

The role of specific membrane lipids in ER-Golgi transport is unclear. Using cell-free assays that measure stages in ER-Golgi transport, a variety of enzyme inhibitors, lipid-modifying enzymes, and lipid ligands were screened. The results indicate that PI(4)P is required for SNARE-dependent fusion of COPII vesicles with the Golgi complex.
Abstract
The role of specific membrane lipids in transport between endoplasmic reticulum (ER) and Golgi compartments is poorly understood. Using cell-free assays that measure stages in ER-to-Golgi transport, we screened a variety of enzyme inhibitors, lipid-modifying enzymes, and lipid ligands to investigate requirements in yeast. The pleckstrin homology (PH) domain of human Fapp1, which binds phosphatidylinositol-4-phosphate (PI(4)P) specifically, was a strong and specific inhibitor of anterograde transport. Analysis of wild type and mutant PH domain proteins in addition to recombinant versions of the Sac1p phosphoinositide-phosphatase indicated that PI(4)P was required on Golgi membranes for fusion with coat protein complex II (COPII) vesicles. PI(4)P inhibition did not prevent vesicle tethering but significantly reduced formation of soluble n-ethylmaleimide sensitive factor adaptor protein receptor (SNARE) complexes between vesicle and Golgi SNARE proteins. Moreover, semi-intact cell membranes containing elevated levels of the ER-Golgi SNARE proteins and Sly1p were less sensitive to PI(4)P inhibitors. Finally, in vivo analyses of a pik1 mutant strain showed that inhibition of PI(4)P synthesis blocked anterograde transport from the ER to early Golgi compartments. Together, the data presented here indicate that PI(4)P is required for the SNARE-dependent fusion stage of COPII vesicles with the Golgi complex.
INTRODUCTION
The secretory pathway is responsible for delivery of proteins and lipids from their site of synthesis at the endoplasmic reticulum (ER) to the cell surface and the many membrane-bound compartments that comprise the endomembrane system. Transport between these compartments is mediated by membrane vesicles and tubules that bud from a donor membrane and selectively target to and fuse with an acceptor membrane. This process is vital for cell growth and for maintenance of intracellular compartments as distinct biochemical environments. Many of the molecular mechanisms that underlie membrane transport are highly conserved between diverse intracellular trafficking events as well as among eukaryotic species (Bonifacino and Glick, 2004).
After synthesis of secretory molecules at the ER, folded cargo proteins are packaged into coat protein complex II (COPII)-derived transport vesicles (Sato and Nakano, 2007). In yeast, vesicles then traffic toward the Golgi apparatus and tether to cis-Golgi membranes in a process that depends on Ypt1p, Uso1p, the Grh1/Bug1p complex, and the transport protein particle (TRAPP)-I complex (Sztul and Lupashin, 2006; Behnia et al., 2007). Vesicle fusion ensues as cognate soluble n-ethylmaleimide sensitive factor adaptor protein receptor (SNARE) proteins assemble into trans-SNARE complexes regulated by the function of the SM protein Sly1p (Sudhof and Rothman, 2009) and the SNARE dissociation chaperones Sec17p (α-SNAP) and Sec18p (NSF) (Wickner and Schekman, 2008). Current evidence indicates that the vesicle-SNAREs involved in this process are Bet1p and Bos1p, whereas Sed5p and Sly1p act on the Golgi compartment (Cao and Barlowe, 2000). The R-SNARE Sec22p can function in this fusion reaction from either vesicles or Golgi membranes (Liu and Barlowe, 2002). A complete understanding of the mechanisms underlying ER-to-Golgi transport is limited in part, however, by a lack of knowledge of the role of membrane lipids in COPII vesicle budding, tethering, and fusion with cis-Golgi compartments.
In addition to well-recognized structural roles in forming membrane bilayers and establishing subcellular compartments within eukaryotic cells, specific lipid species are known to regulate membrane dynamics (van Meer et al., 2008). Among these lipids, diacylglycerol (DAG) and the phosphoinositides (phosphorylated derivatives of phosphatidylinositol [PI]) have been implicated in diverse cellular processes, including membrane deformation and vesicle trafficking (Gomez-Fernandez and Corbalan-Garcia, 2007; Strahl and Thorner, 2007; Mulet et al., 2008). Few studies, however, have addressed the role of specific lipid species in transport between ER and Golgi compartments.
In this study we investigated the lipid requirements for ER-to-Golgi transport in the budding yeast Saccharomyces cerevisiae, focusing on the role of phosphoinositides. In vitro budding, tethering, and fusion assays were used to screen phosphoinositide binding domains, enzymes that modify phosphoinositides, and inhibitors of enzymes involved in phosphoinositide metabolism. We found that the pleckstrin homology (PH) domain of human Fapp1, which binds specifically to PI(4)P (phosphatidylinositol-4-phosphate), was a potent and specific inhibitor of ER-to-Golgi transport. Stage-specific assays demonstrated that PI(4)P was required at the Golgi compartment for fusion of COPII vesicles with Golgi acceptor membranes. Moreover, reduction of PI(4)P levels significantly decreased formation of SNARE complexes between vesicle and Golgi SNARE proteins. Based on the results presented in this study, we propose that PI(4)P is an important regulator of COPII vesicle fusion with Golgi membranes.
RESULTS
Characterization of inhibitors of ER-to-Golgi transport
To identify specific lipid requirements for ER-to-Golgi trafficking, we screened an array of lipid-binding domains, enzymes, and enzyme inhibitors in cell-free assays to monitor influences on COPII vesicle budding, vesicle tethering, or vesicle fusion with Golgi acceptor membranes. The following lipid-binding domains were tested: the PI(4)P-binding PH domain of human Fapp1 (Dowler et al., 2000; Stahelin et al., 2007); a tandem repeat of the PI(3)P-binding FYVE domainof Eea1 (Gillooly et al., 2000); the PI(4,5)P2-binding ENTH domain of epsin (Rosenthal et al., 1999); the PI(4,5)P2-binding marcks-effector domain (MED) (Wang et al., 2001); and the DAG-binding C1b domain of protein kinase C (Johnson et al., 2000). In addition, we tested a PI-specific phospholipase C (PI-PLC) from Bacillus cereus (Ross et al., 1992) that converts PI into DAG and inorganic phosphate; and the PLC inhibitor U73122, which prevents the conversion of PI(4,5)P2 into DAG and IP3 by Plc1p, the only known yeast PLC (Bleasdale et al., 1990; Jun et al., 2004). The concentrations of inhibitors used were in the same range as reported to inhibit other in vitro trafficking reactions (Fratti et al., 2004; Jun et al., 2006).
Inhibitors were initially screened for defects of overall ER-to-Golgi transport. In this assay, washed semi-intact cell membranes containing translocated [35S]glyco-pro-alpha-factor (gpαf) were pretreated with indicated inhibitors for 20 min at 4°C. Reactions were then incubated for 1 h at 23°C with purified transport factors to drive transport of gpαf to the Golgi complex (Barlowe, 1997). Upon delivery to the Golgi complex, gpαf receives outer-chain α1,6-mannose residues that can be immunoprecipitated with anti-1,6-mannose-specific serum to quantify gpαf transport (Baker et al., 1988). As observed in Figure 1A, U73122 and PH strongly inhibited ER-to-Golgi transport, whereas C1b, PI-PLC, and FYVE moderately inhibited the reaction. We next determined the specific stage(s) of ER-to-Golgi transport (budding, tethering, or fusion) that were affected by each inhibitor.
FIGURE 1:

Screen for inhibitors of ER-to-Golgi transport. (A) Washed semi-intact cells containing [35S]gpαf were pretreated with indicated inhibitors for 20 min at 4°C. Transport reactions were then incubated with Recon proteins (COPII, Uso1p, and LMA1) and an ATP regeneration system at 23°C for 1 h. The amount of Golgi-modified [35S]gpαf was measured to determine transport efficiency. NA is the background level of transport in the absence of transport factors. (B) Semi-intact cell acceptor membranes were pretreated with indicated inhibitors for 20 min at 4°C and then incubated with COPII vesicles containing [35S]gpαf in the presence of fusion factors (U/L: Uso1p and LMA1) and an ATP regeneration system at 23°C for 1 h. Golgi-modified [35S]gpαf was measured to determine fusion efficiency. The no Acpt condition represents the background level of fusion in the absence of semi-intact cell acceptor membranes. (C) Pretreated, semi-intact cells as in panel A were incubated with COPII or COPII plus Uso1p for 30 min at 23°C. To measure budding (black bars) or tethering (white bars), diffusible vesicles containing [35S]gpαf were separated from semi-intact cell membranes by centrifugation. (D) Mock-budding reactions as in panel C were treated with trypsin, and total protease protected [35S]gpαf was quantified to assess membrane integrity.
To measure COPII vesicle fusion with Golgi membranes, we performed two-stage fusion reactions. In the first stage, COPII vesicles containing [35S]gpαf were generated from microsomes and separated from donor ER membranes by centrifugation (Cao and Barlowe, 2000). In the second stage, washed semi-intact cell membranes pretreated with indicated inhibitors were incubated with [35S]gpαf containing vesicles in the presence of fusion factors for 1 h at 23°C (Cao and Barlowe, 2000). As shown in Figure 1B, U73122, PH, and PI-PLC strongly inhibited fusion of COPII vesicles with Golgi acceptor membranes, whereas C1b and FYVE moderately inhibited the reaction. The no acceptor membrane control (no Acpt) reflects the level of Golgi membranes that carry over with COPII vesicles from the microsome budding reaction.
We next determined whether the inhibitors influenced COPII vesicle budding or vesicle tethering to Golgi membranes. For this assay, washed semi-intact cell membranes containing [35S]gpαf were pretreated with indicated inhibitors and then incubated for 30 min at 23°C with COPII proteins to drive vesicle budding (Cao et al., 1998). Because freely diffusible vesicles can be separated from ER and Golgi membranes by centrifugation, budding and Uso1p-dependent tethering of budded vesicles to Golgi membranes can be measured by quantifying the level of [35S]gpαf-containing diffusible vesicles in the supernatant fraction (Cao et al., 1998). As shown in Figure 1C, U73122 was a strong inhibitor of budding, whereas FYVE and PI-PLC were moderate inhibitors of budding. None of the inhibitors produced a strong influence on vesicle tethering to Golgi membranes. We observed, however, that the PH and C1b domains caused an increase in diffusible vesicles. We interpret this increase in diffusible vesicles to be caused primarily by a block in vesicle fusion with Golgi membranes. To ensure that these effects did not arise from a loss in membrane integrity, protease protection of luminal [35S]gpαf was assessed after inhibitor treatments. As shown in Figure 1D, none of the inhibitors compromised the integrity of ER membranes at the concentrations tested, including 80 μM U73122 and 1 nM PI-PLC (unpublished data).
