

Abstract
Intensive investigations into the pathophysiological significance of the proteasome in the heart did not start until the beginning of the past decade but exciting progresses have been made and are summarized here as two fronts. First, strong evidence continues to emerge to support a novel hypothesis that proteasome functional insufficiency represents a common pathological phenomenon in a large subset of heart disease, compromises protein quality control in heart muscle cells, and thereby acts as a major pathogenic factor promoting the progression of the subset of heart disease to congestive heart failure. This front is represented by the studies on the ubiquitin-proteasome system (UPS) in cardiac proteinopathy, which have taken advantage of a transgenic mouse model expressing a fluorescence reporter for UPS proteolytic function. Second, pharmacological inhibition of the proteasome has been explored experimentally as a potential therapeutic strategy to intervene some forms of heart disease, such as pressure overload cardiac hypertrophy, viral myocarditis, and myocardial ischemic injury. Not only between the two fronts but also within each one, a multitude of inconsistency and controversy remain to be explained and clarified. At present, the controversy perhaps reflects the sophistication of cardiac proteasomes in terms of the composition, assembly, and regulation, as well as the intricacy and diversity of heart disease in terms of its etiology and pathogenesis. A definitive role of altered proteasome function in the development of various forms of heart disease remains to be established.
Keywords: proteasome, heart disease, desmin-related cardiomyopathy, myocardial ischemia, cardiac hypertrophy, myocarditis, diabetes
1. Introduction
The heart is arguably the only organ in the body that is constantly bearing heavy mechanical work load, a high metabolic rate, a diverse array of neurohumoral regulations, and a variety of stresses under physiological conditions, let alone during disease states. It is a challenge for a polypeptide to attain and maintain its proper conformation in a beating heart. Consequently, the heart is among a few organs in the body that are more susceptible to proteotoxic stress [1]. Therefore, protein quality control (PQC) is vitally important for the heart [2–6]. As in other cells, PQC, both endoplasmic reticulum (ER) dependent and ER-independent, is carried out by an elaborate collaboration between molecular chaperones and targeted proteolysis. The latter is primarily carried out by the ubiquitin-proteasome system (UPS) although the autophagy-lysosomal pathway may play a supplemental role in removing misfolded proteins, especially insoluble ones [1, 7, 8]. Hence, from the PQC point of view, normal proteasome function is essential to cardiac physiology. However, the role of the proteasome in the heart is far more than just PQC. First, proteasome function is important for the balanced turnover of functionally important cardiac proteins, such as gap-junction related proteins and contractile proteins [9, 10]. Second, proteasome affects membrane receptors that regulate cardiac function, such as G protein coupled receptors. Proteasome-mediated degradation can affect the functional states of receptors via degrading components involved in the receptor trafficking [11], signaling coupling [12], and even receptor proteins per se [13]. Third, proteasome function is involved in activation or inactivation of many signal transduction pathways, such as NF-κB and β-catenin mediated pathways [14–16]. These pathways play important roles in cardiac remodeling. Finally, proteasome function is important in regulation of gene expression [17, 18]. In the heart, in addition to NF-κB and β-catenin, the proteasome mediates p53 degradation [19]. Increased proteasome activities have been linked to accumulation of p53 and increased apoptosis in disease hearts [20, 21]. Altered proteasome-mediated degradation of transcription factor GATA4 was found to be involved in cardiomyocyte injury [22].
In cardiac cells, efficient and well controlled protein degradation is critical to the removal of the damaged and terminally misfolded proteins as well as to the regulatory degradation of normal proteins to maintain intracellular protein homeostasis and thus the normal cardiac function. On the other hand, pathological processes, such as ischemia, oxidative stress, inflammation, and aberrant protein aggregation, can alter proteasome activities, which may in turn contribute to the progression of the disease. In the past decade, studies have increasingly revealed the alterations of proteasome function in a variety of heart disease [2–4, 23]. Generally, both increased and decreased proteasome activities have been reported in a number of heart disease animal models and diseased human hearts. These inconsistency and controversy are not surprising in considering the heterogeneity of heart disease, which is usually progressively developed from different primary causes. Nevertheless, the emerging evidence indicates that proteasome functional insufficiency (PFI) occurs in at least a school of heart disease and may play pathogenic roles promoting disease progression [21, 24–30], while proteasome inhibition has shown some promise in pharmacological intervention of a school of cardiac pathology [31, 32].
2. Cardiac proteasomes
Until very recently, the understandings of the architecture, function, and regulation of mammalian cardiac proteasomes are largely based on studies of non-cardiac cells and lower eukaryotes such as yeast. Emerging studies mainly from Ping’s laboratory have provided novel insight into the characteristics of cardiac proteasome composition and regulation [33, 34]. The proteomic analysis of proteasome complexes purified from murine hearts reveals that cardiac proteasomes are much more sophisticated compared with those from yeast and bear distinctions from other mammalian tissues as well [34, 35].
The 20S subcomplex of cardiac proteasomes consists of all regular α and β subunits (α1 through α7 and β1 through β7). In addition, the three inducible β subunits (β1i, β2i, and β5i) are also found in the heart. Therefore, compositions of the cardiac 20S complex can be quite diverse. It is speculated that the diversity of 20S assembly may provide functional specificity and selectivity. The importance of β1i in the heart was recently demonstrated in mice by gene targeting. Mice with germ-line ablation of the β1i gene were shown to lose ischemia-preconditioning (IPC) induced cardioprotection. IPC-mediated degradation of PTEN (phosphatase and tensin homologue deleted on chromosome 10) and activation of the downstream protein kinase Akt were impaired in these mice [36].
In the 19S subcomplex from the heart, in addition to all the regular ATPase subunits (Rpt 1 through Rpt 6) and non-ATPase subunits (Rpn 1 through Rpn 12), a new alternatively spliced isoform of Rpn10 (Rpn10b) is expressed along with its primary isoform Rpn 10a [35].
In addition to 19S being an activator, the 11S and PA200 complexes also can bind to the 20S proteasome to enhance its function [37]. Heart tissues express less 11S proteasome subcomplexes than liver [35]. PA200, although found in total heart lysates, was not detected in the 26S or 20S proteasome preparations from the heart. These data suggest that the 11S and PA200 may not play a major role in regulation of 20S activity in the heart under the baseline condition. However, myocardial expression of 11S proteasomes was found to increase in experimental diabetes [26]. The PI31 complex, an inhibitor of 20S proteasomes [38, 39], also seems to co-exist with the cardiac 26S proteasome complexes [34].
The proteolytic function of cardiac proteasomes is not only determined by the molecular organization and structural assembly, but also regulated via posttranslational modification and partner association of the proteasome. Using a new approach of in-solution isoelectric focusing in a laminar flow, Drews et al. found that cardiac proteasomes exhibit different molecular composition and proteolytic activity from those in the liver. For example, most of cardiac proteasome components show an isoelectric point (pI) of 5.26, compared with the pI of 5.05 for proteasomes from most other tissues, which may reflect the phosphorylation complement of these proteasomes. In addition, cardiac 20S proteasome subpopulations display distinct proteolytic activities [40, 41]. These findings are echoed by a recent report showing that the multiple subpopulations of cardiac proteasomes respond differently to the same proteasome inhibitor [42].
