

Abstract
p56dok-2 acts as a multiple docking protein downstream of receptor or non-receptor tyrosine kinases. However, the role of p56dok-2 in biological functions of cells is not clear. We found that transcription of the p56dok-2 gene in macrophages was increased markedly in response to cytokines such as macrophage colony-stimulating factor (M-CSF), granulocyte/macrophage-CSF and interleukin-3 (IL-3). Forced expression of p56dok–2 inhibited M-CSF-, granulocyte-CSF-, IL-3- and stem cell factor-induced proliferation of myeloid leukemia cells, M-NFS-60. The p56dok-2-overexpressing cells showed an impaired induction of c-myc but not of c-jun, junB or c-fos when stimulated with M-CSF. Consistent with these results, the peritoneal cavity of the hairless (hr/hr) strain of mutant mice, whose cells expressed less p56dok-2 than wild-type mice, contained more macrophages than that of +/hr mice. Moreover, the inhibition of endogenous p56dok-2 expression in macrophage-like tumor cells, J774A.1, by stable expression of antisense p56dok-2 mRNA accelerated cell proliferation. The study identifies a novel role for p56dok-2 as a molecule that negatively regulates signal transduction and cell proliferation mediated by cytokines in a feedback loop.
Keywords: cell proliferation/cytokine/Dok-R/FRIP/p56dok-2
Introduction
The proliferation and differentiation of cells of many lineages are tightly regulated by proteins known as cytokines. The binding of cytokines to their receptors activates multiple tyrosine kinases including receptor tyrosine kinases, JAK family kinases and Src family kinases (reviewed in Ihle, 1995). The receptors for cytokines such as macrophage colony-stimulating factor (M-CSF) and stem cell factor (SCF) are transmembrane tyrosine kinases and the cytoplasmic domain of these receptors encodes a tyrosine kinase (Sherr et al., 1985; Huang et al., 1990; Williams et al., 1990; Zsebo et al., 1990). In contrast, the receptors for the majority of cytokines do not encode a tyrosine kinase catalytic domain. Studies conducted with a variety of cytokine receptors have provided evidence that, regardless of receptor type, the activation of tyrosine kinases and the phosphorylation of substrates drive cell proliferation or differentiation (reviewed in Ihle, 1995).
More recent studies have also elucidated the mechanisms responsible for negatively regulating the cytokine signals. SHP-1 is a tyrosine phosphatase that binds to the erythropoietin (Epo) receptor and the β chain of interleukin-3 (IL-3) receptor (Yi et al., 1993, 1995). A mutation in the SHP-1 gene results in hyper tyrosine phosphorylation of cellular proteins and increased levels of macrophages and erythrocytes (Shultz et al., 1993; Tsui et al., 1993). SHIP, SH2-containing inositol phosphatase, has also been shown to function as a growth inhibitory molecule and its targeted disruption results in hematopoietic perturbations as a consequence of hyper-responsiveness to stimulation by IL-3, M-CSF and granulocyte/macrophage-CSF (GM-CSF) (Liu et al., 1997; Helgason et al., 1998).
The CIS/JAB/SOCS/SSI family consists of relatively small proteins with a centrally located SH2 domain and a unique carboxyl motif termed the SOCS box (Yoshimura et al., 1995; Endo et al., 1997; Naka et al., 1997; Starr et al., 1997). Studies have also implicated these molecules in the negative regulation of cytokine signal transduction. For example, CIS protein binds tyrosine phosphorylated Epo receptor and can block Epo signaling (Yoshimura et al., 1995). Importantly, stimulation with a broad array of cytokines is capable of upregulating mRNA for one or more members of the family (Yoshimura et al., 1995; Starr et al., 1997), suggesting that these molecules act in a classic negative feedback loop to regulate cytokine signal transduction.
Here, we provide evidence that p56dok-2 is also a cytokine-inducible inhibitor of cytokine signal transduction. p56dok-2 (also known as Dok-R or FRIP) is a member of a newly identified subfamily of docking proteins (Di Cristofano et al., 1998; Jones and Dumont, 1998; Nelms et al., 1998), of which p62dok is the prototype (Carpino et al., 1997; Yamanashi and Baltimore, 1997). More recently, the third member of this dok family, Dok-L/Dok-3, has been cloned (Cong et al., 1999; Lemay et al., 2000). Analysis of the expressed sequence tag database reveals the presence of additional members of the family (Di Cristofano et al., 1998). The N-terminal part of dok proteins contains a pleckstrin homology domain thought to be involved in the membrane localization of proteins (Noguchi et al., 1999). There is a potential phosphotyrosine binding domain near the central region (Jones and Dumont, 1998). The C-terminal region contains a number of potential tyrosine phosphorylation and SH2 domain interaction sites (Bhat et al., 1998; Jones and Dumont, 1999; Lock et al., 1999; Noguchi et al., 1999), leading to the suggestion that dok proteins act as multiple docking proteins downstream of tyrosine kinases. In fact, in reconstitution experiments using transiently transfected cells, epitope-tagged p56dok-2 was phosphorylated on tyrosine residues when co-expressed with Bcr–Abl tyrosine kinase (Di Cristofano et al., 1998), an activated form of Tek/Tie2 receptor tyrosine kinase (Jones and Dumont, 1998) or Src family kinases, Lyn, Hck or Src (Lock et al., 1999). Moreover, stimulation of cell lines that stably expressed p56dok-2 with growth factors or cytokines, including epidermal growth factor, insulin, IL-2, IL-3 or IL-4, also resulted in rapid phosphorylation of p56dok-2 (Nelms et al., 1998; Lock et al., 1999). Once phosphorylated, p56dok-2 was capable of interacting via specific binding sites with SH2 domains of rasGTPase-activating protein (rasGAP) (Di Cristofano et al., 1998; Jones and Dumont, 1998, 1999; Nelms et al., 1998; Lock et al., 1999) and with an adapter protein, Nck (Jones and Dumont, 1998; Lock et al., 1999).
Although it was found that ectopic overexpression of p62dok upregulated motility of chinese hamster ovary cells (Noguchi et al., 1999), more recent studies clearly show that p62dok, at least in B cells, is a negative regulator of MAP kinase and cell proliferation (Tamir et al., 2000; Yamanashi et al., 2000). Inactivation of the p62dok gene by homologous recombination has shown that upon B cell receptor cross-linking, p62dok suppresses MAP kinase and is dispensable for FcγRIIB-mediated negative regulation of cell proliferation (Yamanashi et al., 2000). Further more, transfection experiments in a B cell line, A20, suggest that Dok-L/Dok-3 is also a negative regulator of immunoreceptor signaling (Lemay et al., 2000). In contrast to the p62dok and Dok-L/Dok-3 genes, p56dok-2 gene expression is not normally detectable in B cells but is high in T cells and macrophages (Nelms et al., 1998; Lemay et al., 2000). Therefore, p56dok-2 is thought to have a role in T cells and macrophages. In fact, the results of experiments done with the hairless (hr) strain of mutant mice raise the possibility that p56dok-2 negatively regulates the proliferation of T cells but not of B cells: axillary lymph node T cells of hr/hr mice expressed 3- to 5-fold less p56dok-2 than wild-type mice and were hyper-responsive to stimulation with IL-2 and T cell receptor activation (Nelms et al., 1998). However, this result is not necessarily evidence that p56dok-2 functions as a growth inhibitory molecule since the hairless phenotype is thought to result from the disruption of a gene termed hairless, a putative zinc-finger gene, not the p56dok-2 gene (Cachon-Gonzalez et al., 1994). Based on an overexpression study and antisense inhibition of gene expression, we show here that p56dok-2 functions as a negative regulator of cell proliferation induced by a range of cytokines. Importantly, we show that the expression of such a negative regulator is induced by cytokines themselves.
Results
Identification of p56dok-2 as a cytokine-inducible gene
To obtain genes whose induction is mediated by cytokines, we employed the cDNA library subtraction technique (Diatchenko et al., 1996). We prepared a cDNA library of murine bone marrow-derived macrophages cultured in cytokine-free medium and subtracted this cDNA library from that of cells stimulated with a macrophage-specific growth factor, M-CSF, for 3 h. To identify genes induced by M-CSF, clones isolated randomly from the subtracted library were screened by northern blot analysis. The study identifies a number of novel or known genes (S.Suzu, K.Nomaguchi, M.Yamada, H.Hayasawa, F.Kimura and K.Motoyoshi, manuscript in preparation). One of the genes frequently isolated by these procedures was that encoding p56dok-2.