In summary, the tested compounds could be placed into three general classes. The first class contains the PI(4,5)P2 binding domains, ENTH and MED, which did not have a significant effect on ER-to-Golgi transport. The second class comprised the inhibitors U73122, PI-PLC, and FYVE, which targeted both budding and fusion. At higher concentrations, U73122 and PI-PLC were strong inhibitors of budding and fusion, whereas FYVE produced modest inhibition of both of these stages. The last class of inhibitors included the PH and C1b domains that acted specifically at the vesicle fusion stage, with the PH domain acting as a more potent inhibitor. Further study of the inhibitors U73122, PI-PLC, C1b, and FYVE will be necessary to understand their mechanisms of inhibition. We decided to focus on the role of PI(4)P in fusion of COPII vesicles because PI(4)P is prevalent in the early secretory pathway (D’Angelo et al., 2008) and because the PH domain acted potently and specifically at the vesicle fusion stage.
PI(4)P is required at the Golgi complex for SNARE-mediated fusion with COPII vesicles
To determine whether the observed inhibition with the PH domain was specific for PI(4)P, we compared the wild-type PH domain (PHWT) used in Figure 1 to a mutant (PHN,K) that does not bind PI(4)P (Dowler et al., 2000; Blumental-Perry et al., 2006). Titration of these binding domains in one-stage transport reactions showed that PHWT is a dose-dependent transport inhibitor, whereas PHN,K did not inhibit transport even at the highest concentrations tested (Figure 2A). Furthermore, in reactions comparing one-stage transport, vesicle budding, and vesicle tethering (Figure 2B), treatment with 5 μM PHN,K did not have detectable effects on transport, whereas 5 μM PHWT inhibited transport and caused an apparent increase in diffusible vesicles (also observed in Figure 1C). Because tethering was largely unaffected (Figure 2B), we conclude that this effect was caused primarily by inhibition of vesicle fusion.
FIGURE 2:

PHWT inhibits COPII vesicle fusion with Golgi membranes. (A) Titration of PHWT (black squares) and PHN,K (white squares) in one-stage transport experiments was performed as described in Figure 1A. Maximum transport was equivalent to 20% overall ER-to-Golgi transport. NA represents background level of transport in the absence of transport factors. (B) Overall transport (black bars), budding (gray bars) and tethering (white bars) assays were carried out simultaneously as in Figure 1. PHWT and PHN,K were tested at 5 μM. Arrows show background transport in the absence of transport factors. (C) Fusion reactions were conducted as in Figure 1B and monitored for the formation of cross-linked Bet1pI83C-Sec22pD153C heterodimers (white bars and immunoblot) and for Golgi modification of [35S]gpαf (black bars). Reactions were probed with antibodies against Sec22p and Och1p (loading control). Arrow indicates position of cross-linked linked Bet1pI83C-Sec22pD153C heterodimer.
Tethered vesicles could be prevented from fusing with Golgi membranes if formation of trans-SNARE complexes was blocked or if assembled trans-SNARE complexes could not catalyze membrane fusion. To determine whether PHWT treatment prevents formation of trans-SNARE complexes, we used an assay that measures the formation of disulfide cross-links between cognate SNARE protein pairs. In this assay, the SNARE proteins Bet1p and Sec22p contain cysteine residues introduced into their SNARE domains in positions that produce cross-linked adducts when assembled into SNARE complexes (Flanagan and Barlowe, 2006). Briefly, we performed two-stage fusion reactions chasing Bet1pI83C vesicles into Sec22pD153C acceptor membranes and monitored formation of the Bet1p-Sec22p disulfide cross-linked heterodimer in parallel with transport of [35S]gpαf. As shown in Figure 2C, the inhibition of gpαf transport by 5 μM PHWT mirrors the inhibition of SNARE complex formation, whereas 5 μM PHN,K did not cause detectable inhibition in this assay. Taken together, these data indicate that the PHWT domain inhibits fusion of COPII vesicles through specific interactions with PI(4)P and suggests that the role of PI(4)P in fusion is upstream or simultaneous with trans-SNARE complex assembly.
To further explore these results, we sought to biochemically deplete PI(4)P by treating membranes with recombinant Sac1, a phosphoinositide lipid phosphatase that removes the phosphate residue from the inositol head group of PI(4)P. As a control we used the catalytically inactive mutant Sac1C392S (Maehama et al., 2000). Surprisingly, titration of these proteins in two-stage fusion reactions showed that the mutant Sac1C392S was a potent inhibitor of fusion whereas the Sac1WT protein did not display an equal inhibitory effect (Figure 3A). Moreover, in transport, budding, tethering, and two-stage fusion assays, addition of 2.8 μM Sac1C392S inhibited transport, caused COPII vesicles to accumulate, inhibited vesicle fusion, and blocked formation of cross-linked SNARE complexes (Figure 3, B and C). These results are similar to PHWT domain inhibition and suggested that the catalytically inactive mutant Sac1C392S acts as a PI(4)P lipid ligand. To test this hypothesis, we performed liposome-binding assays to monitor binding of Sac1WT and Sac1C392S to PI(4)P liposomes. As shown in Figure 3D, 32% of Sac1C392S bound to PI(4)P liposomes (lane 12) compared to 2% background binding to PI liposomes (lane 10). In contrast, 13% of Sac1WT bound to PI(4)P liposomes (compare lanes 12 and 6) with similar background binding to PI liposomes (compare lanes 10 and 4). Together, these results indicate that the catalytically inactive Sac1C392S mutant inhibits vesicle fusion through tight binding to membrane PI(4)P as observed for the PHWT domain.
FIGURE 3:

Sac1C392S inhibits COPII vesicle fusion with Golgi membranes. (A) Titration of Sac1WT (black squares) and Sac1C392S (white squares) in two-stage fusion reactions was performed as in Figure 1B. Maximum transport was equivalent to 32% fusion. NA shows the background level of fusion in the absence of transport factors and –Acceptor represents fusion level in the absence of added semi-intact cell acceptor membranes. (B) Overall transport (black bars), budding (gray bars), and tethering (white bars) assays were carried out simultaneously as in Figure 2B. Sac1WT and Sac1C392S were tested at 2.8 μM. Arrows show background transport in the absence of transport factors. (C) Fusion reactions were carried out as in Figure 2C and monitored for the formation of cross-linked Bet1pI83C-Sec22pD153C heterodimers (white bars and immunoblot) and for Golgi-modified [35S]gpαf (black bars). (D) Sac1C392S is a PI(4)P lipid ligand. Sac1WT or Sac1C392S (3 μM) were incubated with buffer (lanes 1, 2, 7, and 8), PI liposomes (lanes 3, 4, 9, and 10), or PI(4)P liposomes (lanes 5, 6, 11, and 12) for 30 min at 30°C. Liposomes were recovered by sedimentation, and the amount of protein in the supernatant (S) or co-sedimenting with liposomes in the pellet (P) was determined by Coomassie blue staining of 7.5% SDS–PAGE gels. Liposome molar composition: 50% PC, 30% PE, 10% PS, and 10% PI or PI(4)P.
We were surprised that Sac1WT treatment in the 5 μM range did not produce stronger inhibition of vesicle fusion. We considered, however, the possibility that our assay conditions may not be optimal for Sac1 phosphatase activity to deplete PI(4)P from semi-intact cell membranes. To promote Sac1WT activity, semi-intact cell acceptor membranes were incubated with higher concentrations of Sac1WT for 30 min at 12°C and then were diluted into standard two-stage fusion reactions. As shown in Figure 4A, pretreatment of acceptor membranes with increasing concentrations of Sac1WT caused a dose-dependent inhibition of vesicle fusion with concentrations greater than 25 μM near the minus acceptor control level. It should be noted that after pretreatment of membranes with the indicated concentration of Sac1WT, membranes were diluted approximately eightfold such that maximal final concentrations of Sac1WT in transport reactions were near those used in Figure 3A. Therefore the inhibitory effects of Sac1WT were likely due to catalytic activity of the enzyme and not due to lipid ligand binding. Finally, pretreatment of acceptor membranes with 50 μM Sac1WT also caused an accumulation of diffusible vesicles in budding reactions and strong inhibition of SNARE complex formation (Figure 4, B and C). These findings suggest that PI(4)P present on acceptor membranes is required for efficient SNARE complex assembly and fusion of ER-derived vesicles with Golgi membranes.
FIGURE 4:

Pretreatment of membranes with 50 μM Sac1WT inhibits COPII vesicle fusion with Golgi membranes. (A) Semi-intact cell membranes pretreated with Sac1WT at 12°C for 30 min were used in two-stage fusion reactions as described in Figure 1B. Maximum transport is equivalent to 35% fusion. (B) Semi-intact cells pretreated with 50 μM Sac1WT as in panel A were used in budding (black bars) and tethering (gray bars) assays as in Figure 1C. NA (white bars) shows background budding in the absence of transport factors. (C) Sec22pD153C semi-intact cells were pretreated with 50 μM Sac1WT as in panel A, and then fusion reactions were carried out as in Figure 2C monitoring the formation of cross-linked Bet1pI83C-Sec22pD153C heterodimers (white bars and immunoblot) and Golgi-modified [35S]gpαf fusion (black bars). (D) Reactions were assembled as in panel A and washed twice with excess buffer prior to setting up two-stage fusion reactions. NA shows background fusion in the absence of fusion factors, and the arrow indicates background fusion in the absence of acceptor membranes. Aliquots of the treated membranes were immunoblotted for Sac1p using an anti-penta-His antibody and for the Golgi membrane protein Och1p (loading control). Arrow indicates Och1p; asterisk indicates a cross-reactivity of anti-Och1p with the Sac1WT protein.