Protein kinase A (PKA) and protein phosphatase 2A (PP2A) were found in purified murine cardiac proteasome preparations, suggesting that PKA and PP2A may regulate the 20S complex. Indeed, in vitro functional study revealed that PKA and PP2A can respectively phosphorylate and dephosphorylate serine- and threonine-sites in multiple cardiac 20S subunits. The PKA-induced phosphorylation increased the three peptidase activities of the 20S proteasome in a substrate-specific fashion [43]. A subsequent study by others with non-cardiac cells further demonstrates that PKA positively regulates proteasome function through phosphorylation of Ser120 of Rpt6 subunit of the 19S proteasome [44]. A recent study suggests PKA activation enhances proteasome activities in canine hearts [45, 46].
It is well known that ATP is required for both ubiquitination and proteasomal degradation. Interestingly, Powell et al reported that the optimal ATP concentration for in vitro proteasome peptidase activity assays is often lower than 100 μM and the in vitro proteasome peptidase activities will go down when the ATP concentration used for the assay is beyond the optimal level [47]. The optimal ATP concentrations are apparently lower than the normal intracellular ATP level by a factor of >10 because the latter is in the low millimolar range, depending on the cell types [48, 49]. Geng et al subsequently reported the activation of at least a subset of 26S proteasomes at critically low ATP levels and its contribution to myocardial injury in cold ischemia [50]. These observations led to a provocative hypothesis that the physiological levels of ATP may negatively regulate proteasome activities in the cell, which has recently been tested and supported by a study from Liu and Wang’s laboratories using cultured mammalian cells [51]. The mechanism underlying the negative impact of high ATP on the proteasome remains to be investigated but the implication of this newly unraveled phenomenon is potentially far-reaching in terms of both a better understanding of proteasome functional changes in pathological processes and the development of new strategies to manipulate proteasome function to treat disease.
Collectively, emerging studies have revealed that compositions and organizations of mammalian cardiac proteasomes are highly complex and diverse, suggesting functional complexity, specificity, and selectivity. Cardiac proteasome activities can be regulated through at least three mechanisms: synthesis and assembly of the proteasome complex, interactions between the proteasome proper and associated partners, and posttranslational modifications of proteasome subunits [34]. Because of these multiplicity and complexity in structural composition and functional regulation, it is not surprising that alterations of proteasome activities in heart disease are complex.
3. Proteasome functional insufficiency in cardiac proteinopathy
Like neural degenerative disease, cardiac proteinopathy is coined to describe a family of heart disease with the presence of protein aggregates in heart muscle cells being the pathological feature [1]. The most studied cardiac proteinopathy is unarguably desmin-related cardiomyopathy (DRC). DRC is actually the cardiac manifestation of desmin-related myopathy which is featured by the presence of desmin-positive protein aggregates in muscle cells, including skeletal, cardiac, and sometimes smooth muscle cells. Desmin-related myopathy is caused by mutations in the genes encoding for desmin (the muscle specific intermediate filament protein) or its partner proteins, such as αB-crystallin (CryAB), and myotiolin. These DRC-linked mutations often cause misfolding of their respective gene products and/or affect the assembly and maintenance of desmin filaments [6]. Transgenic (tg) mice with cardiomyocyte-restricted overexpression of human DRM-linked mutations, namely a 7-amino acid (R172 through E178) deletion mutation of desmin (D7-des) or a missense (R120G) mutation of CryAB (CryABR120G), recapitulate key aspects of human DRM [52, 53]. The tg mice carrying 3 copies of murine CryABR120G display no discernible cardiac phenotype at 1 month but develop cardiac hypertrophy and diastolic malfunction at 3 month and CHF at 6 months [53], providing a reliable animal model for investigations into the pathogenesis and experimental intervention of cardiac proteinopathy [54–57].
Ubiquitin-proteasome system proteolytic function was vigorously assessed, and PFI has been revealed, in both D7-des and CryABR120G tg mouse hearts [27, 58], which provides the first unequivocal demonstration in intact animals that expression of misfolded proteins leads to PFI. Notably, UPS function in both cases was assessed using a tg mouse model of ubiquitous expression of a previously validated UPS-specific surrogate substrate engineered by modifying an enhanced green fluorescence protein (GFP) via carboxyl fusion of degron CL1, referred to as GFPdgn [59]. Changes in GFPdgn protein stability reflect inversely the status of UPS proteolytic function. When introduced into CryABR120G mice, GFPdgn accumulation occurred in the heart before 1 month and became more pronounced as the animal was ageing even though all the three proteasome peptidase activities were markedly increased. Further evaluation suggests that insufficient 19S proteasome function may be the immediate cause of PFI developed in DRC mouse hearts. Inhibition of protein aggregation via overexpression of molecular chaperones or administration of pharmacological agents significantly diminished the ability of overexpression of these mutant proteins to accumulate the surrogate substrate in cultured heart muscle cells [58, 60]. This not only confirms that protein aggregation is sufficient to impair proteasome function, as observed previously in other cell types [61], but also proves for the first time that aberrant protein aggregation is required for the mutant desmin or CryABR120G to impair the proteasome in heart muscle cells. It should be pointed out that comparable or even greater overexpression of wild type desmin or CryAB transcripts does not lead to increases in the level of total ubiquitinated proteins, proteasome peptidase activities, or GFPdgn accumulation in mouse hearts [27, 58], indicating that it not simply the amount of protein load but rather the misfolding nature of the protein that causes PFI.
Desmin-related cardiomyopathy is not common but the pathophysiological significance of PFI observed in DRC mice is potentially far reaching. This is because aberrant protein aggregation at least in the form of pre-amyloid oligomers formation has been observed in most failing human hearts with hypertrophic or dilated cardiomyopathy [56]. Aberrant protein aggregation was also implicated in a mouse model of hypertensive cardiomyopathy which is the second most common cause of CHF [62]. Therefore, PFI may occur during development of a large subset of human disease to CHF. Indeed, increased levels of ubiquitinated proteins have been observed in failing human hearts resulting from dilated cardiomyopathy or ischemic heart disease [63]. The latter is the most common cause of CHF. Increased ubiquitinated proteins have been observed virtually in all animal models of primary heart disease no matter whether altered proteasome peptidase activities were measured or not [1]. Ubiquitinated proteins are generally degraded very efficiently by the proteasome in normal cells. Hence, an increase in the steady level of ubiquitinated proteins in the cell is usually indicative of PFI. Interestingly, this notion was challenged by a recent report claiming that the accumulation of ubiquitin conjugates in mouse brains overexpressing disease-linked poly-glutamine happens without other evidence of global impairment of the UPS, based on the failure to detect an increase in the protein level of a co-expressed proteasome reporter substrate (UbG76V-GFP) [64].