The induction of the p56dok-2 gene by M-CSF was observed in murine bone marrow-derived macrophages, murine peritoneal macrophages and factor-dependent murine myeloid leukemia M-NFS-60 cells (Figure 1A, top panel). In contrast to these primary macrophages and M-NFS-60 cells, the immortalized murine macrophage-like tumor cells, J774A.1, expressed p56dok-2 transcript at a high level even when the cells were serum depleted for 24 h (Figure 1A, top panel). The relatively high expression of the p56dok-2 gene in J774A.1 cells was also noted by others (Lemay et al., 2000). No obvious change in p62dok expression was observed in these cells (Figure 1A, middle panel). A major p56dok-2 transcript of 2 kb was induced within 1.5 h after stimulation of bone marrow macrophages with M-CSF and the expression was maintained thereafter (Figure 1B). A minor transcript of 3 kb, which was noted earlier by others (Jones and Dumont, 1998), was also detected (Figure 1B, upper band). p56dok-2 could be induced not only by M-CSF but also by other growth factors for macrophages: GM-CSF and IL-3 induced p56dok-2 expression in bone marrow macrophages (Figure 1B). The induction of p56dok-2 protein was confirmed by an immunoblotting analysis. The induced expression in M-NFS-60 cells was detected within 3 h of stimulation with M-CSF and the level of expression increased until 18 h (Figure 1C). Collectively, these results clearly indicate that p56dok-2 is a cytokine-inducible gene.

Fig. 1. Northern blot (A and B) and immunoblot analyses (C) of p56dok-2. (A) Bone marrow macrophages, peritoneal macrophages or M-NFS-60 cells were factor depleted and then stimulated with M-CSF for the periods indicated. J774A.1 cells were serum depleted for 24 h. Total RNA (5 µg/lane) was blotted to membrane and hybridized with probe to p56dok-2 or p62dok. To verify the amount of RNA loaded, the same blots were hybridized with GAPDH probe. Exposure times: top and middle panels, 14 h; bottom panel, 1 h. (B) Bone marrow macrophages were factor depleted for 12 h and then stimulated with either M-CSF, GM-CSF or IL-3 for the periods indicated. Total RNA (5 µg/lane) was blotted to membrane and hybridized with probes to p56dok-2 or GAPDH. Exposure times: top panel, 12 h; bottom panel, 2 h. (C) M-NFS-60 cells were factor depleted for 4 h and then stimulated with M-CSF for the periods indicated. The cleared cell lysates containing equal amounts of protein (5 µg/lane) were blotted to membrane and probed with anti-p56dok-2 antibody. To verify the amount of protein loaded, the same blot was probed with anti-rasGAP antibody. (A, B and C) Data shown are representative of two independent experiments with similar results.
Suppression of cell proliferation by forced expression of p56dok-2
To prove the proposed hypothesis that p56dok-2 negatively regulates cell proliferation, we attempted to express p56dok-2 cDNA using a strong constitutive promoter (Mizushima and Nagata, 1990; Kimura et al., 1994; Suzu et al., 1998) in factor-dependent murine myeloid leukemia M-NFS-60 cells (Nakoinz et al., 1990; Suzu et al., 1997). The cells were shown to express more p56dok-2 gene in response to M-CSF stimulation (see Figure 1A). The cells were also known to proliferate well in response to M-CSF and weakly in response to SCF, IL-3 or granulocyte-CSF (G-CSF) (Nakoinz et al., 1990). The transfectants were screened for their expression of p56dok-2 protein by immunoblotting. Few of the G418-resistant clones obtained showed an increase in the expression of p56dok-2 but four independent clones that expressed more p56dok-2 than those obtained by the transfection of empty vector were isolated (Figure 2A). As shown in Figure 2B, all clones of M-NFS-60 cells expressing p56dok-2 (NFS-dok2, clones 7, 17, 25 and 26) showed a reduced pro liferation rate in response to M-CSF when compared with parental cells (NFS-wt) or cells transfected with empty vector (NFS-vector, clones 1 and 2). Among p56dok-2-expressing cells, clones 25 and 26, which expressed more p56dok-2 than clone 7 or 17 (see Figure 2A), showed a markedly reduced proliferation rate (Figure 2B). If clone 26 was maintained in the antibiotic G418-free medium, the expression of p56dok-2 was lost with time and the cells regained their responsiveness to M-CSF (data not shown), excluding the possibility of a change that affected the intrinsic ability of the cells to proliferate. The inhibitory effect of the constitutive expression of p56dok-2 was not specific for M-CSF-dependent cell proliferation. Even if the p56dok-2-overexpressing M-NFS-60 cells (clone 26, see Figure 2A and B) were cultured in the presence of SCF, IL-3 or G-CSF, they showed a reduced proliferative response to these cytokines (Figure 2C). These results suggest that p56dok-2 is a negative regulator of cell proliferation mediated by a range of cytokines in M-NFS-60 cells.
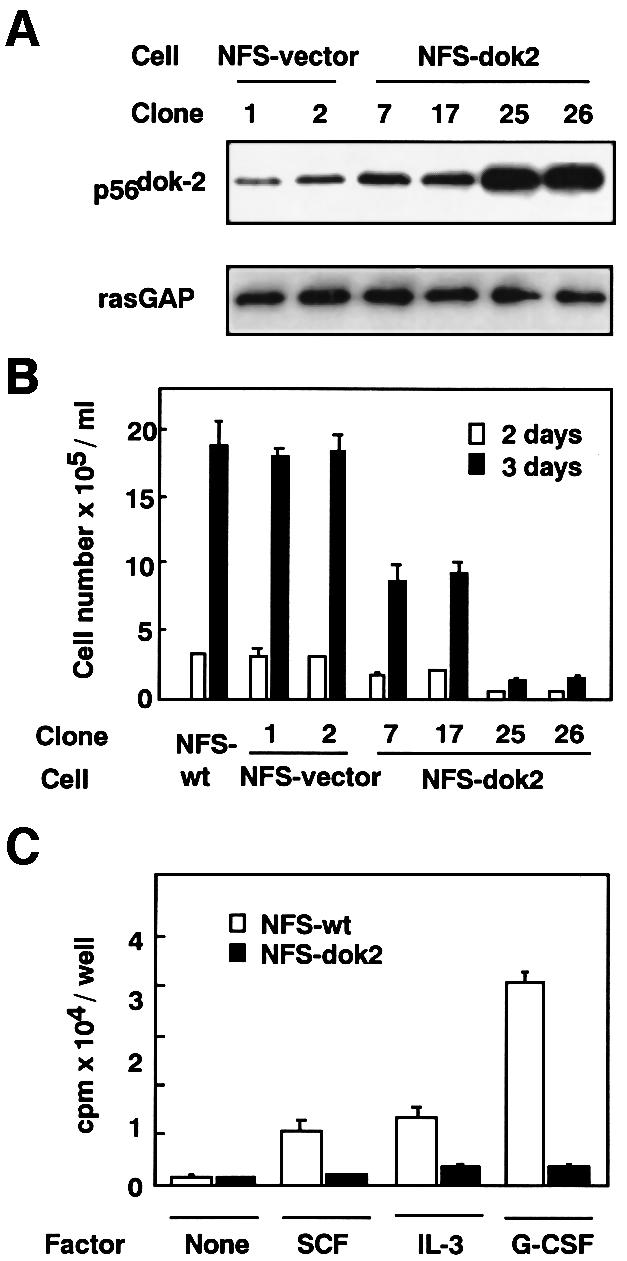
Fig. 2. Establishment of transfectants of M-NFS-60 cells expressing p56dok-2 and their proliferation rates. (A) M-NFS-60 cells were transfected with empty vector (NFS-vector) or with p56dok-2 cDNA (NFS-dok2). The transfectants were analyzed for their expression of p56dok-2 protein by immunoblotting. The M-NFS-60 cell lines were factor depleted for 4 h in RPMI 1640 medium containing 1% BSA and the cell lysates were prepared from the cells. The cleared cell lysates containing equal amounts of protein (5 µg/lane) were blotted to membrane and probed with anti-p56dok-2 antibody. To verify the amount of protein loaded, the same blot was probed with anti-rasGAP antibody. Data shown are representative of two independent experiments with similar results. (B) The proliferation of parental M-NFS-60 cells (NFS-wt), cells transfected with empty vector (NFS-vector) or cells transfected with p56dok-2 cDNA (NFS-dok2) in response to M-CSF. The cells were seeded at a density of 1 × 104 cells/ml and cultured for 2 or 3 days in medium containing 10% FCS and M-CSF. Error bars from triplicate assays are shown. These results are representative of three independent experiments. (C) The proliferation of parental M-NFS-60 cells (open bars) or cells expressing p56dok-2 (clone 26, see A and B) (solid bars) cultured in the absence of factor (None), or the presence of SCF, IL-3 or G-CSF. Cells were seeded at a density of 2 × 103 cells/well and cultured for 72 h. The cells were then pulsed with [3H]thymidine and the incorporated radioactivity was measured. Error bars from triplicate assays are shown. These results are representative of three independent experiments.