To further support the proposal that Sac1WT inhibition was due to the catalytic phosphatase activity of the enzyme and not due to steric hindrance of binding to PI(4)P, Sac1WT-treated membranes were washed prior to two-stage fusion reactions to remove excess Sac1WT. As shown in Figure 4D, this wash did not substantially reduce transport in mock treated membranes (washed vs. unwashed), whereas the 50 μM Sac1WT pretreatment inhibited transport to near the minus acceptor control level and was not rescued by the wash. To show that the wash procedure was effective, immunoblot analysis documented that ∼76% of Sac1WT was removed after washing treated semi-intact cell acceptor membranes (Figure 4D). Moreover, we observed that formation of the Bet1pI83C-Sec22pD153C cross-linked heterodimer was strongly inhibited by Sac1WT pretreatment and was not rescued by the wash (Supplemental Figure S1A). In addition, direct measurement of PI(4)P levels in semi-intact cell membranes demonstrated that the Sac1WT pretreatment was effective. After control incubations, semi-intact cell membranes contained 123 pmol PI(4)P/109 cells, whereas Sac1WT-treated membranes contained 78 pmol PI(4)P/109 cells (Supplemental Figure S1B). Finally, we found that N-ethylmaleimide inactivation of Sac1WT (Maehama et al., 2000) completely prevented its inhibitory activity in two-stage fusion reactions compared to mock treated Sac1WT (Supplemental Figure S1C). Based on the collective evidence, we conclude that pretreatment of membranes with 50 μM Sac1WT inhibits vesicle fusion through reduction of membrane PI(4)P levels.
The fact that preincubation of acceptor membranes with Sac1WT inhibited transport (Figure 4) suggested that PI(4)P is required on Golgi membranes. PI(4)P, however, may also be required on COPII vesicles for fusion with the Golgi compartment. To address this issue, we took advantage of the fact that budding is not inhibited by treatment with 50 μM Sac1WT (Figure 4B). For this experiment, microsomes containing [35S]gpαf were treated with 50 μM Sac1WT or buffer for 30 min at 12°C. Treated microsomes were then used to generate COPII vesicles, and vesicles were purified by flotation on Nycodenz density gradients (Barlowe, 1997). In two-stage fusion reactions, floated vesicles containing [35S]gpαf were chased into untreated acceptor membranes. Notably, vesicles generated in the presence of 50 μM Sac1WT chased into acceptor Golgi membranes as efficiently as control vesicles (Figure 5), indicating that PI(4)P is required only at the Golgi compartment for fusion with COPII vesicles. Together these findings demonstrate that PI(4)P is required on Golgi acceptor membranes for trans-SNARE complex assembly and subsequent fusion with COPII vesicles.
FIGURE 5:
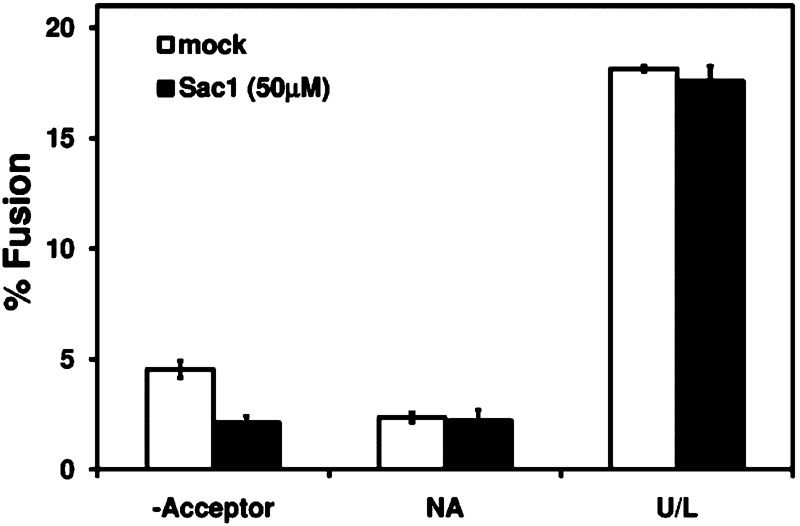
PI(4)P is required at the Golgi for fusion with COPII vesicles. Microsomes containing [35S]gpαf were treated with 50 μM Sac1WT or buffer (mock) at 12°C for 30 min. Treated microsomes were then used to generate COPII vesicles, and vesicles were purified by flotation on Nycodenz density gradients. Isolated vesicles were then incubated in two-stage fusion reactions with untreated semi-intact cell acceptor membranes as in Figure 1B.
ER-Golgi fusion factors in PI(4)P-dependent vesicle fusion
We hypothesized that PI(4)P on Golgi membranes interacted with protein fusion factors to mediate bilayer fusion. If PI(4)P recruits a cytosolic factor for COPII vesicle fusion with Golgi membranes, addition of crude cytosol might be expected to reverse PHWT inhibition. We performed two-stage fusion reactions with increasing concentrations of cytosol in the presence of 3, 5, or 7 μM PHWT. High concentrations of cytosol, however, did not rescue PHWT inhibition of COPII vesicle fusion with Golgi membranes (Supplemental Figure S2) suggesting that such a PI(4)P binding fusion factor was not present in our cytosol preparation.
Next, we treated semi-intact cells with inhibitory concentrations of PHWT, Sac1WT, or Sac1C392S and examined if known proteins involved in vesicle tethering and fusion were displaced from the membranes. We did not detect differences in membrane association of Ypt1p, Uso1p, Sly1p, or subunits of the conserved oligomeric Golgi (COG) and TRAPP complexes. In contrast, low levels of the SNARE disassembly chaperone Sec18p and the Grh1/Bug1 tethering complex were detected in the supernatant fraction after treatment of semi-intact cell membranes (Supplemental Figure S3). This observation suggested that PI(4)P levels influence membrane association of specific fusion factors. We note, however, that addition of a crude cytosol, which contained Grh1p, Bug1p, and Sec18 (Supplemental Figure S2), or the addition of purified Sec18p (see Figure 7 later in the paper) did not compete with PHWT for inhibition of transport.
To further explore the hypothesis that a PI(4)P binding factor promotes COPII vesicle fusion, we took a candidate-based approach to screen viable yeast deletion strains lacking proteins shown to have affinity for PI(4)P (Supplemental Table S1). Previous reports have characterized PI(4)P binding proteins in yeast based on an analysis of PH domain–containing proteins (Yu et al., 2004) and on lipid-binding assays using proteome chips (Zhu et al., 2001). We prepared semi-intact cell acceptor membranes from more than 20 different candidate strains, including the combined osh1Δ osh2Δ osh3Δ deletion mutant (Beh et al., 2001) to account for potential redundancy. In two-stage fusion reactions, all of the acceptor membranes tested displayed wild-type levels of fusion with the exception of gcs1Δ strain, which was reduced to 47% of the wild-type level (Supplemental Table S1). GCS1 encodes an Arf-GAP known to act in COPI-dependent Golgi to ER retrograde transport but is also reported to regulate assembly of ER-Golgi SNARE protein complexes (Poon et al., 1999; Schindler and Spang, 2007). Further experimentation will be required to determine whether the gcs1Δ mutation produces a direct or indirect effect on fusion of COPII vesicles with Golgi membranes. We note, however, that addition of crude cytosol to transport reactions, which contained Gcs1p, did not reverse PHWT inhibition of transport (Supplemental Figure S2).
We also considered the possibility that essential membrane-bound proteins could be involved in PI(4)P-dependent vesicle fusion at Golgi membranes. The ER-Golgi SNARE proteins could interact with PI(4)P as suggested by studies of other SNARE-dependent membrane fusion events (James et al., 2008; Lam et al., 2008; Mendonsa and Engebrecht, 2009; Mima and Wickner, 2009; Murray and Tamm, 2009). To investigate this hypothesis we characterized PI(4)P-dependent vesicle fusion in membranes overexpressing Sly1p and the four ER-Golgi SNARE proteins.
Overexpression of SNARE proteins and Sly1p alleviates the PI(4)P requirement in fusion
Because inhibition of PI(4)P function in ER-to-Golgi transport prevented formation of SNARE complexes but did not have a significant effect on vesicle tethering, we hypothesized that the role of PI(4)P in fusion might be required downstream of tethering but before or simultaneous with SNARE protein function. Thus, we reasoned that overexpression of the fusion machinery (all four ER-Golgi anterograde SNAREs and Sly1p) might yield acceptor membranes that are resistant or less sensitive to the PHWT inhibitor. We constructed the overproducer strains by transforming wild-type yeast with empty vectors (control) or with 2μ plasmids containing the BET1, BOS1, SEC22, SED5, and/or SLY1 under control of their endogenous promoters. Different combinations of SNARE overexpressing acceptor membranes were tested in two-stage fusion reactions. Overexpression of certain combinations produced modest levels of resistance to 3 or 5 μM PHWT (unpublished data). Overexpression of all four anterograde ER-Golgi SNAREs and SLY1, however, provided a higher level of resistance to PHWT compared to the partial overexpressor strains. Therefore we further characterized this overexpressor strain.
To determine fold overexpression and test whether overexpression influenced other ER-to-Golgi transport factors, we prepared semi-intact cell membranes from the strain overexpressing BET1, BOS1, SEC22, SED5, and SLY1 (from here on referred to as the overexpressor) for comparison with control strains. Experiments to assess the distribution of proteins contained in total, soluble, and membrane pellet fractions monitored the overexpressed proteins and a variety of other ER- and Golgi-localized markers (Figure 6A). We observed that Bet1p, Bos1p, Sec22p, Sed5p, and Sly1p were overexpressed three- to ninefold (compare total lanes), whereas the expression level and fractionation behavior of other marker proteins was not detectably altered.
FIGURE 6:

Characterization of membranes that overexpress anterograde ER-Golgi SNARE proteins and Sly1p. (A) Semi-intact cells from the overexpressor strain containing 2μ-BET1, 2μ-BOS1, 2μ-SEC22, 2μ-SED5, and 2μ-SLY1 (CBY3061) and the wild-type strain (CBY3062) were fractionated into soluble (S100) and pellet (P100) fractions for immunoblot analysis. (B) Budding reactions in which CBY3061 and CBY3062 microsomes were incubated in the absence (–) or presence (+) of COPII proteins for 30 min at 23°C. Immunoblots compare indicated proteins in budded vesicle fractions with one-tenth of total (T) budding reactions. Longer exposures (dark) are included for the Sec22p and Bet1p immunoblots.