A major concern we raise on the report by Maynard et al. is that they failed to rule out the possibility that a decrease in the synthesis of the reporter could have rendered the unchanged UbG76V-GFP protein level in the face of UPS impairment. In fact, the data on one of the reporter gene products suggest that the synthesis of the reporter was significantly decreased, which argues against the conclusion of paper, but the author missed a more complete and convincing interpretation of the data in its entirety. This is because as revealed by the western blot analyses, the reporter transgene gives rise to at least two main protein products: a larger one and a smaller one. The former is degraded by the proteasome but the latter is not; therefore, only the larger product can be used as the intended reporter for proteasomal function. Compared with the controls, the level of the larger protein product in the mouse brains expressing tg poly-glutamines remained unchanged but its smaller cousin displayed a consistent and remarkable decrease. The author interprets the decrease of the smaller tg product as a probable result of increased degradation from a unknown proteolytic pathway [64]. However, the data can be more reasonably interpreted as that the transcription and/or translation of the reporter transgene were significantly decreased in the poly-glutamine-expressing brains. This decrease has led to the decrease in the shorter tg protein product whose degradation is not affected by proteasome impairment, whereas the accumulation of the larger tg product by impaired UPS proteolytic function had been masked by the decreased synthesis of the reporter gene. This alternative interpretation is consistent with transcription dysregulation previously described in poly-glutamine disease [65], as well as the known intertwining regulations between the proteasome and gene expression in general [66, 67]. Therefore, the challenge raised by the report of Maynard et al. against considering accumulation of ubiquitinated proteins in the cell as a sign of PFI is very debatable.
In addition to bona fide misfolded proteins, some other mutant proteins such as some familial hypertrophic cardiomyopathy-linked mutations of cardiac myosin binding protein C were also shown to cause PFI in cultured heart muscle cells or the heart [68, 69].
As the primary proteolytic machine to remove misfolded proteins in the cell, the proteasome plays a critical role in PQC. PFI can obviously lead to PQC inadequacy, but this can be compensated to some extent by activation of autophagy. Elegant work from Hill’s laboratory has recently shown that autophagy was activated as a protective mechanism against CryABR120G based DRC in mice [70]. Nevertheless, as mentioned above, the accumulation of aberrant protein aggregates and ubiquitinated proteins seen in CHF resulting from a variety of primary heart disease suggests strongly that the PQC inadequacy as a result of PFI is a common phenomenon in the progression of these diseases to CHF. Inadequate PQC allows more misfolded proteins to escape from surveillance and triggers more protein aggregation which in turn impairs the proteasome, thereby forming a vicious circle. Experimental studies have repeatedly shown that the sufficiency of the expression of misfolded proteins and resultant aberrant protein aggregation to cause the heart to fail [52, 53, 71, 72]. Therefore, PQC inadequacy has been proposed as a major pathogenic factor in the development of CHF [1, 2, 6].
As illustrated in Figure 1, the center of this evolving PQC hypothesis of CHF is PFI. In addition to be supported by the experimental studies described above, this hypothesis is increasingly backed up by clinical evidence, including the development of cardiac dysfunction in some patients receiving proteasome inhibitors for cancer treatment and the positive identification of autoimmunity against proteasome subunits in some dilated cardiomyopathy patients [28, 73–75].
Figure 1. An illustration of the hypothesis that proteasome functional insufficiency contributes to cardiac dysfunction.
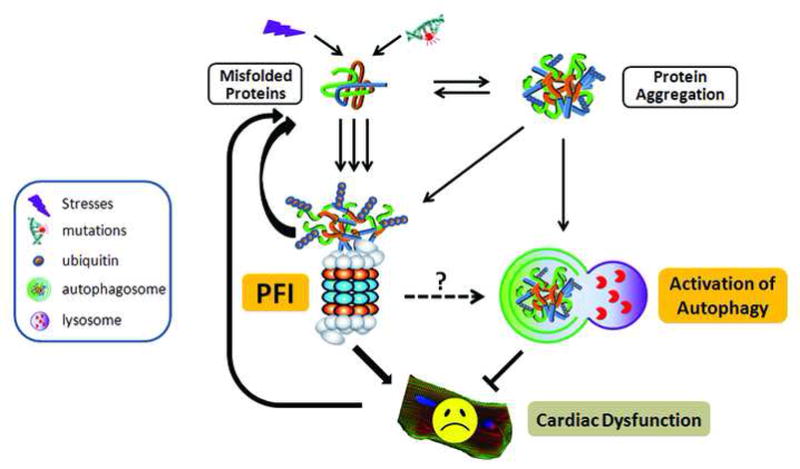
Under stress conditions, the increased production of abnormal proteins overwhelms the proteasome and the resultant aberrant protein aggregation impairs proteasome proteolytic function, leading to proteasome functional insufficiency (PFI). PFI in turn accumulates misfolded proteins, thereby forming a vicious cycle between accumulation of misfolded proteins and PFI. PFI can induce cardiomyocyte dysfunction and/or cardiomyocyte death and the dysfunctional cardiomyocytes produce more misfolded proteins, thereby forming a vicious cycle between cardiac dysfunction and PFI. Aberrant protein aggregation and/or PFI activate the autophagy-lysosome pathway, which may help relieve proteotoxic stress and attenuate PFI induced cardiomyocyte dysfunction. The legend for the symbols is shown in the box at far left. (Adopted from Su and Wang Cardiovasc Res 2010; 85; 253–262. Permission will be sought from the publisher.)
The mechanisms by which PFI induces cardiac dysfunction can be multiple. The imbalance of protein synthesis and degradation and the accumulation of abnormal proteins in the heart can impact heart muscle cell structure, function, and fate. For example, PFI reduced the degradation of p53 and Bax, the pro-apoptotic proteins in the heart, which may contribute to cardiomyocyte apoptosis in heart failure [21, 76]. In addition, PFI may alter the activation of some signaling pathways that are involved in cardiac remodeling and dysfunction. In a recent study [30], for example, we found that PFI activated the calcineurin/NFAT (Nuclear Factor of Activated T-cells) pathway, a pivotal pathway that mediates cardiac remodeling and dysfunction [77]. Our data show that inhibition of 20S proteasomes stimulated NFAT nuclear translocation in mouse hearts and cultured adult mouse cardiomyocytes in a calcineurin-dependent manner. We further revealed that calcineurin protein levels and NFAT transactivation were markedly increased in the heart of a mouse model of DRC and that expression of an aggregation-prone mutant desmin also directly increased calcineurin protein levels in cultured cardiomyocytes. Moreover, we demonstrated that pharmacologically induced systemic proteasome inhibition was sufficient to cause cardiac hypertrophy and facilitated maladaptive remodeling of a pressure overloaded heart [30]. These findings indicate that PFI-induced activation of calcineurin-NFAT signaling pathway may contribute to cardiac remodeling and dysfunction in DRC hearts and other cardiomyopathy in which PFI is present.