Analyses of p56dok-2 expression and peritoneal cells in hr/hr mice
The p56dok-2 gene was shown to be localized to mouse chromosome 14 at a previously characterized locus termed hairless (Nelms et al., 1998), but the hairless phenotype, lymphadenopathy and splenomegaly, is thought to result from the disruption of a putative zinc-finger gene termed hairless, not the p56dok-2 gene (Cachon-Gonzalez et al., 1994). Nevertheless, lymph node T cells from mice homozygous for the hairless allele (hr/hr) have a significantly lower level of p56dok-2 transcript than those from heterozygous (+/hr) mice, which may be due to the defect of the putative zinc-finger gene (Nelms et al., 1998). We also found that both bone marrow cells and spleen cells from hr/hr mice expressed less p56dok-2 transcript than those from +/hr mice (Figure 3A). Thus, the mutant mice are thought to be useful for analyzing the physiological role of p56dok-2.

Fig. 3. Analyses of p56dok-2 expression and peritoneal cells in hr/hr mice. (A) Total RNA from bone marrow cells or spleen cells of 10- or 13-week-old hr/hr mice and heterozygous control (+/hr) mice was prepared, blotted to membrane (5 µg/lane for bone marrow cells and 2.5 µg/lane for spleen cells) and then hybridized with probe to p56dok-2 or GAPDH. Exposure times: top panel, 3 days; bottom panel, 1 h. (B) FACS profiles of peritoneal cells from hr/hr and +/hr mice. Cells were stained with anti-Mac-1 antibody and analyzed by flow cytometry. (C) Wright–Giemsa-stained cytospin preparations of peritoneal cells collected from hr/hr and +/hr mice. Arrows indicate typical cells with macrophage morphology. (B and C) Data shown are representative of an analysis with 8 hr/hr mice and 8 +/hr mice 13 weeks old. The experiments were repeated using 10-week-old mice.
Because we identified p56dok-2 as a cytokine-inducible gene in macrophages (see Figure 1), we attempted to analyze the change in the mononuclear phagocyte system in hr/hr mice. For this purpose, we compared the phenotype of peritoneal cells of hr/hr mice with that of +/hr control mice. Although there was no change in the total number of peritoneal cells between hr/hr mice and +/hr mice (3.9 ± 0.4 × 105 cells for hr/hr and 4.1 ± 1.2 × 105 cells for +/hr), the percentage of cells expressing a surface marker for macrophages, Mac-1, of hr/hr mice was higher than that of +/hr mice (30 ± 6% for hr/hr mice and 14 ± 2% for +/hr mice) (Figure 3B). There was no difference in the median log fluorescence on Mac-1-positive cells between hr/hr mice and +/hr mice (Figure 3B). The fact that the peritoneal cavity of hr/hr mice contained more macrophages than that of +/hr mice was confirmed by enumerating cells with macrophage morphology in cytocentrifuge preparations (Figure 3C). Our data and the result reported previously with hr/hr mice (Nelms et al., 1998) raise the possibility that p56dok-2 is critical in regulating the proliferation of at least macrophages and T cells.
Acceleration of cell proliferation by antisense inhibition of p56dok-2 expression
To prove more convincingly that p56dok-2 is a negative regulator of cell proliferation of macrophages, we attempted to express antisense p56dok-2 mRNA in macrophage-like J774A.1 cells. The cells were shown to constitutively express p56dok-2 transcript at a high level (see Figure 1A). The cells were transfected with a selectable expression plasmid containing full-length p56dok-2 cDNA in the antisense orientation. As a control, the cells were transfected with empty vector. Transfected clones were selected in the presence of G418. A total of four antisense-transfected G418-resistant clones was isolated and a clone (J774-AS-dok2) that showed a significant decrease in the level of p56dok-2 protein compared with two control clones (J774-vector, clones 1 and 2) was obtained (Figure 4A). Among the control clones, clone 1 contained slightly less p56dok-2 protein than clone 2 (Figure 2A), and the level of p56dok-2 protein of clone 2 was comparable to that of parental cells (data not shown). If the antisense clone was maintained in G418-free medium, the level of p56dok-2 protein returned to that of parental cells (data not shown). Thus, the proliferation rate of the antisense clone was compared with that of the control clones maintained in G418-containing medium. Although J774A.1 cells are immortalized tumor cells and proliferate in the absence of exogenously added growth factor (Fan et al., 1993), the antisense clone (J774-AS-dok2) showed an accelerated proliferation compared with two control clones (J774-vector, clones 1 and 2) (Figure 4B). Among clones obtained by transfection with empty vector, clone 1, which contained slightly less p56dok-2 protein than clone 2 (Figure 4A), proliferated slightly more quickly than clone 2 (Figure 4B). These results strongly suggest that p56dok-2 functions as a negative regulator of cell proliferation.
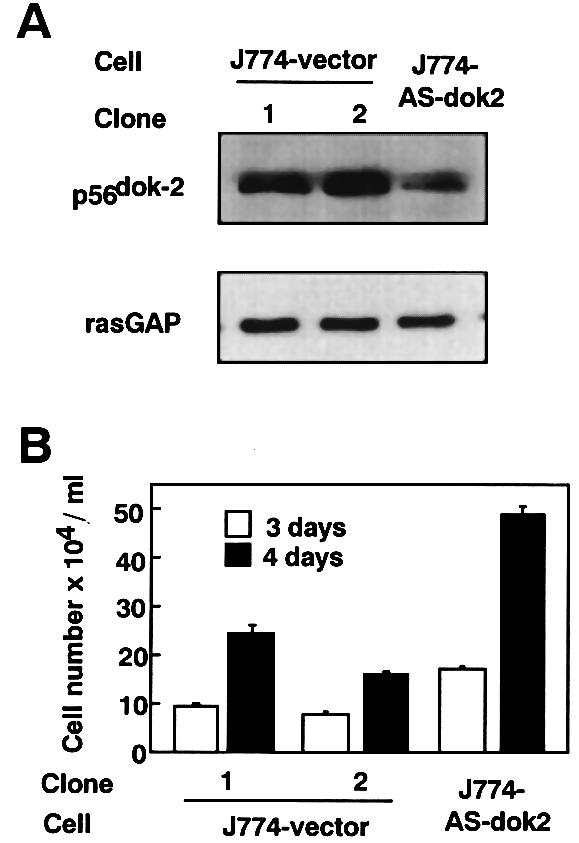
Fig. 4. Establishment of transfectants of J774A.1 cells expressing antisense p56dok-2 mRNA and its proliferation rate. (A) J774A.1 cells were transfected with empty vector (J774-vector) or with vector containing the full-length p56dok-2 cDNA in the antisense orientation (J774-AS-dok2). The transfectants were analyzed for their expression of p56dok-2 protein by immunoblotting. At the exponential growth stage, cells were harvested and lysates were prepared from the cells. The cleared cell lysates containing equal amounts of protein (2.5 µg/lane) were blotted to membrane and probed with anti-p56dok-2 antibody. To verify the amount of protein loaded, the same blot was probed with anti-rasGAP antibody. Data shown are representative of two independent experiments with similar results. (B) The proliferation of J774A.1 cells transfected with empty vector (J774-vector) or cells transfected with antisense p56dok-2 cDNA (J774-AS-dok2). The cells were seeded at a density of 1 × 104 cells/ml and cultured for 3 (open bars) or 4 days (solid bars) in medium containing 10% FCS. Error bars from triplicate assays are shown. These results are representative of three independent experiments.