Bet1p, Bos1p, Sec22p, and Sed5p are known to cycle between the ER and Golgi compartments and are efficiently packaged into COPII vesicles (Cao and Barlowe, 2000); therefore, we expected increased levels of these SNARE proteins in COPII vesicles from overexpressor membranes. Relative COPII packaging efficiencies were measured in budding assays using microsomes from the wild type and overexpressor strains. As shown in Figure 6B, the ER-Golgi SNARE proteins were 8- to 15-fold more abundant in overexpressor microsomes compared to wild type (total lanes). In budding assays, we observed increased levels of Bet1p (1.5-fold), Bos1p (2-fold), and Sec22p (5-fold) in COPII vesicles. Sed5p and Sly1p, however, were not increased in vesicles, which may be explained by the steady-state localization of Sed5p to cis-Golgi membranes (Wooding and Pelham, 1998). As a negative control, the ER resident Sec63p was not packaged into COPII vesicles. The packaging of known COPII vesicle proteins, such as Erv25p and Erv46p, was not changed. Taken together, these data show that COPII vesicles from overexpressor membranes contain more Bet1p, Bos1p, and Sec22p than wild-type membranes and that the distribution of other proteins involved in ER-to-Golgi transport was not sensitive to overexpression of Sly1p and the anterograde ER-Golgi SNARE proteins.
We next investigated how this overexpression condition influenced vesicle fusion. Addition of Sec18p to one-stage and two-stage fusion reactions has a minor effect on fusion rates, probably because of the abundant levels of Sec18p found in association with semi-intact cell membranes (Barlowe, 1997). We reasoned, however, that overexpressor membranes might require additional Sec18p to prime SNARE proteins for maximal fusion. Titration of recombinant Sec18p in one-stage transport reactions using overexpressor membranes indicated that ∼4 μM produced maximal stimulation (unpublished data), similar to previous titrations of Sec18p using washed Golgi membrane preparations (Barlowe, 1997). As shown in Figure 7A, we observed that transport with overexpressor and control membranes was indistinguishable in the absence of Sec18p. Addition of 4 μM Sec18p, however, stimulated transport ∼1.6-fold in overexpressor membranes, whereas wild-type membranes were not stimulated by Sec18p addition. Moreover, there was a decrease in sensitivity to inhibition by 3 μM PHWT in the overexpressor membranes compared to control, which was further enhanced by addition of 4 μM Sec18p. Interestingly, the level of transport in the overexpressor membranes in the presence of Sec18p and PHWT was similar to that of wild-type membranes in the absence of PHWT (Figure 7A).
FIGURE 7:

Overexpression of anterograde ER-Golgi SNAREs and Sly1p partially rescues PHWT inhibition. (A) Transport in wild-type (CBY3062) and overexpressor (CBY3061) semi-intact cells incubated for 20 min at 4°C with 3 μM PHWT and/or 4 μM Sec18p followed by transport for 1 h at 23°C. (B) Microsomes treated as in panel A were mock treated (NA) or incubated with COPII proteins for 30 min at 23°C to monitor budding. (C) Indicated semi-intact cell acceptor membranes were pretreated with PHWT and/or 4 μM Sec18p for 20 min at 4°C, then were incubated with COPII vesicles containing [35S]gpαf in the presence of fusion factors for 1 h at 23°C. The –Acpt condition indicates background fusion in the absence of added acceptor membranes. The data in panel C are plotted relative to control transport levels in Supplemental Figure S4.
Sec18p likely stimulates transport at the vesicle fusion stage by resolving abundant cis-SNARE complexes in overexpressor membranes, although there could be an influence on vesicle budding. To test this possibility, we performed [35S]gpαf budding reactions from wild type and overexpressor microsomes and found that there were no significant differences in gpαf budding between the wild type and overexpressor membranes with or without added Sec18p (Figure 7B). These data indicate that Sec18p treatment potentiates fusion of COPII vesicles and that transport in the presence of 4 μM Sec18p with the overexpressor membranes is significantly less sensitive to the PHWT inhibitor.
Finally, acceptor membranes from the overexpressor strain were used in two-stage fusion experiments using wild-type vesicles in the presence or absence of recombinant Sec18p and increasing concentrations of PHWT. We observed that PHWT produced the expected dose-dependent inhibition of transport when chasing wild-type vesicles into wild-type membranes (Figure 7C, compare to Figure 2A) and that addition of Sec18p to this reaction displayed partial rescue of the PHWT inhibition. Interestingly, transport was significantly less sensitive to increasing concentrations of PHWT inhibitor when wild-type vesicles were chased into overexpressor membranes, and the addition of Sec18p significantly enhanced this rescue (Figure 7C and Supplemental Figure S4). Together, these data indicate that increasing the amount of anterograde ER-Golgi SNARE proteins and Sly1p does not bypass but alleviates the requirement for PI(4)P on Golgi membranes for vesicle fusion.
PI(4)P is required for ER-to-Golgi transport in vivo
A central role for PI(4)P in Golgi secretory function was first reported in sec14 and pik1 mutants, which show reduced levels of cellular PI(4)P and kinetic defects in Golgi transport at restrictive temperatures (Hama et al., 1999; Walch-Solimena and Novick, 1999; Audhya et al., 2000). At steady state, Pik1p localizes to trans-Golgi and inner-nuclear compartments (Walch-Solimena and Novick, 1999; Strahl et al., 2005). In both yeast and mammalian cells, Pik1p-generated Golgi PI(4)P is required to recruit clathrin adaptor proteins for export from trans-Golgi membranes (D’Angelo et al., 2008; Clague et al., 2009). Therefore a reduction in PI(4)P at Golgi membranes caused by shifting pik1 mutants to the restrictive temperature strongly inhibits anterograde transport from the Golgi compartment. It should be noted, however, that ER-to-Golgi transport in these experiments was also kinetically delayed (Walch-Solimena and Novick, 1999; Audhya et al., 2000).
On the basis of these reports and our in vitro data, we hypothesized that a pool of Golgi-localized PI(4)P is required for proper trafficking into cis-Golgi compartments as well as out of the trans-Golgi complex. We also considered the possibility that a PI(4)P gradient across the Golgi complex, with highest requirements at trans-compartments, could cause inhibition of trans-Golgi export first, followed by inhibition of ER-to-Golgi transport. To test this hypothesis, we modified the protocol from Audhya and colleagues to increase the amount of time mutant cells were incubated at the restrictive temperature prior to initiating the pulse-chase experiment (Audhya et al., 2000). As shown in Figure 8A, pik1–83 cells displayed a dramatic block in maturation of the ER-form (p1) of carboxypeptidase Y (CPY) into the Golgi-form (p2) compared to wild-type cells, indicative of a block in ER-to-Golgi transport. Taken together, these results suggest that traffic out of the trans-Golgi complex is more sensitive to PI(4)P depletion (Audhya et al., 2000) but that continued deficiency in Pik1p activity blocks transport to the cis-Golgi compartment. In addition, it is also possible that PI(4)P could be required for additional transport steps within the Golgi complex.
FIGURE 8:

Depletion of PI(4)P in vivo causes a block in ER-to-Golgi transport and influences Golgi morphology. (A) Wild-type (SEY6210) and pik1–83 (AAY104) strains grown to a mid-log stage at 26°C were shifted to 37°C for 45 min, pulsed for 7 min at 37°C, and chased at 37°C for the indicated times. The ER (p1), Golgi (p2), and vacuolar (m; mature) forms of CPY are indicated. Note the block in maturation of p1 CPY in the mutant. (B–E) Wild-type (CBY2970, CBY2973, CBY2974) and pik1–83 (CBY2980, CBY2983, and CBY2984) cells expressing Sec63p-GFP (C), Sec21p-GFP (D), or Sec7p-GFP (E) were grown on YPD at 26°C to 0.4 OD600/ml and shifted to 37°C. (B) Cultures were analyzed for growth over time by absorbance. (C-E) pik1–83 cells retain ER (C), cis-Golgi (D), and late-Golgi (E) structures. Scale bars: 5 μm. A quantitative analysis of ER and Golgi morphologies observed in a population of cells at the 1-h time point is provided in Supplemental Figure S5.
Given the highly dynamic nature of the Golgi complex, we investigated an alternative possibility that loss of Pik1p caused a general disassembly of Golgi structures. To address this possibility, we compared growth rates and Golgi morphologies in pik1–83 and wild-type cells after shift to the restrictive temperature. As shown in Figure 8B, both strains displayed similar growth kinetics and remained metabolically active up to 1 h after the shift to 37°C. At the 2- and 3-h time points, however, the pik1–83 cells ceased growth. We note that our chase was initiated at a time point that roughly corresponded to the last chase time point of the published data (Audhya et al., 2000) and that the 5- and 15-min time points in our pulse-chase experiment corresponded to a total time at the restrictive temperature of 57 and 67 min, respectively, suggesting that the observed block in ER-to-Golgi transport was not caused by a general metabolic defect.
To inspect organelle morphologies after the temperature shift, we monitored ER, cis-Golgi, and trans-Golgi markers by fluorescence microscopy of Sec63p-GFP, Sec21p-GFP, and Sec7p-GFP, respectively. As shown in Figure 8, C–E, ER and Golgi structures were visible in mutant cells after 1 h and even 2 h at the restrictive temperature. Based on the time frame of our pulse-chase experiment, we quantified morphologies 1 h after shift to the restrictive temperature. ER morphology was not significantly altered at the 1-h time point as observed by a ∼15% reduction in the number of cells with wild-type structures (Figure 8C and Supplemental Figure S5C). In addition, we did not detect noticeable alterations in ER exit site morphology when Sec13p-GFP was observed in pik1–83 cells compared to wild type (unpublished data). Both cis- and trans-Golgi membrane markers, however, showed an increase in the number of smaller Golgi puncta accompanied by larger ring-like structures that presumably correspond to Berkeley bodies also observed in arf1Δ, gea1Δ, sec7–1, and pik1–101 mutants (Gaynor et al., 1998; Peyroche et al., 2001; Bruinsma et al., 2004; Karhinen and Makarow, 2004; Demmel et al., 2008). Quantification of Golgi morphologies demonstrated acute defects at trans-Golgi compartments with a ∼64% reduction the number of cells with wild-type trans-Golgi structures and moderate defects at cis-Golgi compartments with a ∼36% reduction in the number of cells with wild-type cis-Golgi structures (Figure 8, D and E, and Supplemental Figure S5C). These observations indicate that Pik1p deficiency does not cause a general loss of Golgi membranes and is consistent with a block in both export from trans-Golgi compartments and transport into cis-Golgi compartments.
DISCUSSION
The study of anterograde ER-to-Golgi traffic has yielded a comprehensive list of proteins and mechanisms involved in vesicle budding, cargo sorting, and fusion of COPII vesicles with the Golgi complex. We know little, however, about the role that specific lipids play in these processes. To advance our understanding, we performed the first screen for phosphoinositide requirements in anterograde ER-to-Golgi transport in yeast using a number of lipid-binding domains, enzymes, and enzyme inhibitors in cell-free budding, tethering, and fusion assays. We classified the effects of these compounds into distinct categories. The first group comprised the PI(4,5)P2 binding domains (ENTH and MED), which did not have a significant effect on ER-to-Golgi transport. A second group comprised inhibitors that targeted both budding and fusion and included the PLC inhibitor U73122, a PI-specific PLC, and the PI(3)P binding domain (FYVE). A third group comprised inhibitors that specifically targeted fusion of COPII vesicles and included the PI(4)P and DAG binding domains (PH and C1b, respectively).