4. Proteasomes in ischemic heart disease
It has been reported that myocardial ischemia causes loss of proteasome activities. In vivo, after 30 minutes of occlusion of the anterior descending branch of left coronary artery, the trypsin-like activity of 20S proteasomes in heart tissues was significantly reduced [24]. Furthermore, the loss of proteasome activity was also found in isolated heart with ischemia/reperfusion (I/R) [78].
It is well known that myocardial I/R causes a robust increase in the level of reactive oxygen species (ROS) that is responsible for I/R injury. There is a good body of evidence that ROS can inhibit proteasome function [79]. A recent in vitro study showed that oxidative modification of the 20S subunits resulted in changes in the 2D gel electrophoresis pattern of proteasome subunits that were associated with reduced 20S proteasomal activity [80]. In addition to the 20S, it was also found that the 19S ATPase subunit Rpt6 was very sensitive to oxidative stress [81, 82]. Therefore, the oxidative modification of proteasome subunits is at least one of the mechanisms of proteasome impairment observed in myocardial ischemia and I/R injury.
In addition, myocardial ischemia results in ATP depletion in the heart tissue [83]. Since proteasome-mediated proteolysis is ATP-dependent, ATP depletion could well be a cause of reduced proteasome function in ischemic heart disease [4, 47].
The proteasome is responsible for degrading apoptosis associated proteins [84]. It has been found that inhibition of proteasome accumulates pro-apoptotic proteins, including p53 [20, 21, 85], Bax [21], and PKCδ [86]. Indeed, proteasome inhibition increases apoptosis in ischemic hearts [87]. Therefore, the inhibition of proteasomal function during ischemia and I/R injury may be detrimental, at least partially due to increases in cardiomyocyte apoptosis. This possibility is well demonstrated by recent studies using cardiac IPC models [88, 89]. Data from these studies show that IPC, known as its protective effect against cardiac I/R injury, can prevent oxidative inhibition of 26S proteasomes in the heart, which is accompanied with reduced accumulation of pro-apoptotic proteins. Conversely, pretreatments with proteasome inhibitors diminish the cardio-protection of IPC, associated with increased accumulation of pro-apoptotic proteins, in I/R hearts. These data further suggest that preservation and restoration of proteasomal function is important during cardiac ischemia and I/R.
Conversely, it has been found that treatments with proteasome inhibitors reduced infarct size and preserved cardiac function in myocardial I/R [90, 91]. This protective effect was thought to be associated with the inhibition of inflammatory response [29, 92]. It is known that during I/R, the NF-κB pathway is activated, resulting in the increased inflammatory cytokines, such as TNFα and IL-6, which deteriorate tissue damage [93]. UPS-mediated degradation of inhibitory protein IκB is required for the activation of NF-κB [15, 94]. Therefore, the beneficial effect of proteasome inhibition seen in I/R may reflect an indirect effect via inhibition of the activation of NF-κB and thus the reduction of inflammatory response in the lesion area.
It is unclear how to unify these contrary findings, the inhibited proteasome activities versus the protective effect of proteasome inhibition in myocardial I/R. Interestingly, a study showed that treatments with the low or high concentrations of an oxidative reagent, peroxynitrite, respectively increased or decreased the activities of purified 20S proteasomes in vitro. Furthermore, in the in vivo experiment, the treatment with an oxidative reagent peroxynitrite for 6 hours induced an increase in proteasome function, while after 24 hours of the treatment, the proteasome activity was inhibited [95]. These data suggest that the different extents and/or durations of oxidative exposure to the proteasome in myocardial I/R may cause a dual-effect on proteasome function. Based on this notion, it is possible that at the early stage of myocardial ischemia, oxidative modification may increase proteasome activities, which may be involved in activation of the NF-κB signaling pathway and thus inflammatory responses. With a longer time and more severe oxidative modification, on the other hand, the proteasome activity is inhibited. It is reasonable to presume that proteasome inhibition could serve as a compensatory mechanism in response to the robust activation of NF-κB and inflammatory reactions in I/R hearts. However, given the broad and critical function of proteasomes on protein degradation, the long term inhibition of the proteasome could be detrimental, particularly when damaged proteins are increased in I/R. Indeed, a recent study indicates that pre-perfusion with the tocotrienol-rich fraction of palm oil produced protective effect in isolated hearts with I/R via proteasome stabilization [96].
5. Altered proteasome function in load-dependent heart disease
Being terminally differentiated, adult heart muscle cells are unable to divide. Instead, they grow bigger in individual cell size via a process known as cardiac hypertrophy, in response to increased workload. By the Law of Laplace, the increased ventricular pressure due to hypertension will increase the wall tension (stress), whereas an increase in ventricular wall thickness (hypertrophy) can reduce the wall tension to counter ventricular pressure overload. While physiological cardiac hypertrophy induced by exercise is considered a adaptive response, pathological hypertrophy, as seen in hypertension, heart valve disorders, and other cardiovascular illness, are maladaptive and will ultimately lead to CHF [97]. Cardiac hypertrophy involves alterations in both protein synthesis and degradation [98]. It remains controversial whether proteasome function during pressure-overloaded cardiac hypertrophy is insufficient or not.
It was found in an earlier study using transverse aortic constriction (TAC) induced cardiac hypertrophy in mice that all 3 peptidase activities of the proteasome began decreasing at 2 weeks after TAC, before the onset of cardiac dysfunction. At 4 weeks when heart failure occurred, the decrease in proteasome activities became more pronounced. Consequently, there were progressive increases in ubiquitinated proteins in the heart from 2 weeks to 4 weeks after TAC [21]. Through immunohistochemistry analyses, this study also showed increased ubiquitinated proteins in human failing hearts. It is therefore suggested that decreased proteasome function may be involved in the development of cardiac hypertrophy and heart failure, supporting the PQC hypothesis [1].
It is not clear how proteasome function is impaired in the load-dependent heart disease. Studies using mRNA microarrays showed a down-regulation of some α and β subunits of the 20S proteasome in heart tissues from CHF patients compared with those from healthy donors [99, 100], suggesting that the impaired proteasome function can occur at the transcriptional level. Interestingly, recent clinical studies show that the treatment to reduce heart load (unloading) in CHF patients increases 20S proteasome expression and activities and decreases ubiquitinated protein concentration compared with before treatment [101–103], suggesting that the cardiac overload inhibits proteasome expression and function. In addition, the increased abnormal protein aggregates may contribute to the inhibition of proteasome activity in load-dependent heart diseases [23]. Moreover, it is known that ROS are increased and play a role in development of CHF. It is conceivable that the increased ROS may modify and inhibit proteasome subunits in failing hearts, as seen in other conditions such as ageing and Alzheimer’s disease [104, 105]. This possibility is supported by a recent study that investigated proteasomal function in human heart failure and hypertrophic cardiomyopathy [106]. Data from this study show that proteasome activities were markedly reduced in failing and hypertrophic hearts, which were partially restored after mechanical unloading. While there were no changes in the protein contents of 11S, 19S, and 20S proteasomes, total protein carbonyls, 4-hydroxynonenylated proteins, and oxidative modification of 19S ATPase subunit Rpt5 were increased in failing hearts compared with the non-failing hearts. These data provide evidence that posttranslational modifications to proteasome subunits may account for proteasome inhibition and defective protein degradation in human failing hearts.