Effects of overexpression of p56dok-2 on M-CSF-induced activation of the signaling pathway
We next attempted to clarify the mechanism by which the constitutive overexpression of p56dok-2 inhibited the proliferation of M-NFS-60 cells in response to M-CSF. M-CSF receptor, encoded by the c-fms proto-oncogene, is a member of the receptor tyrosine kinase family (Sherr et al., 1985). Ligand binding leads to the autophosphorylation of M-CSF receptor and the assembly of phosphotyrosine-dependent signaling complexes (reviewed in Rohrschneider, 1995). Proteins interacting directly with M-CSF receptor have been described. Of these, Src family kinases are shown to associate with phosphotyrosine 559 in the juxtamembrane region of M-CSF receptor (Alonso et al., 1995). Furthermore, the activation of Src kinases and the subsequent induction of an immediately early response gene, c-myc, have been shown to be critical for M-CSF-dependent cell proliferation (Roussel et al., 1991; Roche et al., 1995). We therefore assumed that the constitutive overexpression of p56dok-2 affected the Src–c-myc pathway.
M-CSF stimulation induced the expression of the immediate-early response genes, c-myc, c-jun, junB and c-fos in parental M-NFS-60 cells (Figure 5). However, induction of the c-myc gene by M-CSF in p56dok-2-overexpressing M-NFS-60 cells was severely impaired (Figure 5). Induction of junB- or c-fos mRNA in the two cell lines was comparable and induction of c-jun in p56dok-2-overexpressing cells was slightly higher than in parental cells (Figure 5). In any case, the finding that induction of c-myc is severely impaired in p56dok-2-overexpressing cells, which show a reduced proliferation rate (see Figure 2), is consistent with the notion that c-myc is a key regulator of M-CSF-dependent cell proliferation.
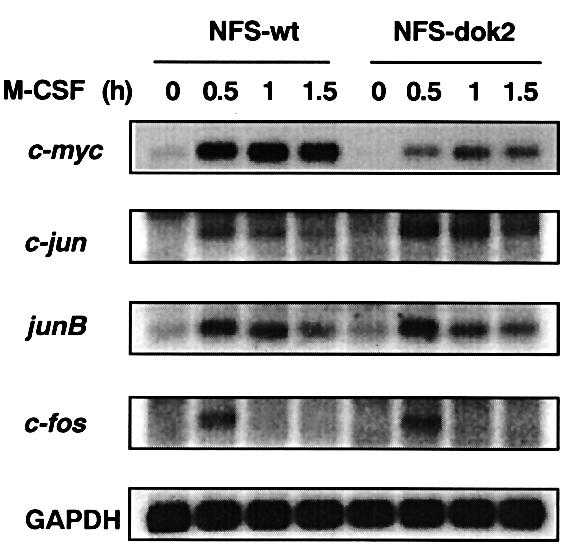
Fig. 5. Expression of immediate-early response genes in M-NFS-60 cells. Parental M-NFS-60 cells (NFS-wt) or p56dok-2-overexpressing M-NFS-60 cells (NFS-dok2) (clone 26, see Figure 2) were factor depleted for 4 h in RPMI 1640 medium containing 1% BSA and then stimulated with M-CSF for the periods indicated. Total RNA (5 µg/lane) was blotted to membrane and hybridized with probe to c-myc, c-jun, junB, c-fos or GAPDH. Exposure times: c-myc and GAPDH, 4 h; c-jun, junB and c-fos, 8 h. Data shown are representative of two independent experiments with similar results.
We next examined whether overexpression of p56dok-2 affected the activation of Src family kinases. While p56dok-2 was originally identified as a substrate of Bcr–Abl tyrosine kinase (Di Cristofano et al., 1998) and a protein capable of interacting with the intracellular domains of IL-4 receptor (Nelms et al., 1998) and with Tek/Tie2 receptor tyrosine kinase (Jones and Dumont, 1998), it was recently identified as a substrate of Src family kinases (Lock et al., 1999). We found that M-CSF stimulation was able to induce the tyrosine phosphorylation of p56dok-2 in M-NFS-60 cells and that the phosphorylation was severely reduced when the cells were pretreated with Src family kinase inhibitor, PP2 (Figure 6A), confirming that p56dok-2 was a substrate of Src family kinases in the cells. Furthermore, we found that p56dok-2 could be phosphorylated in p56dok-2-overexpressing cells upon M-CSF stimulation and that the level of the phosphorylation in the cells was equivalent to that in parental cells (Figure 6B). It therefore seems likely that constitutive overexpression of p56dok-2 affected the cascade downstream of Src family kinases but not the activation of the kinases itself.
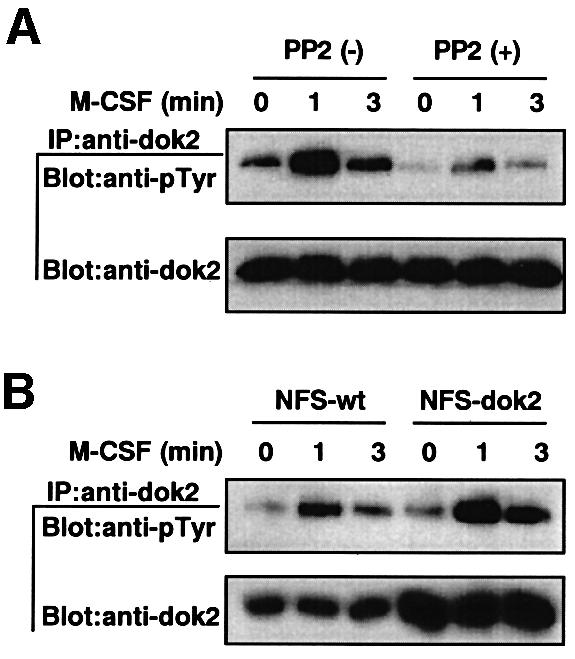
Fig. 6. Tyrosine phosphorylation of p56dok-2 in M-NFS-60 cells. (A) Parental M-NFS-60 cells were factor depleted for 4 h in RPMI 1640 medium containing 1% BSA, treated with PP2 for the last 30 min or left untreated, and then stimulated with M-CSF for the periods indicated. (B) Parental M-NFS-60 cells (NFS-wt) or p56dok-2-overexpressing M-NFS-60 cells (NFS-dok2) (clone 26, see Figure 2) were factor depleted for 4 h in RPMI 1640 medium containing 1% BSA and then treated with M-CSF for the periods indicated. (A and B) Immunoprecipitates (IP) were obtained with anti-p56dok-2 antibody (goat polyclonal antibody) and analyzed by immunoblotting (Blot) with anti-phosphotyrosine antibody (pTyr). The blots were reprobed with anti-p56dok-2 antibody (rabbit antiserum). Data shown are representative of two independent experiments with similar results.
The Src family kinase-mediated signaling pathway that regulates the expression of c-myc gene has remained largely unresolved. However, a recent study raises the possibility that the induction of c-myc by Src family kinases is in part dependent on activation of the ras/MAP kinase pathway (Cheng et al., 1999). An adapter molecule, Shc, is a good candidate that links Src family kinases to the ras/MAP kinase pathway (reviewed in Corey and Anderson, 1999). We found that Shc was tyrosine phosphorylated rapidly in response to M-CSF stimulation in parental cells but the phosphorylation was severely reduced in p56dok-2-overexpressing cells (Figure 7A). Consistent with the reduction in Shc phosphorylation, the association between Shc and Grb2 in p56dok-2-overexpressing cells was reduced (Figure 7A). Similarly, there was a diminution in the activation of ERK2, one of the MAP kinases, in p56dok-2-overexpressing cells (Figure 7B). However, the diminution in the activation of ERK2 was modest when compared with that in Shc phosphorylation (Figure 7A) and in c-myc induction (see Figure 5). Therefore, although the inhibitory effect of p56dok-2 on the induction of the c-myc gene might be in part dependent on the ras/MAP kinase pathway, it is obvious that other Src kinase-mediated signaling pathways that regulate the induction of c-myc are affected by p56dok-2.