PI(4)P is the most abundant cellular phosphoinositide (Lemmon, 2008) and is prevalent at the Golgi complex with additional pools detected at the plasma and nuclear membranes (D’Angelo et al., 2008). In both yeast and mammalian cells, the Golgi pool of PI(4)P is required for Golgi secretory function (D’Angelo et al., 2008), and was recently shown to be required for retention of cis-, medial-, and trans-Golgi glycosyltransferases in yeast (Tu et al., 2008; Wood et al., 2009). Golgi levels of PI(4)P are thought to be regulated by multiple activities, including the PI-transfer protein Sec14p (Mousley et al., 2007), the trans-Golgi localized PI 4-kinase Pik1p, and the ER-localized phosphoinositide-phosphatase Sac1p (Mayinger, 2009). Using a PI(4)P-specific PH domain lipid ligand and a recombinant Sac1p-phosphatase to reduce the levels of PI(4)P in vitro, we observed that PI(4)P was not involved in budding or tethering of COPII vesicles, but was required on Golgi acceptor membranes for fusion with COPII vesicles. More specifically, we found that inhibition of PI(4)P function reduced formation of the SNARE complexes that catalyze fusion of vesicles with Golgi membranes.
In vitro COPII vesicle budding from synthetic liposomes was shown to require low concentrations of PI(4)P and PI(4,5)P2 to achieve efficient binding of Sec23/Sec24 and to support vesicle budding (Matsuoka et al., 1998). Our data indicate that neither phosphoinositide is required for COPII vesicle budding on ER membranes, although other acidic phospholipids on the ER surface could satisfy this requirement. Alternatively, recruitment of Sec23/Sec24 by protein cargo molecules on the surface of ER membranes may be sufficient to assemble COPII coats for vesicle budding, as observed on synthetic proteoliposomes (Sato and Nakano, 2005).
In mammalian cells, evidence suggests that PI(4)P is required for remodeling ER exit sites and regulating COPII vesicle budding (Blumental-Perry et al., 2006). In vitro, we did not detect a requirement for PI(4)P in COPII budding from yeast ER, and in vivo pik1–83 yeast cells shifted to a restrictive temperature did not accumulate ER membranes or cause disassembly of Golgi compartments as would be expected if COPII budding were inhibited (Wooding and Pelham, 1998; Prinz et al., 2000; Heidtman et al., 2003; Kamena et al., 2008; Lorente-Rodriguez et al., 2009). This contrast may be explained by differences in the structural organization of ER export sites between yeast and mammalian cells. We speculate, however, that PI(4)P functions in both yeast and mammalian cells for fusion of COPII vesicles with Golgi membranes based on the evidence that, in both species, Golgi PI(4)P is required for Golgi secretory function (D’Angelo et al., 2008) and that Golgi PI(4)P levels are tightly regulated by PI 4-kinases and PI(4)P 4-phosphatases in response to growth conditions (Mayinger, 2009). In addition, PI(4)P was recently observed to significantly colocalize with cis-Golgi tethering factors in mammalian cells (Hammond et al., 2009). Further experimentation will be needed, however, to demonstrate whether PI(4)P mediates fusion at early Golgi compartments in mammalian cells.
On the basis of the collective findings, we hypothesize that a trafficking-dependent gradient of PI(4)P is established across the Golgi complex with its highest levels at the trans-Golgi and lowest levels at the cis-Golgi compartments. This PI(4)P gradient would be necessary for transport into and out of the Golgi complex. Because Pik1p and Sac1p have been implicated in regulating Golgi PI(4)P levels in yeast and mammalian cells (Mayinger, 2009), we propose that this gradient is regulated by Pik1p and Sac1p activities. This hypothesis is supported by our observation that pik1–83 cells shifted to a restrictive temperature for 1 h displayed morphological defects in trans-Golgi structures, accompanied by moderate defects in cis-Golgi structures. In this framework, inactivation of Pik1p would initially inhibit anterograde transport from trans-Golgi membranes (Walch-Solimena and Novick, 1999; Audhya et al., 2000). As retrograde transport continues, depletion of PI(4)P from cis-Golgi compartments would ultimately inhibit fusion of ER-derived vesicles with Golgi membranes. This proposal is consistent with our analysis of pik1–83 semi-intact cell membranes in vitro, which did not display thermosensitive defects in cell-free transport assays (Supplemental Figure S6). By increasing the time of pik1–83 inactivation compared to published reports (Audhya et al., 2000), we detected a strong block in ER-to-Golgi transport. It is interesting to note that mutations that mislocalize Sac1p to the Golgi also produce kinetic delays in ER-to-Golgi transport in addition to anterograde transport defects from the Golgi (Schorr et al., 2001; Konrad et al., 2002). Taken together, these data are consistent with an in vivo requirement for PI(4)P in fusion of COPII vesicles with the Golgi apparatus.
What is the molecular function of PI(4)P in fusion? We observed that overexpression of the fusion factors BET1, BOS1, SEC22, SED5, and SLY1 partially rescued PHWT-mediated inhibition of COPII vesicle fusion with Golgi membranes. This result indicates that PI(4)P may act concomitantly with SNARE protein function. We hypothesize that cis-Golgi localized PI(4)P interacts directly with SNARE proteins and increases their local concentrations at specific fusion sites to promote COPII vesicle fusion with the Golgi. There is precedent for this type of association. SNARE proteins are known to associate with membranes through phosphoinositide-binding domains, as is the case for the soluble SNARE Vam7p, which is recruited to the vacuolar membrane by PI(3)P (Cheever et al., 2001; Fratti et al., 2004). Alternatively, SNARE proteins often contain polybasic juxtamembrane regions, which directly interact with negatively charged lipids (i.e., phosphatidic acid and phosphoinositides), as is the case for yeast and mammalian exocytic SNARE proteins. Moreover, these SNARE–lipid interactions are required for secretion and viability (Van Komen et al., 2005; Vicogne et al., 2006; James et al., 2008; Lam et al., 2008; Mendonsa and Engebrecht, 2009; Murray and Tamm, 2009; Williams et al., 2009). Similar interactions between Golgi-localized PI(4)P and specific motifs in ER-Golgi SNARE proteins may be required to catalyze efficient fusion of ER-derived vesicles with Golgi membranes.
In summary, this study establishes a new requirement for PI(4)P in SNARE-dependent fusion of COPII vesicles with Golgi membranes. Given the level of conservation in ER-to-Golgi transport machinery, PI(4)P function in COPII vesicle fusion with the Golgi complex is likely to be conserved and therefore constitutes an additional mechanism to control transport through the early secretory pathway. A refined cell-free fusion assay and facile genetic approaches should allow us to determine if specific ER-to-Golgi SNARE proteins or potentially other fusion factors are recruited by PI(4)P to membrane fusion sites.
MATERIALS AND METHODS
Yeast strains and media
Yeast strains used in this study are listed in Table 1. Unless noted otherwise, cultures were grown at 26°C (for temperature-sensitive mutant stains) or 30°C in rich medium (YPD: 1% yeast extract, 2% peptone, 2% dextrose) or in minimal medium (YMD: 0.7% yeast nitrogen base without amino acids, 2% dextrose, and appropriate amino acid supplements). Standard yeast (Sherman, 1991) and cloning protocols (Ausubel et al., 1987) were used. Yeast cells were transformed by the lithium acetate method (Elble, 1992).
TABLE 1: Strains used in the study.
| Strain | Genotype | Source |
|---|---|---|
| AAY104 | SEY6210; pik1Δ::HIS3 [pRS314-pik1–83] | Audhya et al., 2000 |
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Brachmann et al., 1998 |
| CBY1584 | BY4742; sec22Δ::KANR [pRS313-SEC22(D153C)] | Flanagan and Barlowe, 2006 |
| CBY1676 | BY4742; bet1Δ::KANR [pRS315-BET1(I83C)] | Flanagan and Barlowe, 2006 |
| CBY2970 | SEY6210; [pSEC63-GFP, URA3, CEN (pJK59)] | This study |
| CBY2973 | SEY6210; SEC21-GFPx3 | This study |
| CBY2974 | SEY6210; SEC7-GFPx3 | This study |
| CBY2980 | AAY104; [pSEC63-GFP, URA3, CEN (pJK59)] | This study |
| CBY2983 | AAY104; SEC21-GFPx3 | This study |
| CBY2984 | AAY104; SEC7-GFPx3 | This study |
| CBY 3061 | FY834; [pRS423-SEC22, HIS3] [pRS424-SED5, TRP1] [YEp511-BET1, LEU2] [pRS426-BOS1, URA3] [pRS327-SLY1, LYS2] | This study |
| CBY3062 | FY834; [pRS423] [pRS424] [pRS425] [pRS426] [pRS327] | This study |
| FY834 | MATα his3Δ200 ura3–52 leu2Δ1 lys2Δ202 trp1Δ63 | Winston et al., 1995 |
| SEY6210 | MATα leu2–3,112 ura3–52 his3-Δ200 trp1-Δ901 lys2–801 suc2-Δ9 | Robinson et al., 1988 |
Plasmids pRS426-BOS1 and YEp511-BET1(LEU2) have been described (Dascher et al., 1991; Duden et al., 1994). Plasmid pRS423-SEC22 was constructed by subcloning a ∼1.5-kb BamHI-EcoRV fragment of YEp511-SEC22 (Dascher et al., 1991) into pRS423 (Christianson et al., 1992). Plasmids pRS424-SED5 and pRS327-SLY1 were generated by amplifying BY4742 genomic DNA including 300–500 bp upstream and downstream sequences and cloning into pRS424 (Christianson et al., 1992) or pRS327 (Eriksson et al., 2004). Plasmids generated in this study were confirmed by sequencing. FY834 was transformed with appropriate empty vectors or with the BET1, BOS1, SEC22, SED5, and SLY1 2μ vectors to generate CBY3062 and CBY3061, respectively. Overexpression was confirmed by immunoblot (Figure 6).
Wild-type (SEY6210) and pik1–83 (AAY104) strains were transformed with pJK59 [CEN SEC63-GFP URA3] (Prinz et al., 2000), or a chromosomal GFPx3 tag was introduced into the SEC21 and SEC7 loci as described (Rossanese et al., 2001) to generate CBY2970, CBY2973, CBY2974, CBY2980, CBY2983, and CBY2984.