Notably, there are also experimental reports showing that proteasome activity is increased in pressure-overloaded cardiac hypertrophy. In a study using a canine model with chronic pressure overload induced by aortic banding, the transcript and protein levels of some proteasome subunits, as well as proteasome activities were shown to increase in the inner layer but not the outer layer of the hypertrophic left ventricle wall [107]. Furthermore, pharmacologically induced proteasome inhibition was shown in rodents to reduce pressure-overload cardiac hypertrophy and protect against cardiac remodeling [108–111]. Understandably, cardiac hypertrophy/remodeling requires proteolysis to tear down the existing structure, namely sarcomeres, for building up new ones in heart muscle cells. The degradation of most sarcomeric proteins is via the UPS [2, 112]. Moreover, the UPS functioning properly is necessary for efficient transcription and translation that are increased during cardiac hypertrophy. Therefore, it is not surprising that proteasome inhibition can block or slow down the hypertrophy and remodeling processes. What was unexpected is that these reports present data seemingly arguing against the importance of PQC in pressure-overloaded hearts. They observed that chronic and quite severe proteasome inhibition did not seem to adversely affect cardiac function but rather in some cases appeared to be beneficial to the diseased heart. As mentioned earlier, proteasome function is critical to the activation of the NF-κB signaling pathway at multiple steps [15, 113]. The activation of NF-κB is involved in cardiac hypertrophy and remodeling [114, 115]. Hence, one proposed mechanism for the protection of proteasome inhibition is reducing NF-κB activation during the maladaptive hypertrophy and heart failure [32]. Another potential explanation is that chronic proteasome inhibition activates autophagy which in turn compensates for the reduced proteasome function to maintain PQC. This attractive hypothesis has not been directly tested but is consistent with previous findings from a similar and a different cardiac pathological condition [62, 70]. It should be pointed out that a true outcome study on chronic proteasome inhibition treatment to load-dependent cardiomyopathy or most other cardiomyopathies using mortality or lifespan as the endpoint has not been reported. It is also noted that the no-harm or beneficial effects of whole-body pharmacological inhibition of proteasomes observed in rodent models provide valuable information for therapeutic exploration but may not necessarily stand against a potential pathogenic role of cardiomyocyte-restricted PFI implicated in a large subset of heart disease. To define the latter, proteasome inhibition targeted specifically to cardiomyocytes is required but has not been reported yet. As indicated earlier, many studies on humans do suggest non-desirable cardiac effects of proteasome inhibition, especially when the patients are elderly or have preexisting heart conditions [28, 73–75].
6. Enhanced proteasome proteolytic function in doxorubicin cardiotoxicity
Doxorubicin (Dox) is a potent anticancer agent that belongs to the anthracycline family [116]. Its major side effect is cardiotoxicity when used as anti-malignance agent, preventing Dox from being more effectively or broadly used [117, 118]. To date the mechanism by which Dox induces cardiotoxicity remains poorly understood. Recent studies indicate that Dox can increase proteasome activities in cultured heart muscle cells and the heart of intact animals, raising a possibility that the excessive proteasome activation may be a mediating factor for Dox cardiotoxicity [119]. Using the GFPdgn reporter mice, Wang’s laboratory was able to show that the administration of Dox significantly reduced GFPdgn protein level compared with the control group. Consistent with the in vivo study, treatment with Dox in culture cardiomyocytes from adult GFPdgn mice significantly reduced GFPdgn protein in a dose-dependent manner. This effect was completely abolished by proteasome inhibitors [59]. In line with this study, Liu et al. have investigated further the mechanism underlying the effect of Dox on proteasome function [120]. In a 3T3 cell line stably expressing a proteasome function reporter (GFPu) that was engineered similarly to GFPdgn [61], treatment with Dox enhanced the degradation of GFPu via increasing proteasome activities. Moreover, Dox treatment increased E3 ligase CHIP (carboxyl-terminus of heat shock protein cognate 70) via a posttranscriptional mechanism [120]. These data suggest that Dox increases UPS function by acting on both the ubiquitination apparatus and the proteasome. Dox-induced increase in proteasome activities were also suggested by other studies. Nuclear factor-activated T cells 5 (NFAT5) is a ubiquitously expressed transcriptional factor. Dox treatment was found to decrease the protein level, but not the mRNA level, of NFAT5 in cultured cardiomyocytes. This effect was prevented by the proteasome inhibitor, MG-132, or proteasome-specific inhibitor 1, suggesting that Dox increases NFAT5 degradation via enhancing proteasome activities [121]. Another study indicated that Dox treatment induced apoptosis in K562 erythroleukemic cells via enhancing proteasome activities. This study further revealed that Dox increased 26S proteasome function by inducing the changes in composition and phosphorylation of proteasome subunits [122].
It will be interesting and important to test whether proteasome inhibition prevents or attenuates Dox cardiotoxicity in intact animals. Further investigation of the mechanisms of Dox-mediated activation of the proteasome may not only provide insight into the Dox cardiotoxicity but also shed light on the development of benign pharmacological agents to enhance proteasome function. The latter have a great therapeutic potential to disease with PFI being a major pathogenic factor although Dox itself obviously cannot be used for that purpose.
7. Proteasomes in viral myocarditis and other cardiac disorders
As discussed in a recent comprehensive review by Luo et al. [123], the role that proteasome-mediated proteolysis plays in the pathogenesis of viral myocarditis is quite unique, compared with that in most other types of heart disease. Viral myocarditis is caused by persistent viral infection in the heart. The infection can cause cardiac inflammation, remodeling, dysfunction, and even CHF [124, 125]. Proteasome function is involved in viral replication and therefore important to the effectiveness of viral infection [123]. One of the major viruses that cause myocarditis is coxsackie virus B3 (CVB3) [126]. In a CVB3-induced murine myocarditis model, the formation and activity of immunoproteasomes was observed in the heart, which was believed to contribute to the increased cardiac inflammatory response [127]. Treatment with proteasome inhibitor attenuated CVB3-induced myocardial damage [128], suggesting that the abnormal proteasome function may play a pathogenic role in the myocardial damage of viral myocarditis.
