, Wippich FM, Luxenhofer G, Plattner H, Stuermer CA. Trafficking of the microdomain scaffolding protein reggie-1/flotillin-2. Eur J Cell Biol. 2008;87:211–226. doi: 10.1016/j.ejcb.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Lawrence CJ. A standardized kinesin nomenclature. J Cell Biol. 2004;167:19–22. doi: 10.1083/jcb.200408113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S. The matrix corroded: podosomes and invadopodia in extra cellular matrix degradation. Trends Cell Biol. 2007;17:107–117. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Linder S. Invadosomes at a glance. J Cell Sci. 2009;122:3009–3013. doi: 10.1242/jcs.032631. [DOI] [PubMed] [Google Scholar]
- Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–385. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- Linder S, Higgs H, Hufner K, Schwarz K, Pannicke U, Aepfelbacher M. The polarization defect of Wiskott-Aldrich syndrome macrophages is linked to dislocalization of the Arp2/3 complex. J Immunol. 2000a;165:221–225. doi: 10.4049/jimmunol.165.1.221. [DOI] [PubMed] [Google Scholar]
- Linder S, Hufner K, Wintergerst U, Aepfelbacher M. Microtubule-dependent formation of podosomal adhesion structures in primary human macrophages. J Cell Sci. 2000b;113:4165–4176. doi: 10.1242/jcs.113.23.4165. [DOI] [PubMed] [Google Scholar]
- Linder S, Nelson D, Weiss M, Aepfelbacher M. Wiskott-Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc Natl Acad Sci USA. 1999;96:9648–9653. doi: 10.1073/pnas.96.17.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machuy N, Thiede B, Rajalingam K, Dimmler C, Thieck O, Meyer TF, Rudel T. A global approach combining proteome analysis and phenotypic screening with RNA interference yields novel apoptosis regulators. Mol Cell Proteomics. 2005;4:44–55. doi: 10.1074/mcp.M400089-MCP200. [DOI] [PubMed] [Google Scholar]
- McMichael BK, Cheney RE, Lee BS. Myosin X regulates sealing zone patterning in osteoclasts through linkage of podosomes and microtubules. J Biol Chem. 2010;285:9506–9515. doi: 10.1074/jbc.M109.017269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory S, Brazier H, Pawlak G, Blangy A. Rho GTPases in osteoclasts: orchestrators of podosome arrangement. Eur J Cell Biol. 2008;87:469–477. doi: 10.1016/j.ejcb.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Osiak AE, Zenner G, Linder S. Subconfluent endothelial cells form podosomes downstream of cytokine and RhoGTPase signaling. Exp Cell Res. 2005;307:342–353. doi: 10.1016/j.yexcr.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Pfaff M, Jurdic P. Podosomes in osteoclast-like cells: structural analysis and cooperative roles of paxillin, proline-rich tyrosine kinase 2 (Pyk2) and integrin alphaVbeta3. J Cell Sci. 2001;114:2775–2786. doi: 10.1242/jcs.114.15.2775. [DOI] [PubMed] [Google Scholar]
- Piddini E, Schmid JA, de MR, Dotti CG. The Ras-like GTPase Gem is involved in cell shape remodelling and interacts with the novel kinesin-like protein KIF9. EMBO J. 2001;20:4076–4087. doi: 10.1093/emboj/20.15.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci. 2009;122:3015–3024. doi: 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- Redondo-Muñoz J, Ugarte-Berzal E, Terol MJ, Van Den Steen PE, Hernandez del CM, Roderfeld M, Roeb E, Opdenakker G, Garcia-Marco JA, Garcia-Pardo A. Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia b cell survival through its hemopexin domain. Cancer Cell. 2010;17:160–172. doi: 10.1016/j.ccr.2009.12.044. [DOI] [PubMed] [Google Scholar]
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Sack S, Kull FJ, Mandelkow E. Motor proteins of the kinesin family. Structures, variations, and nucleotide binding sites. Eur J Biochem. 1999;262:1–11. doi: 10.1046/j.1432-1327.1999.00341.x. [DOI] [PubMed] [Google Scholar]
- Sakurai-Yageta M, Recchi C, Le DG, Sibarita JB, Daviet L, Camonis J, D’Souza-Schorey C, Chavrier P. The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J Cell Biol. 2008;181:985–998. doi: 10.1083/jcb.200709076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte T, Paschke KA, Laessing U, Lottspeich F, Stuermer CA. Reggie-1 and reggie-2, two cell surface proteins expressed by retinal ganglion cells during axon regeneration. Development. 1997;124:577–587. doi: 10.1242/dev.124.2.577. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Solis GP, Hoegg M, Munderloh C, Schrock Y, Malaga-Trillo E, Rivera-Milla E, Stuermer CA. Reggie/flotillin proteins are organized into stable tetramers in membrane microdomains. Biochem J. 2007;403:313–322. doi: 10.1042/BJ20061686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounni NE, et al. Up-regulation of vascular endothelial growth factor-A by active membrane-type 1 matrix metalloproteinase through activation of Src-tyrosine kinases. J Biol Chem. 2004;279:13564–13574. doi: 10.1074/jbc.M307688200. [DOI] [PubMed] [Google Scholar]
- Steffen A, Le DG, Poincloux R, Recchi C, Nassoy P, Rottner K, Galli T, Chavrier P. MT1-MMP-dependent invasion is regulated by TI-VAMP/VAMP7. Curr Biol. 2008;18:926–931. doi: 10.1016/j.cub.2008.05.044. [DOI] [PubMed] [Google Scholar]
- Stuermer CA. The reggie/flotillin connection to growth. Trends Cell Biol. 2010;20:6–13. doi: 10.1016/j.tcb.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- Wade RH, Kozielski F. Structural links to kinesin directionality and movement. Nat Struct Biol. 2000;7:456–460. doi: 10.1038/75850. [DOI] [PubMed] [Google Scholar]
- Webb BA, Eves R, Mak AS. Cortactin regulates podosome formation: roles of the protein interaction domains. Exp Cell Res. 2006;312:760–769. doi: 10.1016/j.yexcr.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Wiesner C, Faix J, Himmel M, Bentzien F, Linder S. KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding and extracellular matrix degradation in primary macrophages. Blood. 2010;116:1559–1569. doi: 10.1182/blood-2009-12-257089. [DOI] [PubMed] [Google Scholar]
- Woehlke G, Schliwa M. Directional motility of kinesin motor proteins. Biochim Biophys Acta. 2000;1496:117–127. doi: 10.1016/s0167-4889(00)00013-6. [DOI] [PubMed] [Google Scholar]
- Zambonin-Zallone A, Teti A, Grano M, Rubinacci A, Abbadini M, Gaboli M, Marchisio PC. Immunocytochemical distribution of extracellular matrix receptors in human osteoclasts: a beta 3 integrin is colocalized with vinculin and talin in the podosomes of osteoclastoma giant cells. Exp Cell Res. 1989;182:645–652. doi: 10.1016/0014-4827(89)90266-8. [DOI] [PubMed] [Google Scholar]
- Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114:3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- Zhao Y, He D, Saatian B, Watkins T, Spannhake EW, Pyne NJ, Natarajan V. Regulation of lysophosphatidic acid-induced epidermal growth factor receptor transactivation and interleukin-8 secretion in human bronchial epithelial cells by protein kinase Cdelta, Lyn kinase, and matrix metalloproteinases. J Biol Chem. 2006;281:19501–19511. doi: 10.1074/jbc.M511224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.