Reagents
Purified lipid-binding domains GST-ENTH (Rosenthal et al., 1999), C1b (C-terminal glutathione S-transferase [GST] cleaved) (Johnson et al., 2000), GST-FYVE2 (Gillooly et al., 2000), MED (Wang et al., 2001), HIS6-Sec18p (Thorngren et al., 2004), and U73122 were the gifts of Bill Wickner (Dartmouth).
Plasmids expressing the PH domain of human Fapp1 (PI(4)P adaptor protein-1; residues 1–99) as a wild-type construct (PHWT) (Dowler et al., 2000) or as the W15N, R18K mutant (PHN,K) (Weixel et al., 2008) cloned as N-terminal fusions with maltose binding protein (MBP) were the gifts of Christopher Stroupe (Dartmouth). MBP fusion proteins were purified according to New England Biolabs (Ipswich, MA) recommendations and dialyzed into buffer 88 (20 mM HEPES-KOH pH 7.0, 250 mM sorbitol, 150 mM KOAc, 5 mM MgOAc) without sorbitol. Plasmids expressing the soluble domain of Sac1p (residues 1–507) as a wild-type HIS6-Sac1 construct (Sac1WT) or the catalytically inactive C392S mutant (Sac1C392S) were the gifts of Gregory Taylor (University of Nebraska Medical Center). His6 fusion proteins were purified according to QIAGEN (Valencia, CA) recommendations and dialyzed into buffer 88 without sorbitol containing 0.5 mM dithiothreitol (DTT). The activity of these Sac1 recombinant proteins was confirmed using synthetic liposomes as described (Maehama et al., 2000) and using semi-intact cell membranes (Supplemental Figure S1B).
PI-PLC was purchased from Invitrogen (Carlsbad, CA). Nycodenz, deuterium oxide (D2O), and methylene blue were purchased from Sigma Chemical (St. Louis, MO). Coomassie brilliant blue R-250 was purchased from Bio-Rad (Hercules, CA). All membrane transport inhibitors were diluted with buffer 88 to the appropriate concentration and screened for inhibition of in vitro transport at the indicated concentrations.
Lipids and liposome-binding assay
POPC, POPE, POPS, soy PI (Avanti Polar Lipids, Alabaster, AL), Rhodamine-labeled DHPE (Invitrogen, Carlsbad, CA) and diC16 D-myo-PI(4)P (Echelon Biosciences, Salt Lake City, UT), were purchased from the indicated sources. Liposomes were prepared as described (Patki et al., 1997) with minor modifications. Lipids were mixed in glass tubes at the following molar ratios of 50% POPC, 29.75% POPE, 0.25% Rhodamine labeled DHPE, 10% POPS, and 10% soy PI or diC16 D-myo-PI(4)P, then dried to a thin film under a stream of nitrogen gas and resuspended to a final concentration of 2 mg total lipid / ml Sac1 phosphatase buffer (Maehama et al., 2000): 100 mM NaOAc, 50 mM Bis-Tris, 50 mM Tris-HCl, pH 6.0, 2 mM DTT. Resuspended lipids were sonicated for 5 min at 30°C, separated into 50-μl aliquots, flash-frozen, and stored at −70°C.
Liposome-binding assays were as described (Loyet et al., 1998) with minor modifications. Sac1WT and Sac1C392S proteins were precleared by centrifugation at 270,000 × g for 10 min. Binding reactions (70 μl) containing 3 μM precleared Sac1WT or Sac1C392S protein in Sac1 phosphatase buffer with or without 50 μl of PI or PI(4)P liposomes were incubated for 30 min at 30°C. Bovine serum albumin (BSA) was added to 1 mg/ml to minimize nonspecific binding. Liposomes were recovered by sedimentation at 270,000 × g for 10 min, and an aliquot of the supernatant was collected. Pellets were washed with excess Sac1 phosphatase buffer containing BSA at 1 mg/ml and resuspended to the initial volume with wash buffer. Equivalent volumes were resolved on 7.5% SDS–PAGE gels and stained with 0.05% Coomassie brilliant blue R-250. Densitometric analysis was performed using ImageJ (Rasband, 1997–2005). Fluorometric quantification of Rhodamine labeled DHPE (λex/λem 560 nm/580 nm) indicated that 87% ± 6.7% (SD) of the starting liposomes were recovered in pellet fractions.
Antibodies and immunoblotting
Antibodies against CPY (Rothblatt et al., 1989); Grh1p (Behnia et al., 2007); Kar2p (Brodsky and Schekman, 1993); Och1p, Erv46p (Otte et al., 2001); Sly1p (Flanagan and Barlowe, 2006); Sec22p (Liu and Barlowe, 2002); Bet1p, Bos1p (Sogaard et al., 1994); α1,6-mannose linkages, Sed5p (Cao et al., 1998); Sec63p (Feldheim et al., 1992); Uso1p, Bet3p, Cog2p/Sec35p (Ballew et al., 2005); Erv25p (Belden and Barlowe, 1996); Gdi1p (Garrett et al., 1994); Sec13p (Salama et al., 1993); Ypt1p (Rexach et al., 1994); Sec17p, Sec18p (Haas and Wickner, 1996); and Sec61p (Stirling et al., 1992) have been described. Polyclonal antibodies were raised against full-length untagged Bug1p as follows: Bug1p was cloned into an Impact-CN protein expression vector, purified according to the manufacturer’s specifications (New England Biolabs), and used to immunize rabbits by standard procedures (Covance, Denver, PA). For Western blots, anti-Bug1p serum was diluted 1:1,000. Monoclonal anti-5HIS was purchased from QIAGEN. Western blot analysis was performed with nitrocellulose membranes by using the SuperSignal West Pico chemiluminescent substrate (Pierce Chemical, Rockford, IL) and developed with a UVP Bioimaging System (Upland, CA). Densitometric analysis of immunoblots was performed using ImageJ (Rasband, 1997–2005).
In vitro vesicle budding, tethering, and transport assays
Yeast semi-intact cell membranes and ER microsomes were prepared as described (Baker et al., 1988; Wuestehube and Schekman, 1992). Prior to assembling in vitro reactions, semi-intact cell membranes or microsomes were incubated with indicated inhibitors and/or recombinant Sec18p for 20 min at 4°C, or with Sac1WT for 30 min at 12°C as described in the text and figure legends. In vitro assays to measure overall transport, vesicle budding, vesicle tethering, two-stage fusion reactions, and SNARE cross-linking assays using [35S]gpαf were performed as described (Barlowe, 1997; Cao et al., 1998; Flanagan and Barlowe, 2006).
For vesicle purification on Nycodenz gradients, microsomes containing [35S]gpαf were treated with 50 μM Sac1WT for 30 min at 12°C and then used to generate COPII vesicles (Cao et al., 1998). ER-derived vesicles were purified by flotation on Nycodenz density gradients prepared in D2O to strip peripherally associated membrane proteins, and then floated vesicles were chased into washed semi-intact cell membranes at 23°C for 1 h (Barlowe, 1997).
Pulse-chase experiments
Wild type (SEY6210) and pik1–83 (AAY104) strains were grown at 26°C in minimal medium to 0.4 OD600/ml (Beckman DU®-64 Spectrophotometer; Fullerton, CA), and then shifted to 37°C for 25 min. These cultures were washed in 37°C-warm pulse-chase medium lacking cysteine, methionine, and sulfate, concentrated to 4 OD600/ml (Audhya et al., 2000) and further incubated at 37°C. In total, cultures were shifted to 37°C for 45 min prior to the pulse. Cultures were pulsed for 7 min by the addition of EasyTag EXPRESS35S Protein Labeling Mix (Perkin Elmer-Cetus, Waltham, MA) to a final concentration of ∼25 μCi/OD600. Cells were chased by the addition of cysteine, methionine, and (NH4)2SO4 at a final concentration of 2.5 mM. Sample collection, lysis, and immunoprecipitations were performed as described (Belden and Barlowe, 1996).
Microscopy
Samples at the indicated time points were concentrated to 14 OD600/ml in the presence of 0.008% methylene blue to identify live versus dead cells (Teparic et al., 2004), and 3 ml was applied to slides. Fluorescent and bright field images were acquired using a microscope (model BX51; Olympus) equipped with a 100-W mercury arc lamp, Plan Apochromat 60× objective (1.4 NA) and a Sensicam QE CCD camera (Cooke Inc., Romulus, MI). The camera and microscope were controlled by the IP Lab system (Scanalytics, Sunnyvale, CA). All images were processed in Openlab software (Improvision, Lexington, MA).
Supplementary Material
Acknowledgments
We thank Joshua Wilson, Polina Shindiapina, Christopher Hickey, and Russell Monds for scientific discussions; Christine Bentivoglio for purifying Bug1p used for antibody production; Bill Wickner for the many reagents and microscope access; Duane Compton for help with image processing; Christopher Stroupe (University of Virginia) and Gregory Taylor (University of Nebraska Medical Center) for bacterial constructs; Jasper Rine (University of California at Berkeley) and Scott Emr (Cornell University) for yeast strains; and Anne Spang (University of Basel) for Gcs1p anti-serum This work was supported by National Institutes of Health Grant GM052549.
Abbreviations used:
- COPII
coat protein complex II
- SNARE
soluble n-ethylmaleimide sensitive factor adaptor protein receptor
- TRAPP
transport protein particle
- gpαf
glyco-pro-alpha-factor
- DAG
diacylglycerol
- PI
phosphatidylinositol
- PI(4)P
phosphatidylinositol-4-phosphate
- PH
pleckstrin homology
- FYVE domain
zinc finger domain found in Fab1p
- YOTB
Vac1p and EEA1
- ENTH
Epsin N-terminal homology
- MED
marcks (myristoylated alanine-rich C-Kinase substrate)-effector domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-04-0317) on November 30, 2010.