Fig. 7. Effect of constitutive overexpression of p56dok-2 on tyrosine phosphorylation of Shc and its association with Grb2 (A) and on activation of ERK2 (B). (A and B) Parental M-NFS-60 cells (NFS-wt) or p56dok-2-overexpressing M-NFS-60 cells (NFS-dok2) (clone 26, see Figure 2) were factor depleted for 4 h in RPMI 1640 medium containing 1% BSA and then treated with M-CSF for the periods indicated. (A) Immunoprecipitates (IP) were obtained with anti-Shc antibody (rabbit polyclonal) and analyzed by immunoblotting (Blot) with antibodies to phosphotyrosine (pTyr), Grb2 and Shc (mouse monoclonal IgG). Alternatively, immunoprecipitates were obtained with anti-Grb2 antibody (rabbit polyclonal IgG) and analyzed by immunoblotting with antibodies to Shc and Grb2 (mouse monoclonal IgG). Data shown are representative of two independent experiments with similar results. (B) The cleared cell lysates containing equal amounts of protein (5 µg/lane) were blotted to membrane and probed with antibodies to phosphorylated ERK (pERK) and total ERK (ERK). Data shown are representative of four independent experiments with similar results.
Discussion
In this study, we have provided evidence that p56dok-2 functions as a negative regulator of cell proliferation. Forced expression of p56dok-2 suppressed M-CSF-, SCF-, IL-3- and G-CSF-dependent proliferation of M-NFS-60 cells (Figure 2). Moreover, the inhibition of endogenous p56dok-2 expression in J774A.1 cells by stable expression of antisense p56dok-2 mRNA accelerated proliferation of the cells (Figure 4). Recently, Yamanashi et al. (2000) generated mice that lack p62dok, which is closely related to p56dok-2, and clearly demonstrated that p62dok, at least in B cells, is a negative regulator of cell proliferation. In contrast to the p62dok gene, the p56dok-2 gene is not normally detectable in B cells but is present in T cells and macrophages (Nelms et al., 1998; Lemay et al., 2000). Therefore, p56dok-2 might function as a negative regulator of cell proliferation in T cells or macrophages. Studies with hr/hr mice support this hypothesis. Axillary lymph node T cells, bone marrow cells and spleen cells of hr/hr mice expressed less p56dok-2 transcript than those of +/hr mice (Nelms et al., 1998; Figure 3A). The mice have lymphadenopathy of axillary lymph nodes (Nelms et al., 1998) and the number of macrophages in the peritoneal cavity is significantly higher in hr/hr mice than in +/hr mice (Figure 3), in both of which the increase might result from cellular hyperproliferation due to the impaired negative regulation of p56dok-2. However, once again, we must mention that there is no direct evidence that the immunologic/hematologic phenotype of hr/hr mice is caused by the altered expression of p56dok-2 since the hairless phenotype is thought to result from the disruption of a putative zinc-finger gene, hairless (Cachon-Gonzalez et al., 1994). The precise physiological role of p56dok-2 will be clarified further by generating mice that lack this gene.
Another noteworthy finding of our study is that p56dok-2 is a cytokine-inducible gene. Expression of p56dok-2 in macrophages is induced by M-CSF, GM-CSF and IL-3 (Figure 1B). Similarly, p56dok-2 is induced in M-NFS-60 cells by M-CSF (Figure 1A). None of the dok family genes has been shown to be transcriptionally regulated by extracellular stimuli including cytokine stimulation. Because p56dok-2 suppresses cytokine-mediated cell proliferation, it becomes clear that p56dok-2 can act in a feedback loop that negatively regulates cytokine-mediated cell proliferation. Although we found that p62dok was not induced in macrophages by M-CSF (see Figure 1A), the assessment of p62dok or Dok-L/Dok-3 gene as a cytokine-inducible gene will be clarified further by analyses with other cell types and other cytokines.
Although the induction of p56dok-2 expression in M-NFS-60 cells by M-CSF was obvious (Figure 1A), that by either IL-3, G-CSF or SCF was detectable but slight (data not shown). Since M-NFS-60 cells proliferate well in response to M-CSF and weakly in response to IL-3, G-CSF or SCF (Nakoinz et al., 1990), the expression of the p56dok-2 gene may be regulated by a signal transduction pathway that transmits proliferative signal.
Based on its structural features, p56dok-2 has been thought to act as a multiple docking protein downstream of receptor or non-receptor tyrosine kinases. However, the role of p56dok-2 in tyrosine kinase-mediated signal transduction is largely unknown. p56dok-2 and p62dok were first noted for their association with rasGAP upon phosphorylation (Carpino et al., 1997; Yamanashi and Baltimore, 1997; Di Cristofano et al., 1998; Jones and Dumont, 1998; Nelms et al., 1998). Since rasGAP is capable of negatively regulating ras by enhancing its intrinsic GTPase activity, one attractive model might be that p56dok-2 links rasGAP to the attenuation of ras signaling. However, in contrast to p56dok-2 and p62dok, Dok-L/Dok-3, the third member of the dok family, does not bind rasGAP (Cong et al., 1999; Lemay et al., 2000). Furthermore, p56dok-2 was shown to attenuate epidermal growth factor-stimulated activation of MAP kinase independently of its association with rasGAP (Jones and Dumont, 1999). Thus, the inhibitory effect of p56dok-2 on cell proliferation may be independent of its association with rasGAP and ras signaling. In this study, we found that the induction of c-jun or c-fos in response to M-CSF was not inhibited by the constitutive overexpression of p56dok-2 (Figure 5). Since the induction of c-jun or c-fos has been shown to be dependent on ras function (Barone and Courtneidge, 1995), p56dok-2 is thought to be involved in a signaling pathway other than the ras pathway.
We found that the induction of c-myc in response to M-CSF was severely impaired in p56dok-2-overexpressing cells (Figure 5). The induction of c-myc has been shown to be required for M-CSF-dependent cell proliferation based on the following observations. One of the major autophosphorylation sites within ligand-stimulated M-CSF receptor is tyrosine 809, which is located in the activation loop of the kinase domain (Roussel et al., 1990). Substitution of this residue with phenylalanine (Y809F) leads to reduction but not abolition of receptor protein tyrosine kinase activity (Davis et al., 1997). However, NIH 3T3 cells expressing the mutant M-CSF receptor (Y809F) fail to proliferate in response to M-CSF (Roussel et al., 1990) and to induce c-myc (Roussel et al., 1991). Finally, enforced expression of c-myc restores the ability of the cells expressing the mutant M-CSF receptor to proliferate in response to M-CSF (Roussel et al., 1991). Therefore, the finding that the induction of c-myc is severely impaired in p56dok-2-overexpressing cells, which show a reduced proliferation rate, is consistent with the notion that c-myc is a key regulator of M-CSF-dependent cell proliferation.
Src family kinases have been shown to activate a pathway that mediates the induction of c-myc and to be required for M-CSF-dependent cell proliferation based on the following observations: (i) the constitutive expression of c-myc rescues a block to DNA synthesis elicited by a dominant-negative form of Src (Barone and Courtneidge, 1995); (ii) Src kinases associate with and are activated by the M-CSF receptor (Courtneidge et al., 1993); and (iii) the microinjection of antibodies to Src kinases inhibits DNA synthesis in response to M-CSF (Roche et al., 1995). However, Src family kinase-mediated signaling pathways that regulate the expression of the c-myc gene have remained largely unresolved. Our data indicate that the overexpression of p56dok-2 affected the cascade downstream of Src family kinases rather than the activation of the kinases itself (Figure 6). Biochemical and genetic evidence has also indicated the importance of Shc in initiating the signaling cascade by Src kinases. For example, there is a loss of tyrosine phosphorylation of Shc in lyn-deficient cells (Nagai et al., 1995; Ptasznik et al., 1995) and overexpression of Fyn is associated with the formation of a complex between Fyn and Shc (Li et al., 1996). Shc participates in early signaling complexes of the M-CSF receptor but does not bind directly to the receptor (Lioubin et al., 1994), suggesting that Shc is a substrate of Src kinases activated by the M-CSF receptor but not of the receptor itself. p56dok-2 is also a substrate of Src kinases (Lock et al., 1999; Figure 6A). We found that tyrosine phosphorylation of Shc was severely reduced in p56dok-2-overexpressing cells (Figure 7A). These findings raise the possibility that p56dok-2 competes with Shc for Src kinases and thereby inhibits the Src kinase-initiating pathway. However, more studies will be needed to clarify the inhibitory mechanism of p56dok-2 in the Src kinase–c-myc pathway.