Diabetes is a growing risk factor of heart disease [129–131]. Diabetic cardiomyopathy is characterized as cardiac remodeling and dysfunction that are independent of diabetes-induced coronary arterial diseases, hypertension, or renal dysfunction [131]. Hyperglycemia, increased ROS, and perhaps other altered metabolic factors present in diabetes can cause protein glycation, oxidation, and cross-linking. Accumulation of these abnormal proteins in the heart may contribute to diabetic cardiomyopathy. It is unknown if the accumulation of these abnormal proteins is associated with inadequate protein degradation. An in-depth study on proteasome alterations in diabetic hearts has not been reported yet but both increased and decreased proteasome function were implicated in diabetes. It appears that hyperglycemia and hyperinsulinemia of diabetes can increase UPS function, which may be responsible for endothelial damage and atherosclerosis as well as muscle wasting in diabetes [132]. On the other hand, it was reported that proteasome activities were inhibited in the heart of rats with streptozotocin induced diabetes [26]. Increased ROS is also a major pathogenic factor in diabetes. Therefore, it is conceivable that oxidative modification of proteasome subunits may play a role in altering proteasome function in diabetes. The role of proteasome dysfunction in diabetic cardiomyopathy remains to be determined.
Ageing related cardiac malfunction is characterized as cardiac hypertrophy, fibrosis, and increased ventricular stiffness [133]. As with other organs and systems, ageing related decreases in proteasome function in the heart have been described [134]. It is believed that the attenuated proteasome function is responsible for accumulation of oxidized proteins in the aged heart [134]. Notably, a recent study indicates that reduction of oxidized proteins in the heart by dietary restriction is due to preservation of proteasome function [135]. Ageing is associated with an increased ROS [136], which is involved in aging associated degenerative diseases [105]. The previously described ROS-induced modifications of proteasome subunits may play a role in the alteration of proteasome function in ageing [137]. Intracellular accumulation of large aggregates of oxidized proteins (e.g., lipofuscin) increases in aged hearts. Powell et al. demonstrated that exposure of heart muscle cells to lipofuscin-like materials in culture causes apoptosis as a result of impaired proteasome function [138].
8. A summary and future prospects
Although intensive investigation in the (patho)physiological significance of the proteasome in the heart has not begun until very recently, exciting progresses have been made. On one hand, evidence is mounting to support the central hypothesis that PFI represents a common pathological phenomenon in a large subset of heart disease, compromises PQC in the heart, and thereby acts as a major pathogenic factor in the progression of the heart disease to CHF. On the other hand, pharmacological inhibition of the proteasome is experimentally implicated as a potential therapeutic strategy to intervene several categories of heart disease, such as pressure overload cardiac hypertrophy, viral myocarditis, doxorubicin cardiotoxicity, and perhaps myocardial ischemic injury. Not only between the two fronts but also within each one exists a multitude of unexplained inconsistency and controversy, which may eventually be clarified in many cases as the field is being advanced but, at the present time, may just be a natural reflection of the sophistication of cardiac proteasomes in terms of the composition, assembly, and regulation, as well as the intricacy and diversity of heart disease in terms of its etiology and pathogenesis.
The research into the role of the proteasome in heart disease is still in its infancy. Advances in fundamental understanding on the mechanisms that harmonize or disharmonize proteasome function and heart function will no doubt open new chapters of cardiac physiology and pathophysiology and promote the search for new measures that prevent or more effectively treat heart disease. Practically, two imminent hurdles to experimentally addressing some of the most important questions are the lack of a reliable genetic model of heart-restricted proteasome inhibition as well as a benign method to enhance proteasome function. Lifting these hurdles will surely accelerate research in this important area and bring great potential to develop new ways to fight more effectively against heart disease, the number one cause of death in the developed countries and a major health problem of mankind.
Acknowledgments
This work is supported in part by NIH grants R01HL072166, R01HL085629, and R01HL068936 (to X.W.), R03AG033291 (to Y-F. L.) and American Heart Association grants 0740025N (to X. W.) and 0950088Z (to Y-F. L.), as well as by the Physician Scientists Program of the University of South Dakota.
Abbreviations
- CHF
congestive heart failure
- CryAB
αB-crystallin
- CVB3
coxsackie virus B3
- D7-des
a 7-amino acid (R172 through E178) deletion mutation of desmin
- CryABR120G
a missense (R120G) mutation of CryAB
- Dox
doxorubicin
- DRC
desmin-related cardiomyopathy
- ER
endoplasmic reticulum
- GFP
green fluorescence protein
- GFPdgn
GFP with carboxyl fusion of degron CL1
- IPC
ischemia-preconditioning
- I/R
ischemia/reperfusion
- NFAT
Nuclear factor-activated T cells
- PFI
proteasome functional insufficiency
- PKA
protein kinase A
- PP2A
protein phosphatase 2A
- PQC
protein quality control
- ROS
reactive oxygen species
- TAC
transverse aortic constriction
- UPS
ubiquitin-proteasome system
Footnotes
Disclosure
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Su H, Wang X. The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res. 2010;85:253–262. doi: 10.1093/cvr/cvp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 3.Willis MS, Patterson C. Into the heart: the emerging role of the ubiquitin-proteasome system. J Mol Cell Cardiol. 2006;41:567–579. doi: 10.1016/j.yjmcc.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 4.Powell SR. The ubiquitin-proteasome system in cardiac physiology and pathology. Am J Physiol Heart Circ Physiol. 2006;291:H1–H19. doi: 10.1152/ajpheart.00062.2006. [DOI] [PubMed] [Google Scholar]
- 5.Gomes AV, Zong C, Ping P. Protein degradation by the 26S proteasome system in the normal and stressed myocardium. Antioxid Redox Signal. 2006;8:1677–1691. doi: 10.1089/ars.2006.8.1677. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Su H, Ranek MJ. Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol. 2008;45:11–27. doi: 10.1016/j.yjmcc.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Q, Li J, Wang X. Interplay between the ubiquitin-proteasome system and autophagy in proteinopathies. Int J Physiol Pathophysiol Pharmacol. 2009;1:127–142. [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng Q, Wang X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 2010;52:9–25. [PMC free article] [PubMed] [Google Scholar]