REFERENCES
- Audhya A, Foti M, Emr SD. Distinct roles for the yeast phosphatidylinositol 4-kinases, Stt4p and Pik1p, in secretion, cell growth, and organelle membrane dynamics. Mol Biol Cell. 2000;11:2673–2689. doi: 10.1091/mbc.11.8.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel RM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. New York: Greene Publishing Associates and Wiley-Interscience; 1987. Current Protocols in Molecular Biology. [Google Scholar]
- Baker D, Hicke L, Rexach M, Schleyer M, Schekman R. Reconstitution of SEC gene product-dependent intercompartmental protein transport. Cell. 1988;54:335–344. doi: 10.1016/0092-8674(88)90196-1. [DOI] [PubMed] [Google Scholar]
- Ballew N, Liu Y, Barlowe C. A Rab requirement is not bypassed in SLY1–20 suppression. Mol Biol Cell. 2005;16:1839–1849. doi: 10.1091/mbc.E04-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C. Coupled ER to Golgi transport reconstituted with purified cytosolic proteins. J Cell Biol. 1997;139:1097–1108. doi: 10.1083/jcb.139.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beh CT, Cool L, Phillips J, Rine J. Overlapping functions of the yeast oxysterol-binding protein homologues. Genetics. 2001;157:1117–1140. doi: 10.1093/genetics/157.3.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnia R, Barr FA, Flanagan JJ, Barlowe C, Munro S. The yeast orthologue of GRASP65 forms a complex with a coiled-coil protein that contributes to ER to Golgi traffic. J Cell Biol. 2007;176:255–261. doi: 10.1083/jcb.200607151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belden WJ, Barlowe C. Erv25p, a component of COPII-coated vesicles, forms a complex with Emp24p that is required for efficient endoplasmic reticulum to Golgi transport. J Biol Chem. 1996;271:26939–26946. doi: 10.1074/jbc.271.43.26939. [DOI] [PubMed] [Google Scholar]
- Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ, Bunting S. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther. 1990;255:756–768. [PubMed] [Google Scholar]
- Blumental-Perry A, Haney CJ, Weixel KM, Watkins SC, Weisz OA, Aridor M. Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev Cell. 2006;11:671–682. doi: 10.1016/j.devcel.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Schekman R. A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J Cell Biol. 1993;123:1355–1363. doi: 10.1083/jcb.123.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma P, Spelbrink RG, Nothwehr SF. Retrograde transport of the mannosyltransferase Och1p to the early Golgi requires a component of the COG transport complex. J Biol Chem. 2004;279:39814–39823. doi: 10.1074/jbc.M405500200. [DOI] [PubMed] [Google Scholar]
- Cao X, Ballew N, Barlowe C. Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO J. 1998;17:2156–2165. doi: 10.1093/emboj/17.8.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Barlowe C. Asymmetric requirements for a Rab GTPase and SNARE proteins in fusion of COPII vesicles with acceptor membranes. J Cell Biol. 2000;149:55–66. doi: 10.1083/jcb.149.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M. Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol. 2001;3:613–618. doi: 10.1038/35083000. [DOI] [PubMed] [Google Scholar]
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- Clague MJ, Urbe S, de Lartigue J. Phosphoinositides and the endocytic pathway. Exp Cell Res. 2009;315:1627–1631. doi: 10.1016/j.yexcr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- D’Angelo G, Vicinanza M, Di Campli A, De Matteis MA. The multiple roles of PtdIns(4)P—not just the precursor of PtdIns(4,5)P2. J Cell Sci. 2008;121:1955–1963. doi: 10.1242/jcs.023630. [DOI] [PubMed] [Google Scholar]
- Dascher C, Ossig R, Gallwitz D, Schmitt HD. Identification and structure of four yeast genes (SLY) that are able to suppress the functional loss of YPT1, a member of the RAS superfamily. Mol Cell Biol. 1991;11:872–885. doi: 10.1128/mcb.11.2.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmel L, et al. The clathrin adaptor Gga2p is a phosphatidylinositol 4-phosphate effector at the Golgi exit. Mol Biol Cell. 2008;19:1991–2002. doi: 10.1091/mbc.E06-10-0937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowler S, Currie RA, Campbell DG, Deak M, Kular G, Downes CP, Alessi DR. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem J. 2000;351:19–31. doi: 10.1042/0264-6021:3510019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duden R, Hosobuchi M, Hamamoto S, Winey M, Byers B, Schekman R. Yeast beta-and beta′-coat proteins (COP). Two coatomer subunits essential for endoplasmic reticulum-to-Golgi protein traffic. J Biol Chem. 1994;269:24486–24495. [PubMed] [Google Scholar]
- Elble R. A simple and efficient procedure for transformation of yeasts. Biotechniques. 1992;13:18–20. [PubMed] [Google Scholar]
- Eriksson P, Thomas LR, Thorburn A, Stillman DJ. pRS yeast vectors with a LYS2 marker. Biotechniques. 2004;36:212–213. doi: 10.2144/04362BM01. [DOI] [PubMed] [Google Scholar]
- Feldheim D, Rothblatt J, Schekman R. Topology and functional domains of Sec63p, an endoplasmic reticulum membrane protein required for secretory protein translocation. Mol Cell Biol. 1992;12:3288–3296. doi: 10.1128/mcb.12.7.3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JJ, Barlowe C. Cysteine-disulfide cross-linking to monitor SNARE complex assembly during endoplasmic reticulum-Golgi transport. J Biol Chem. 2006;281:2281–2288. doi: 10.1074/jbc.M511695200. [DOI] [PubMed] [Google Scholar]
- Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W. Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol. 2004;167:1087–1098. doi: 10.1083/jcb.200409068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett MD, Zahner JE, Cheney CM, Novick PJ. GDI1 encodes a GDP dissociation inhibitor that plays an essential role in the yeast secretory pathway. EMBO J. 1994;13:1718–1728. doi: 10.1002/j.1460-2075.1994.tb06436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor EC, Chen CY, Emr SD, Graham TR. ARF is required for maintenance of yeast Golgi and endosome structure and function. Mol Biol Cell. 1998;9:653–670. doi: 10.1091/mbc.9.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000;19:4577–4588. doi: 10.1093/emboj/19.17.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Fernandez JC, Corbalan-Garcia S. Diacylglycerols, multivalent membrane modulators. Chem Phys Lipids. 2007;148:1–25. doi: 10.1016/j.chemphyslip.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Haas A, Wickner W. Homotypic vacuole fusion requires Sec17p (yeast alpha-SNAP) and Sec18p (yeast NSF). EMBO J. 1996;15:3296–3305. [PMC free article] [PubMed] [Google Scholar]
- Hama H, Schnieders EA, Thorner J, Takemoto JY, DeWald DB. Direct involvement of phosphatidylinositol 4-phosphate in secretion in the yeast Saccharomyces cerevisiae. J Biol Chem. 1999;274:34294–34300. doi: 10.1074/jbc.274.48.34294. [DOI] [PubMed] [Google Scholar]
- Hammond GR, Schiavo G, Irvine RF. Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem J. 2009;422:23–35. doi: 10.1042/BJ20090428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidtman M, Chen CZ, Collins RN, Barlowe C. A role for Yip1p in COPII vesicle biogenesis. J Cell Biol. 2003;163:57–69. doi: 10.1083/jcb.200306118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James DJ, Khodthong C, Kowalchyk JA, Martin TF. Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J Cell Biol. 2008;182:355–366. doi: 10.1083/jcb.200801056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, Giorgione J, Newton AC. The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochemistry. 2000;39:11360–11369. doi: 10.1021/bi000902c. [DOI] [PubMed] [Google Scholar]
- Jun Y, Fratti RA, Wickner W. Diacylglycerol and its formation by phospholipase C regulate Rab- and SNARE-dependent yeast vacuole fusion. J Biol Chem. 2004;279:53186–53195. doi: 10.1074/jbc.M411363200. [DOI] [PubMed] [Google Scholar]
- Jun Y, Thorngren N, Starai VJ, Fratti RA, Collins K, Wickner W. Reversible, cooperative reactions of yeast vacuole docking. EMBO J. 2006;25:5260–5269. doi: 10.1038/sj.emboj.7601413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamena F, Diefenbacher M, Kilchert C, Schwarz H, Spang A. Ypt1p is essential for retrograde Golgi-ER transport and for Golgi maintenance in S. cerevisiae. J Cell Sci. 2008;121:1293–1302. doi: 10.1242/jcs.016998. [DOI] [PubMed] [Google Scholar]
- Karhinen L, Makarow M. Activity of recycling Golgi mannosyltransferases in the yeast endoplasmic reticulum. J Cell Sci. 2004;117:351–358. doi: 10.1242/jcs.00858. [DOI] [PubMed] [Google Scholar]
- Konrad G, Schlecker T, Faulhammer F, Mayinger P. Retention of the yeast Sac1p phosphatase in the endoplasmic reticulum causes distinct changes in cellular phosphoinositide levels and stimulates microsomal ATP transport. J Biol Chem. 2002;277:10547–10554. doi: 10.1074/jbc.M200090200. [DOI] [PubMed] [Google Scholar]
- Lam AD, Tryoen-Toth P, Tsai B, Vitale N, Stuenkel EL. SNARE-catalyzed fusion events are regulated by Syntaxin1A-lipid interactions. Mol Biol Cell. 2008;19:485–497. doi: 10.1091/mbc.E07-02-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Liu Y, Barlowe C. Analysis of Sec22p in endoplasmic reticulum/Golgi transport reveals cellular redundancy in SNARE protein function. Mol Biol Cell. 2002;13:3314–3324. doi: 10.1091/mbc.E02-04-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente-Rodriguez A, Heidtman M, Barlowe C. Multicopy suppressor analysis of thermosensitive YIP1 alleles implicates GOT1 in transport from the ER. J Cell Sci. 2009;122:1540–1550. doi: 10.1242/jcs.042457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyet KM, Kowalchyk JA, Chaudhary A, Chen J, Prestwich GD, Martin TF. Specific binding of phosphatidylinositol 4,5-bisphosphate to calcium-dependent activator protein for secretion (CAPS), a potential phosphoinositide effector protein for regulated exocytosis. J Biol Chem. 1998;273:8337–8343. doi: 10.1074/jbc.273.14.8337. [DOI] [PubMed] [Google Scholar]
- Maehama T, Taylor GS, Slama JT, Dixon JE. A sensitive assay for phosphoinositide phosphatases. Anal Biochem. 2000;279:248–250. doi: 10.1006/abio.2000.4497. [DOI] [PubMed] [Google Scholar]
- Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- Mayinger P. Regulation of Golgi function via phosphoinositide lipids. Semin Cell Dev Biol. 2009;20:793–800. doi: 10.1016/j.semcdb.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonsa R, Engebrecht J. Phosphatidylinositol-4,5-bisphosphate and phospholipase D-generated phosphatidic acid specify SNARE-mediated vesicle fusion for prospore membrane formation. Eukaryot Cell. 2009;8:1094–1105. doi: 10.1128/EC.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima J, Wickner W. Phosphoinositides and SNARE chaperones synergistically assemble and remodel SNARE complexes for membrane fusion. Proc Natl Acad Sci USA. 2009;106:16191–16196. doi: 10.1073/pnas.0908694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousley CJ, Tyeryar KR, Vincent-Pope P, Bankaitis VA. The Sec14-superfamily and the regulatory interface between phospholipid metabolism and membrane trafficking. Biochim Biophys Acta. 2007;1771:727–736. doi: 10.1016/j.bbalip.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulet X, Templer RH, Woscholski R, Ces O. Evidence that phosphatidylinositol promotes curved membrane interfaces. Langmuir. 2008;24:8443–8447. doi: 10.1021/la801114n. [DOI] [PubMed] [Google Scholar]
- Murray DH, Tamm LK. Clustering of syntaxin-1A in model membranes is modulated by phosphatidylinositol 4,5-bisphosphate and cholesterol. Biochemistry. 2009;48:4617–4625. doi: 10.1021/bi9003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte S, Belden WJ, Heidtman M, Liu J, Jensen ON, Barlowe C. Erv41p and Erv46p: new components of COPII vesicles involved in transport between the ER and Golgi complex. J Cell Biol. 2001;152:503–518. doi: 10.1083/jcb.152.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patki V, Virbasius J, Lane WS, Toh BH, Shpetner HS, Corvera S. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1997;94:7326–7330. doi: 10.1073/pnas.94.14.7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyroche A, Courbeyrette R, Rambourg A, Jackson CL. The ARF exchange factors Gea1p and Gea2p regulate Golgi structure and function in yeast. J Cell Sci. 2001;114:2241–2253. doi: 10.1242/jcs.114.12.2241. [DOI] [PubMed] [Google Scholar]
- Poon PP, Cassel D, Spang A, Rotman M, Pick E, Singer RA, Johnston GC. Retrograde transport from the yeast Golgi is mediated by two ARF GAP proteins with overlapping function. EMBO J. 1999;18:555–564. doi: 10.1093/emboj/18.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA, Grzyb L, Veenhuis M, Kahana JA, Silver PA, Rapoport TA. Mutants affecting the structure of the cortical endoplasmic reticulum in Saccharomyces cerevisiae. J Cell Biol. 2000;150:461–474. doi: 10.1083/jcb.150.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband WS. 1997–2005. ImageJ, Bethesda, MD: National Institutes of Health.