The phosphorylation of Shc by Src family kinases leads to the association of Shc with Grb2-Sos guanine nucleotide exchange complex (Rozakis-Adcock et al., 1992; Myers et al., 1994; Salcini et al., 1994). This molecular linkage results in the accumulation of activated ras-GTP, which activates raf kinase, which in turn initiates the MAP kinase cascade. In fact, although an earlier study suggested that Src kinases induced c-myc in a ras/MAP kinase pathway-independent manner (Barone and Courtneidge, 1995), a more recent study raises the possibility that the induction of c-myc by Src family kinases is in part dependent on the ras/MAP kinase pathway (Cheng et al., 1999). Consistent with this notion, we observed a reduction in the association of Shc with Grb2 (Figure 7A) and in the activation of ERK2 in M-CSF-stimulated p56dok-2-overexpressing cells (Figure 7B). However, the reduction in the activation of ERK2 was modest when compared with the reduction in c-myc induction (Figure 5). Therefore, it is obvious that unidentified Src kinase-mediated signaling pathways that regulate the induction of c-myc are also affected by p56dok-2.
In this study, we identify p56dok-2 as a molecule that negatively regulates cell proliferation and signal transduction mediated by cytokines in a feedback loop. The CIS/JAB/SOCS/SSI family proteins are also known as inhibitors that act in a similar manner. The inhibitory effect of these proteins appeared to be specific for the JAK/STAT pathway (Yoshimura et al., 1995; Endo et al., 1997; Naka et al., 1997; Starr et al., 1997). A recent study showed that the overexpression of CIS-1, SOCS-1, SOCS-2 or SOCS-3 in murine myeloid leukemia M1 cells had no effect on M-CSF-mediated cellular response such as tyrosine phosphorylation of STAT3, whereas the overexpression of SOCS-1 or SOCS-3 in the cells inhibited IL-6-mediated cellular response (Novak et al., 1999). The result is in contrast to our finding that overexpression of p56dok-2 inhibited M-CSF-mediated cell proliferation and signal transduction. Therefore, the inhibitory effect of p56dok-2 may be different from that of CIS/JAB/SOCS/SSI family proteins. More studies will be needed to understand fully the inhibitory mechanism of p56dok-2.
Materials and methods
Library subtraction
The murine bone marrow macrophages were prepared by culturing femoral bone marrow cells from C57Bl/6 mice (Charles River) with 100 ng/ml M-CSF for 7 days (Suzu et al., 1997). Cells were factor depleted for 12 h in RPMI 1640 medium containing 10% fetal calf serum (FCS) and then treated with 100 ng/ml M-CSF for 3 h. Poly(A) RNA from untreated cells or from those treated with M-CSF was prepared using a mRNA separator kit (Clontech). cDNA library construction and library subtraction were performed according to the manufacturer’s instructions (PCR-Select cDNA subtraction kit, Clontech). The cDNA fragments of the subtracted cDNA library were cloned into pCR2.1 vector (Invitrogen). Randomly isolated clones were further analyzed by direct sequencing and by northern hybridization using total RNA from unstimulated and M-CSF-stimulated bone marrow macrophages.
Cell culture and transfection
The factor-dependent murine myeloid leukemia M-NFS-60 cells were maintained in RPMI 1640 medium supplemented with 10% FCS and 100 ng/ml M-CSF (Nakoinz et al., 1990; Suzu et al., 1997). The immortalized macrophage-like tumor J774A.1 cells were maintained in RPMI 1640 medium supplemented with 10% FCS (Fan et al., 1993). The murine p56dok-2 cDNA was obtained by RT–PCR using the primers 5′-AGGGCAGAGGCAATGGCAGTGGGGA and 5′-CCACCA TTTCCCCTCCACAGCTTCC (DDBJ/EMBL/GenBank accession No. AF030627). The murine p56dok-2 cDNA containing the entire coding region was subcloned into an expression vector, pCEF, which was made by inserting the promoter region of polypeptide chain elongation factor-1 (Mizushima and Nagata, 1990; Kimura et al., 1994; Suzu et al., 1998) into pRc/CMV vector (Invitrogen). The resulting plasmid DNA or pCEF vector containing no insert was transfected into M-NFS-60 cells by using Lipofectamine reagent (Gibco-BRL). The transfected cells were selected in medium containing 200 µg/ml G418 (Geneticin; Gibco-BRL) and M-CSF, and were screened by immunoblotting using anti-p56dok-2 antibody (kindly provided by Dr W.E.Paul). Alternatively, the murine p56dok-2 cDNA containing the entire coding region was subcloned into pCEF vector in the antisense orientation. Then, the plasmid or pCEF vector containing no insert was transfected into J774A.1 cells as described above. The transfected cells were selected in medium containing 400 µg/ml G418. Expression levels of p56dok-2 were determined by immunoblot analysis.
Cell proliferation assay
M-NFS-60 cells were seeded into 6-well 35 mm plates at a density of 1 × 104 cells/ml and cultured for 2 or 3 days in RPMI 1640 medium containing 10% FCS and 100 ng/ml M-CSF. At the end of the cultures, cells were harvested and counted by the trypan blue dye exclusion method. Alternatively, the cells were seeded into 96-well plates (2 × 103 cells/well) and cultured for 72 h in the presence of SCF (100 ng/ml; Genzyme), G-CSF (10 ng/ml; R&D Systems) or IL-3 (10 ng/ml; Gibco-BRL), or in the absence of factor. Cells were then pulsed with [3H]thymidine for 4 h and the incorporated radioactivity was measured by liquid scintillation counting (Suzu et al., 1998). J774A.1 cells were seeded into 6-well 35 mm plates at a density of 1 × 104 cells/ml and cultured for 3 or 4 days in RPMI 1640 medium containing 10% FCS. At the end of the cultures, cells were harvested and counted.
Northern blot analysis
The bone marrow macrophages were prepared as described above. Murine peritoneal macrophages were prepared by culturing peritoneal cells from C57Bl/6 mice with RPMI 1640 medium containing 10% FCS and 100 ng/ml M-CSF for 7 days. These cells were factor depleted for 12 h in RPMI medium containing 10% FCS. M-NFS-60 cells were factor depleted for 4 h in RPMI medium containing 1% bovine serum albumin (BSA). Cells were then treated with either M-CSF (100 ng/ml), GM-CSF (10 ng/ml) or IL-3 (10 ng/ml) for various periods. J774A.1 cells were serum depleted for 24 h in RPMI 1640 medium. Total RNAs from these cells were isolated using RNazol B reagent (Tel-Test), electrophoresed on agarose gels and transferred to a nylon membrane (Hybond N+, Amersham). The membrane was hybridized with radiolabeled cDNA probes (Suzu et al., 1998). The murine p56dok-2 cDNA containing the entire coding region described above was used as a probe. A murine p62dok cDNA was prepared by RT–PCR using the primers 5′-ATGGACGGGGCTGTGATGGAGGGTC and 5′-CACCCCATTGG ACCTCCTATCAGCA (DDBJ/EMBL/GenBank accession No. U78818). cDNAs for murine c-myc, c-jun, junB and c-fos were also prepared by RT–PCR and used as probes. Primers for c-myc and c-jun were purchased from Clontech, and primers for c-fos were from Stratagene. Primers for junB were 5′-AACAGCCTTTCTATCACGACGACTC and 5′-GGTTCA TCTTGTGCAGGTCGTCCAG (DDBJ/EMBL/GenBank accession No. NM008416). A cDNA probe for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Clontech.