- 9.Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res. 1998;83:629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- 10.Eble DM, Spragia ML, Ferguson AG, Samarel AM. Sarcomeric myosin heavy chain is degraded by the proteasome. Cell Tissue Res. 1999;296:541–548. doi: 10.1007/s004410051315. [DOI] [PubMed] [Google Scholar]
- 11.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 12.Penela P, Ruiz-Gomez A, Castano JG, Mayor F., Jr Degradation of the G protein-coupled receptor kinase 2 by the proteasome pathway. J Biol Chem. 1998;273:35238–35244. doi: 10.1074/jbc.273.52.35238. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Armando I, Yu P, Escano C, Mueller SC, Asico L, Pascua A, Lu Q, Wang X, Villar VA, Jones JE, Wang Z, Periasamy A, Lau YS, Soares-da-Silva P, Creswell K, Guillemette G, Sibley DR, Eisner G, Gildea JJ, Felder RA, Jose PA. Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J Clin Invest. 2008;118:2180–2189. doi: 10.1172/JCI33637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Traenckner EB, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. Embo J. 1994;13:5433–5441. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–796. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- 16.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 17.Auld KL, Silver PA. Transcriptional regulation by the proteasome as a mechanism for cellular protein homeostasis. Cell Cycle. 2006;5:1503–1505. doi: 10.4161/cc.5.14.2979. [DOI] [PubMed] [Google Scholar]
- 18.Salghetti SE, Muratani M, Wijnen H, Futcher B, Tansey WP. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc Natl Acad Sci U S A. 2000;97:3118–3123. doi: 10.1073/pnas.050007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue T, Geyer RK, Howard D, Yu ZK, Maki CG. MDM2 can promote the ubiquitination, nuclear export, and degradation of p53 in the absence of direct binding. J Biol Chem. 2001;276:45255–45260. doi: 10.1074/jbc.M107477200. [DOI] [PubMed] [Google Scholar]
- 20.Birks EJ, Latif N, Enesa K, Folkvang T, Luong le A, Sarathchandra P, Khan M, Ovaa H, Terracciano CM, Barton PJ, Yacoub MH, Evans PC. Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc Res. 2008;79:472–480. doi: 10.1093/cvr/cvn083. [DOI] [PubMed] [Google Scholar]
- 21.Tsukamoto O, Minamino T, Okada K, Shintani Y, Takashima S, Kato H, Liao Y, Okazaki H, Asai M, Hirata A, Fujita M, Asano Y, Yamazaki S, Asanuma H, Hori M, Kitakaze M. Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem Biophys Res Commun. 2006;340:1125–1133. doi: 10.1016/j.bbrc.2005.12.120. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi S, Mao K, Zheng H, Wang X, Patterson C, O’Connell TD, Liang Q. Diminished GATA4 protein levels contribute to hyperglycemia-induced cardiomyocyte injury. J Biol Chem. 2007;282:21945–21952. doi: 10.1074/jbc.M703048200. [DOI] [PubMed] [Google Scholar]
- 23.Zolk O, Schenke C, Sarikas A. The ubiquitin-proteasome system: focus on the heart. Cardiovasc Res. 2006;70:410–421. doi: 10.1016/j.cardiores.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 24.Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–30063. doi: 10.1074/jbc.M100142200. [DOI] [PubMed] [Google Scholar]
- 25.Gurusamy N, Goswami S, Malik G, Das DK. Oxidative injury induces selective rather than global inhibition of proteasomal activity. J Mol Cell Cardiol. 2008;44:419–428. doi: 10.1016/j.yjmcc.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Powell SR, Samuel SM, Wang P, Divald A, Thirunavukkarasu M, Koneru S, Wang X, Maulik N. Upregulation of myocardial 11S-activated proteasome in experimental hyperglycemia. J Mol Cell Cardiol. 2008;44:618–621. doi: 10.1016/j.yjmcc.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R, Wang X. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. Faseb J. 2006;20:362–364. doi: 10.1096/fj.05-4869fje. [DOI] [PubMed] [Google Scholar]
- 28.Voortman J, Giaccone G. Severe reversible cardiac failure after bortezomib treatment combined with chemotherapy in a non-small cell lung cancer patient: a case report. BMC Cancer. 2006;6:129. doi: 10.1186/1471-2407-6-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pye J, Ardeshirpour F, McCain A, Bellinger DA, Merricks E, Adams J, Elliott PJ, Pien C, Fischer TH, Baldwin AS, Jr, Nichols TC. Proteasome inhibition ablates activation of NF-kappa B in myocardial reperfusion and reduces reperfusion injury. Am J Physiol Heart Circ Physiol. 2003;284:H919–926. doi: 10.1152/ajpheart.00851.2002. [DOI] [PubMed] [Google Scholar]
- 30.Tang M, Li J, Huang W, Su H, Liang Q, Tian Z, Horak KM, Molkentin JD, Wang X. Proteasome functional insufficiency activates the calcineurin-NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc Res. 2010 doi: 10.1093/cvr/cvq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, Kem DC. Proteasome inhibition during myocardial infarction. Cardiovasc Res. 2010;85:312–320. doi: 10.1093/cvr/cvp309. [DOI] [PubMed] [Google Scholar]
- 32.Hedhli N, Depre C. Proteasome inhibitors and cardiac cell growth. Cardiovasc Res. 2010;85:321–329. doi: 10.1093/cvr/cvp226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomes AV, Zong C, Edmondson RD, Berhane BT, Wang GW, Le S, Young G, Zhang J, Vondriska TM, Whitelegge JP, Jones RC, Joshua IG, Thyparambil S, Pantaleon D, Qiao J, Loo J, Ping P. The murine cardiac 26S proteasome: an organelle awaiting exploration. Ann N Y Acad Sci. 2005;1047:197–207. doi: 10.1196/annals.1341.018. [DOI] [PubMed] [Google Scholar]
- 34.Young GW, Wang Y, Ping P. Understanding proteasome assembly and regulation: importance to cardiovascular medicine. Trends Cardiovasc Med. 2008;18:93–98. doi: 10.1016/j.tcm.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomes AV, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, Jones RC, Thyparambil S, Wang GW, Qiao X, Bardag-Gorce F, Ping P. Mapping the murine cardiac 26S proteasome complexes. Circ Res. 2006;99:362–371. doi: 10.1161/01.RES.0000237386.98506.f7. [DOI] [PubMed] [Google Scholar]
- 36.Cai ZP, Shen Z, Van Kaer L, Becker LC. Ischemic preconditioning-induced cardioprotection is lost in mice with immunoproteasome subunit low molecular mass polypeptide-2 deficiency. Faseb J. 2008;22:4248–4257. doi: 10.1096/fj.08-105940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rechsteiner M, Hill CP. Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 2005;15:27–33. doi: 10.1016/j.tcb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 38.Kirk R, Laman H, Knowles PP, Murray-Rust J, Lomonosov M, Meziane el K, McDonald NQ. Structure of a conserved dimerization domain within the F-box protein Fbxo7 and the PI31 proteasome inhibitor. J Biol Chem. 2008;283:22325–22335. doi: 10.1074/jbc.M709900200. [DOI] [PubMed] [Google Scholar]
- 39.Zaiss DM, Standera S, Kloetzel PM, Sijts AJ. PI31 is a modulator of proteasome formation and antigen processing. Proc Natl Acad Sci U S A. 2002;99:14344–14349. doi: 10.1073/pnas.212257299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drews O, Zong C, Ping P. Exploring proteasome complexes by proteomic approaches. Proteomics. 2007;7:1047–1058. doi: 10.1002/pmic.200600574. [DOI] [PubMed] [Google Scholar]