- Rexach MF, Latterich M, Schekman RW. Characteristics of endoplasmic reticulum-derived transport vesicles. J Cell Biol. 1994;126:1133–1148. doi: 10.1083/jcb.126.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta LM, Emr SD. Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal JA, Chen H, Slepnev VI, Pellegrini L, Salcini AE, Di Fiore PP, De Camilli P. The epsins define a family of proteins that interact with components of the clathrin coat and contain a new protein module. J Biol Chem. 1999;274:33959–33965. doi: 10.1074/jbc.274.48.33959. [DOI] [PubMed] [Google Scholar]
- Ross TS, Wang FP, Majerus PW. Mammalian cells that express Bacillus cereus phosphatidylinositol-specific phospholipase C have increased levels of inositol cyclic 1:2-phosphate, inositol 1-phosphate, and inositol 2-phosphate. J Biol Chem. 1992;267:19919–19923. [PubMed] [Google Scholar]
- Rossanese OW, Reinke CA, Bevis BJ, Hammond AT, Sears IB, O’Connor J, Glick BS. A role for actin, Cdc1p, and Myo2p in the inheritance of late golgi elements in Saccharomyces cerevisiae. J Cell Biol. 2001;153:47–62. doi: 10.1083/jcb.153.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothblatt JA, Deshaies RJ, Sanders SL, Daum G, Schekman R. Multiple genes are required for proper insertion of secretory proteins into the endoplasmic reticulum in yeast. J Cell Biol. 1989;109:2641–2652. doi: 10.1083/jcb.109.6.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama NR, Yeung T, Schekman RW. The Sec13p complex and reconstitution of vesicle budding from the ER with purified cytosolic proteins. EMBO J. 1993;12:4073–4082. doi: 10.1002/j.1460-2075.1993.tb06091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Nakano A. Reconstitution of cargo-dependent COPII coat assembly on proteoliposomes. Methods Enzymol. 2005;404:83–94. doi: 10.1016/S0076-6879(05)04009-7. [DOI] [PubMed] [Google Scholar]
- Sato K, Nakano A. Mechanisms of COPII vesicle formation and protein sorting. FEBS Lett. 2007;581:2076–2082. doi: 10.1016/j.febslet.2007.01.091. [DOI] [PubMed] [Google Scholar]
- Schindler C, Spang A. Interaction of SNAREs with ArfGAPs precedes recruitment of Sec18p/NSF. Mol Biol Cell. 2007;18:2852–2863. doi: 10.1091/mbc.E06-08-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorr M, Then A, Tahirovic S, Hug N, Mayinger P. The phosphoinositide phosphatase Sac1p controls trafficking of the yeast Chs3p chitin synthase. Curr Biol. 2001;11:1421–1426. doi: 10.1016/s0960-9822(01)00449-3. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Sogaard M, Tani K, Ye RR, Geromanos S, Tempst P, Kirchhausen T, Rothman JE, Sollner T. A rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell. 1994;78:937–948. doi: 10.1016/0092-8674(94)90270-4. [DOI] [PubMed] [Google Scholar]
- Stahelin RV, Karathanassis D, Murray D, Williams RL, Cho W. Structural and membrane binding analysis of the Phox homology domain of Bem1p: basis of phosphatidylinositol 4-phosphate specificity. J Biol Chem. 2007;282:25737–25747. doi: 10.1074/jbc.M702861200. [DOI] [PubMed] [Google Scholar]
- Stirling CJ, Rothblatt J, Hosobuchi M, Deshaies R, Schekman R. Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol Biol Cell. 1992;3:129–142. doi: 10.1091/mbc.3.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl T, Hama H, DeWald DB, Thorner J. Yeast phosphatidylinositol 4-kinase, Pik1, has essential roles at the Golgi and in the nucleus. J Cell Biol. 2005;171:967–979. doi: 10.1083/jcb.200504104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl T, Thorner J. Synthesis and function of membrane phosphoinositides in budding yeast, Saccharomyces cerevisiae. Biochim Biophys Acta. 2007;1771:353–404. doi: 10.1016/j.bbalip.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztul E, Lupashin V. Role of tethering factors in secretory membrane traffic. Am J Physiol Cell Physiol. 2006;290:C11–26. doi: 10.1152/ajpcell.00293.2005. [DOI] [PubMed] [Google Scholar]
- Teparic R, Stuparevic I, Mrsa V. Increased mortality of Saccharomyces cerevisiae cell wall protein mutants. Microbiology. 2004;150:3145–3150. doi: 10.1099/mic.0.27296-0. [DOI] [PubMed] [Google Scholar]
- Thorngren N, Collins KM, Fratti RA, Wickner W, Merz AJ. A soluble SNARE drives rapid docking, bypassing ATP and Sec17/18p for vacuole fusion. EMBO J. 2004;23:2765–2776. doi: 10.1038/sj.emboj.7600286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu L, Tai WC, Chen L, Banfield DK. Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science. 2008;321:404–407. doi: 10.1126/science.1159411. [DOI] [PubMed] [Google Scholar]
- Van Komen JS, Bai X, Rodkey TL, Schaub J, McNew JA. The polybasic juxtamembrane region of Sso1p is required for SNARE function in vivo. Eukaryot Cell. 2005;4:2017–2028. doi: 10.1128/EC.4.12.2017-2028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicogne J, Vollenweider D, Smith JR, Huang P, Frohman MA, Pessin JE. Asymmetric phospholipid distribution drives in vitro reconstituted SNARE-dependent membrane fusion. Proc Natl Acad Sci USA. 2006;103:14761–14766. doi: 10.1073/pnas.0606881103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walch-Solimena C, Novick P. The yeast phosphatidylinositol-4-OH kinase pik1 regulates secretion at the Golgi. Nat Cell Biol. 1999;1:523–525. doi: 10.1038/70319. [DOI] [PubMed] [Google Scholar]
- Wang J, Arbuzova A, Hangyas-Mihalyne G, McLaughlin S. The effector domain of myristoylated alanine-rich C kinase substrate binds strongly to phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2001;276:5012–5019. doi: 10.1074/jbc.M008355200. [DOI] [PubMed] [Google Scholar]
- Weixel KM, Blumental-Perry A, Watkins SC, Aridor M, Weisz OA. Distinct Golgi populations of phosphatidylinositol 4-phosphate regulated by phosphatidylinositol 4-kinases. J Biol Chem 280. 2008:10501–10508. doi: 10.1074/jbc.M414304200. [DOI] [PubMed] [Google Scholar]
- Wickner W, Schekman R. Membrane fusion. Nat Struct Mol Biol. 2008;15:658–664. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D, Vicogne J, Zaitseva I, McLaughlin S, Pessin JE. Evidence that electrostatic interactions between VAMP2 and acidic phospholipids may modulate the fusion of transport vesicles with the plasma membrane. Mol Biol Cell. 2009;20:4910–4919. doi: 10.1091/mbc.E09-04-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston F, Dollard C, Ricupero-Hovasse SL. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast. 1995;11:53–55. doi: 10.1002/yea.320110107. [DOI] [PubMed] [Google Scholar]
- Wood CS, Schmitz KR, Bessman NJ, Setty TG, Ferguson KM, Burd CG. PtdIns4P recognition by Vps74/GOLPH3 links PtdIns 4-kinase signaling to retrograde Golgi trafficking. J Cell Biol. 2009;187:967–975. doi: 10.1083/jcb.200909063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooding S, Pelham HR. The dynamics of golgi protein traffic visualized in living yeast cells. Mol Biol Cell. 1998;9:2667–2680. doi: 10.1091/mbc.9.9.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuestehube LJ, Schekman RW. Reconstitution of transport from endoplasmic reticulum to Golgi complex using endoplasmic reticulum-enriched membrane fraction from yeast. Methods Enzymol. 1992;219:124–136. doi: 10.1016/0076-6879(92)19015-x. [DOI] [PubMed] [Google Scholar]
- Yu JW, Mendrola JM, Audhya A, Singh S, Keleti D, DeWald DB, Murray D, Emr SD, Lemmon MA. Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol Cell. 2004;13:677–688. doi: 10.1016/s1097-2765(04)00083-8. [DOI] [PubMed] [Google Scholar]
- Zhu H, et al. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.