Immunoblot analysis
M-NFS-60 cells were factor depleted for 4 h in RPMI 1640 medium containing 1% BSA. In one experiment, cells were treated with the Src kinase inhibitor, PP2 (10 µM; Calbiochem), for the last 30 min. Cells were then treated with 100 ng/ml M-CSF at 37°C for various periods or left untreated, and were solubilized with lysis buffer (1% Nonidet P-40, 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 µg/ml aprotinin, 1 µg/ml leupeptin and 1 µg/ml pepstatin). J774A.1 cells were also solubilized with the lysis buffer. The cell lysates were centrifuged, and the resulting supernatants were subjected to immunoblot analysis and immunoprecipitation. The protein concentrations of the cleared lysates were determined using the BCA protein assay reagent (Pierce). The cleared cell lysates containing equal amounts of protein were resolved by SDS–PAGE under reducing conditions and the proteins were transferred to a nylon membrane (Hybond-P, Amersham). The membrane was probed with antibodies to p56dok-2 (provided by Dr W.E.Paul), rasGAP (B4F8; Santa Cruz), total ERK (K-23; Santa Cruz) or activated ERK (E-4; Santa Cruz). The antibodies were visualized with horseradish peroxidase-coupled anti-immunoglobulin (Santa Cruz) using the Enhanced Chemiluminescence Western Blotting Detection System (Amersham) according to the manufacturer’s instructions. For immunoprecipitation, the cleared cell lysates containing equal amounts of protein were precleared by treatment with excess protein A/G–agarose (Santa Cruz), then incubated with antibodies to p56dok-2 (M-20; Santa Cruz), Shc (H-108; Santa Cruz) or Grb2 (C-23; Santa Cruz). The immune complexes were precipitated with protein A/G–agarose, washed four times with the lysis buffer, and then subjected to immunoblot analysis with antibodies to phosphotyrosine (PY99; Santa Cruz), p56dok-2 (provided by Dr W.E.Paul), Shc (clone 30; Transduction Laboratories) or Grb2 (clone 24; Transduction Laboratories).
Peritoneal cell analysis of hr/hr mice
hr/hr and +/hr HRS/J mice were obtained from The Jackson Laboratory. Peritoneal cells were collected from mice by lavage with phosphate-buffered saline. Cells were stained for FACS by treatment with phycoerythrin-conjugated anti-Mac-1 antibody (clone M1/70.15; Caltag Laboratories) and analyzed using EPICS PROFILE II (Coulter). Alternatively, cytocentrifuge preparations of peritoneal cells were stained with Wright–Giemsa, and cells with macrophage morphology were enumerated. Total bone marrow cells and spleen cells were obtained from hr/hr and +/hr mice. Total RNAs were isolated from the cells using RNazol B reagent and analyzed for p56dok-2 gene expression by northern hybridization.
Acknowledgments
Acknowledgements
We thank Dr W.E.Paul for providing anti-p56dok-2 antibodies and N.Wakimoto for her excellent technical assistance.
References
- Alonso G., Koegl,M., Mazurenko,N. and Courtneidge,S.A. (1995) Sequence requirements for binding of Src family tyrosine kinases to activated growth factor receptors. J. Biol. Chem., 270, 9840–9848. [DOI] [PubMed] [Google Scholar]
- Barone M.V. and Courtneidge,S.A. (1995) Myc but not Fos rescue of PDGF signalling block caused by kinase-inactive Src. Nature, 378, 509–512. [DOI] [PubMed] [Google Scholar]
- Bhat A., Johnson,K.J., Oda,T., Corbin,A.S. and Druker,B.J. (1998) Interactions of p62dok with p210bcr–abl and Bcr–Abl-associated proteins. J. Biol. Chem., 273, 32360–32368. [DOI] [PubMed] [Google Scholar]
- Cachon-Gonzalez M.B., Fenner,S., Coffin,J.M., Moran,C., Best,S. and Stoye,J.P. (1994) Structure and expression of the hairless gene of mice. Proc. Natl Acad. Sci. USA, 91, 7717–7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpino N., Wisniewski,D., Strife,A., Marshak,D., Kobayashi,R., Stillman,B. and Clarkson,B. (1997) p62dok: a constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell, 88, 197–204. [DOI] [PubMed] [Google Scholar]
- Cheng M., Wang,D. and Roussel,M.F. (1999) Expression of c-Myc in response to colony-stimulating factor-1 requires mitogen-activated protein kinase kinase-1. J. Biol. Chem., 274, 6553–6558. [DOI] [PubMed] [Google Scholar]
- Cong F., Yuan,B. and Goff,S.P. (1999) Characterization of a novel member of the DOK family that binds and modulates Abl signaling. Mol. Cell. Biol., 19, 8314–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey S.J. and Anderson,S.M. (1999) Src-related protein kinases in hematopoiesis. Blood, 93, 1–14. [PubMed] [Google Scholar]
- Courtneidge S.A., Dhand,R., Pilat,D., Twamley,G.M., Waterfield,M.D. and Roussel,M.F. (1993) Activation of Src family kinases by colony-stimulating factor-1 and their association with its receptor. EMBO J., 12, 943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.N., Rock,C.O., Cheng,M., Watson,J.B., Ashmun,R.A., Kirk,H., Kay,R.J. and Roussel,M.F. (1997) Complementation of growth factor receptor-dependent mitogenic signaling by a truncated type I phosphatidylinositol 4-phosphate 5-kinase. Mol. Cell. Biol., 17, 7398–7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diatchenko L. et al. (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl Acad. Sci. USA, 93, 6025–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A. et al. (1998) Molecular cloning and characterization of p56dok-2 defines a new family of RasGAP-binding proteins. J. Biol. Chem., 273, 4827–4830. [DOI] [PubMed] [Google Scholar]
- Endo T.A. et al. (1997) A new protein containing an SH2 domain that inhibits JAK kinases. Nature, 387, 921–924. [DOI] [PubMed] [Google Scholar]
- Fan K., Barendsen,N., Sensenbrenner,L. and Chen,B.D.M. (1993) Deregulation of granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor in murine macrophage cell line J774A.1. J. Cell. Physiol., 154, 535–542. [DOI] [PubMed] [Google Scholar]
- Helgason C.D. et al. (1998) Targeted disruption of SHIP leads to hematopoietic perturbations, lung pathology and a shortened life span. Genes Dev., 12, 1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E., Nocka,K., Beier,D.R., Chu,T., Buck,J., Lahm,H., Wellner,D., Leder,P. and Bessmer,P. (1990) The hematopoietic growth factor KL is encoded by the SI locus and is the ligand of the c-kit receptor, the gene product of the W locus. Cell, 63, 225–233. [DOI] [PubMed] [Google Scholar]
- Ihle J.N. (1995) Cytokine receptor signalling. Nature, 377, 591–594. [DOI] [PubMed] [Google Scholar]
- Jones N. and Dumont,D.J. (1998) The Tek/Tie2 receptor signals through a novel Dok-related docking protein, Dok-R. Oncogene, 17, 1097–1108. [DOI] [PubMed] [Google Scholar]
- Jones N. and Dumont,D.J. (1999) Recruitment of dok-R to the EGF receptor through its PTB domain is required for attenuation of erk MAP kinase activation. Curr. Biol., 9, 1057–1060. [DOI] [PubMed] [Google Scholar]
- Kimura F., Suzu,S., Nakamura,Y., Wakimoto,N., Kanatani,Y., Yanai,N., Nagata,N. and Motoyoshi,K. (1994) Structural analysis of proteoglycan macrophage colony-stimulating factor. J. Biol. Chem., 269, 19751–19756. [PubMed] [Google Scholar]
- Lemay S., Davidson,D., Latour,S. and Veillette,A. (2000) Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol. Cell. Biol., 20, 2743–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.-Q., Subleski,M., Fusaki,N., Yamamoto,T., Copeland,T., Princler,G.L., Kung,H.-F. and Kamata,T. (1996) Catalytic activity of the mouse guanine nucleotide exchanger mSOS is activated by Fyn tyrosine protein kinase and the T-cell antigen receptor in T cells. Proc. Natl Acad. Sci. USA, 93, 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioubin M.N., Myles,G.M., Carlberg,K., Bowtell,D. and Rohrschneider,L.R. (1994) Shc, Grb2, Sos1 and a 150-kilodalton tyrosine-phosphorylated protein form complexes with Fms in hematopoietic cells. Mol. Cell. Biol., 14, 5682–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Damen,J.E., Hughes,M.R., Babic,I., Jirik,F.R. and Krystal,G. (1997) The Src homology 2 (SH2) domain of SH2-containing inositol phosphatase (SHIP) is essential for tyrosine phosphorylation of SHIP, its association with Shc and its induction of apoptosis. J. Biol. Chem., 272, 8983–8988. [DOI] [PubMed] [Google Scholar]
- Lock P., Casagranda,F. and Dunn,A.R. (1999) Independent SH2-binding sites mediate interaction of Dok-related protein with RasGTPase-activating protein and Nck. J. Biol. Chem., 274, 22775–22784. [DOI] [PubMed] [Google Scholar]
- Mizushima S. and Nagata,S. (1990) pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res., 18, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers M.G. Jr, Sun,X.J. and White,M.F. (1994) The IRS-1 signaling system. Trends Biochem. Sci., 19, 289–293. [DOI] [PubMed] [Google Scholar]
- Nagai K., Takata,M., Yamamura,H. and Kurosaki,T. (1995) Tyrosine phosphorylation of Shc is mediated through Lyn and Syk in B cell receptor signaling. J. Biol. Chem., 270, 6824–6829. [DOI] [PubMed] [Google Scholar]
- Naka T. et al. (1997) Structure and function of a new STAT-induced STAT inhibitor. Nature, 387, 924–929. [DOI] [PubMed] [Google Scholar]
- Nakoinz I., Lee,M.-T., Weaver,J.F. and Ralph,P. (1990) Differentiation of the IL-3-dependent NFS-60 cell line and adaption to growth in macrophage colony-stimulating factor. J. Immunol., 145, 860–864. [PubMed] [Google Scholar]
- Nelms K., Snow,A.L., Hu-Li,J. and Paul,W.E. (1998) FRIP, a hematopoietic cell-specific rasGAP-interacting protein phosphoryl ated in response to cytokine stimulation. Immunity, 9, 13–24. [DOI] [PubMed] [Google Scholar]
- Noguchi T. et al. (1999) Tyrosine phosphorylation of p62Dok induced by cell adhesion and insulin: possible role in cell migration. EMBO J., 18, 1748–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak U., Marks,D., Nicholson,S.E., Hilton,D. and Paradiso,L. (1999) Differential ability of SOCS proteins to regulate IL-6 and CSF-1 induced macrophage differentiation. Growth Factors, 16, 305–314. [DOI] [PubMed] [Google Scholar]
- Ptasznik A., Traynor-Kaplan,A. and Bokoch,G.M. (1995) G protein-coupled chemoattractant receptors regulate Lyn tyrosine kinase⋅Shc adapter protein signaling complexes. J. Biol. Chem., 270, 19969–19973. [DOI] [PubMed] [Google Scholar]
- Roche S., Koegl,M., Barone,M.V., Roussel,M.F. and Courtneidge,S.A. (1995) DNA synthesis induced by some but not all growth factors requires Src family protein tyrosine kinases. Mol. Cell. Biol., 15, 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrschneider L.R. (1995) The macrophage-colony stimulating factor (M-CSF) receptor. In Nicola,N.A. (ed.), Guidebook to Cytokines and Their Receptors. Oxford University Press, Oxford, UK, pp. 168–170. [Google Scholar]
- Roussel M.F., Shurtleff,S.A., Downing,J.R. and Sherr,C.J. (1990) A point mutation at tyrosine-809 in the human colony-stimulating factor 1 receptor impairs mitogenesis without abrogating tyrosine kinase activity, association with phosphatidylinositol 3-kinase, or induction of c-fos and junB genes. Proc. Natl Acad. Sci. USA, 87, 6738–6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel M.F., Cleveland,J.L., Shurtleff,S.A. and Sherr,C.J. (1991) Myc rescue of a mutant CSF-1 receptor impaired in mitogenic signalling. Nature, 353, 361–363. [DOI] [PubMed] [Google Scholar]
- Rozakis-Adcock M. et al. (1992) Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature, 360, 689–692. [DOI] [PubMed] [Google Scholar]
- Salcini A.E., McGlade,J., Pelicci,G., Nicoletti,I., Pawson,T. and Pelicci,P.G. (1994) Formation of Shc–Grb2 complexes is necessary to induce neoplastic transformation by overexpression of Shc proteins. Oncogene, 9, 2827–2836. [PubMed] [Google Scholar]
- Sherr C.J., Rettenmier,C.W., Sacca,R., Roussel,M.F., Look,A.T. and Stanley,E.R. (1985) The c-fms proto-oncogene product is related to the receptor for the mononuclear phogocyte growth factor, CSF-1. Cell, 41, 665–676. [DOI] [PubMed] [Google Scholar]
- Shultz L.D., Schweitzer,P.A., Rajan,T.V., Yi,T., Ihle,J.N., Matthews,R.J., Thomas,M.L. and Beier,D.R. (1993) Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell, 73, 1445–1454. [DOI] [PubMed] [Google Scholar]
- Starr R. et al. (1997) A family of cytokine-inducible inhibitors of signalling. Nature, 387, 917–921. [DOI] [PubMed] [Google Scholar]
- Suzu S., Kimura,F., Ota,J., Motoyoshi,K., Itoh,T., Mishima,Y., Yamada,M. and Shimamura,S. (1997) Biologic activity of proteo glycan macrophage colony-stimulating factor. J. Immunol., 159, 1860–1867. [PubMed] [Google Scholar]
- Suzu S., Hatake,K., Ota,J., Mishima,Y., Yamada,M., Shimamura,S., Kimura,F. and Motoyoshi,K. (1998) Identification of alternative spliced transcripts encoding murine macrophage colony-stimulating factor. Biochem. Biophys. Res. Commun., 245, 120–126. [DOI] [PubMed] [Google Scholar]
- Tamir I., Stolpa,J.C., Helgason,C.D., Nakamura,K., Bruhns,P., Daeron,M. and Cambier,J.C. (2000) The RasGAP-binding protein p62dok is a mediator of inhibitory FcγRIIB in B cells. Immunity, 12, 347–358. [DOI] [PubMed] [Google Scholar]
- Tsui H.W., Siminovitch,K.A., de Souza,L. and Tsui,F.W. (1993) Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nature Genet., 4, 124–129. [DOI] [PubMed] [Google Scholar]
- Williams D.E. et al. (1990) Identification of a ligand for the c-kit proto-oncogene. Cell, 63, 167–174. [DOI] [PubMed] [Google Scholar]
- Yamanashi Y. and Baltimore,D. (1997) Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell, 88, 205–211. [DOI] [PubMed] [Google Scholar]
- Yamanashi Y., Tamura,T., Kanamori,T., Yamane,H., Nariuchi,H., Yamamoto,T. and Baltimore,D. (2000) Role of the rasGAP-associated docking protein p62dok in negative regulation of B cell receptor-mediated signaling. Genes Dev., 14, 11–16. [PMC free article] [PubMed] [Google Scholar]
- Yi T., Mui,A.L., Krystal,G. and Ihle,J.N. (1993) Hematopoietic cell phosphatase associates with the interleukin-3 (IL-3) receptor β chain and down-regulates IL-3-induced tyrosine phosphorylation and mitogenesis. Mol. Cell. Biol., 13, 7577–7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi T., Zhang,J., Miura,O. and Ihle,J.N. (1995) Hematopoietic cell phosphatase associates with erythropoietin (Epo) receptor after Epo-induced receptor tyrosine phosphorylation: identification of potential binding sites. Blood, 85, 87–95. [PubMed] [Google Scholar]
- Yoshimura A., Ohkubo,T., Kiguchi,T., Jenkins,N.A., Gilbert,D.J., Copeland,N.G., Hara,T. and Miyajima,A. (1995) A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J., 14, 2816–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsebo K.M. et al. (1990) Stem cell factor is encoded at the SI locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell, 63, 213–224. [DOI] [PubMed] [Google Scholar]