- 41.Drews O, Wildgruber R, Zong C, Sukop U, Nissum M, Weber G, Gomes AV, Ping P. Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol Cell Proteomics. 2007;6:2021–2031. doi: 10.1074/mcp.M700187-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Kloss A, Meiners S, Ludwig A, Dahlmann B. Multiple cardiac proteasome subtypes differ in their susceptibility to proteasome inhibitors. Cardiovasc Res. 2010;85:367–375. doi: 10.1093/cvr/cvp217. [DOI] [PubMed] [Google Scholar]
- 43.Zong C, Gomes AV, Drews O, Li X, Young GW, Berhane B, Qiao X, French SW, Bardag-Gorce F, Ping P. Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ Res. 2006;99:372–380. doi: 10.1161/01.RES.0000237389.40000.02. [DOI] [PubMed] [Google Scholar]
- 44.Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J Biol Chem. 2007;282:22460–22471. doi: 10.1074/jbc.M702439200. [DOI] [PubMed] [Google Scholar]
- 45.Asai M, Tsukamoto O, Minamino T, Asanuma H, Fujita M, Asano Y, Takahama H, Sasaki H, Higo S, Asakura M, Takashima S, Hori M, Kitakaze M. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J Mol Cell Cardiol. 2009;46:452–462. doi: 10.1016/j.yjmcc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 46.Akazawa H, Komuro I. “Change can happen” by PKA: proteasomes in in vivo hearts. J Mol Cell Cardiol. 2009;46:445–447. doi: 10.1016/j.yjmcc.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 47.Powell SR, Davies KJ, Divald A. Optimal determination of heart tissue 26S-proteasome activity requires maximal stimulating ATP concentrations. J Mol Cell Cardiol. 2007;42:265–269. doi: 10.1016/j.yjmcc.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gribble FM, Loussouarn G, Tucker SJ, Zhao C, Nichols CG, Ashcroft FM. A novel method for measurement of submembrane ATP concentration. J Biol Chem. 2000;275:30046–30049. doi: 10.1074/jbc.M001010200. [DOI] [PubMed] [Google Scholar]
- 49.Kawashima S. Inhibition of rat liver transglutaminase by nucleotides. Experientia. 1991;47:709–712. doi: 10.1007/BF01958822. [DOI] [PubMed] [Google Scholar]
- 50.Geng Q, Romero J, Saini V, Baker TA, Picken MM, Gamelli RL, Majetschak M. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem Biophys Res Commun. 2009;390:1136–1141. doi: 10.1016/j.bbrc.2009.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang H, Zhang X, Li S, Liu N, Lian W, McDowell E, Zhou P, Zhao C, Guo H, Zhang C, Yang C, Wen G, Dong X, Lu L, Ma N, Dong W, Dou QP, Wang X, Liu J. Physiological levels of ATP negatively regulate proteasome function. Cell Res. 2010 doi: 10.1038/cr2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Osinska H, Dorn GW, 2nd, Nieman M, Lorenz JN, Gerdes AM, Witt S, Kimball T, Gulick J, Robbins J. Mouse model of desmin-related cardiomyopathy. Circulation. 2001;103:2402–2407. doi: 10.1161/01.cir.103.19.2402. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 54.Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe CG, Kayed R, Robbins J. Reversal of amyloid-induced heart disease in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2005;102:13592–13597. doi: 10.1073/pnas.0503324102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112:3451–3461. doi: 10.1161/CIRCULATIONAHA.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng H, Tang M, Zheng Q, Kumarapeli ARK, Horak KM, Wang X. Doxycycline attenuates protein aggregation in cardiomyocytes and improves survival of a mouse model of cardiac proteinopathy. J Am Coll Cardiol. 2010 doi: 10.1016/j.jacc.2010.01.075. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, Li F, Gerdes AM, Wawrousek EF, Wang X. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res. 2005;97:1018–1026. doi: 10.1161/01.RES.0000189262.92896.0b. [DOI] [PubMed] [Google Scholar]
- 59.Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, Zheng H, Wang X. A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. Faseb J. 2005;19:2051–2053. doi: 10.1096/fj.05-3973fje. [DOI] [PubMed] [Google Scholar]
- 60.Liu J, Tang M, Mestril R, Wang X. Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J Mol Cell Cardiol. 2006;40:451–454. doi: 10.1016/j.yjmcc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 61.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 62.Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ, Jr, Rothermel BA, Hill JA. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–3078. doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–216. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 64.Maynard CJ, Bottcher C, Ortega Z, Smith R, Florea BI, Diaz-Hernandez M, Brundin P, Overkleeft HS, Li JY, Lucas JJ, Dantuma NP. Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc Natl Acad Sci U S A. 2009;106:13986–13991. doi: 10.1073/pnas.0906463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 66.Mittenberg AG, Moiseeva TN, Barlev NA. Role of proteasomes in transcription and their regulation by covalent modifications. Front Biosci. 2008;13:7184–7192. doi: 10.2741/3220. [DOI] [PubMed] [Google Scholar]
- 67.Leung A, Geng F, Daulny A, Collins G, Guzzardo P, Tansey WP. Transcriptional control and the ubiquitin-proteasome system. Ernst Schering Found Symp Proc. 2008:75–97. [PubMed] [Google Scholar]
- 68.Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, Eschenhagen T, Zolk O. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 69.Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, Igawa O, Takashima S, Mizuta E, Miake J, Yamamoto Y, Shirayoshi Y, Kitakaze M, Carrier L, Hisatome I. Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol. 2008;384:896–907. doi: 10.1016/j.jmb.2008.09.070. [DOI] [PubMed] [Google Scholar]
- 70.Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105:9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pattison JS, Sanbe A, Maloyan A, Osinska H, Klevitsky R, Robbins J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation. 2008;117:2743–2751. doi: 10.1161/CIRCULATIONAHA.107.750232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trifilo MJ, Yajima T, Gu Y, Dalton N, Peterson KL, Race RE, Meade-White K, Portis JL, Masliah E, Knowlton KU, Chesebro B, Oldstone MB. Prion-induced amyloid heart disease with high blood infectivity in transgenic mice. Science. 2006;313:94–97. doi: 10.1126/science.1128635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hacihanefioglu A, Tarkun P, Gonullu E. Acute severe cardiac failure in a myeloma patient due to proteasome inhibitor bortezomib. Int J Hematol. 2008;88:219–222. doi: 10.1007/s12185-008-0139-7. [DOI] [PubMed] [Google Scholar]
- 74.Enrico O, Gabriele B, Nadia C, Sara G, Daniele V, Giulia C, Antonio S, Mario P. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br J Haematol. 2007;138:396–397. doi: 10.1111/j.1365-2141.2007.06659.x. [DOI] [PubMed] [Google Scholar]
- 75.Voigt A, Bartel K, Egerer K, Trimpert C, Feist E, Gericke C, Kandolf R, Klingel K, Kuckelkorn U, Stangl K, Felix SB, Baumann G, Kloetzel PM, Staudt A. Humoral anti-proteasomal autoimmunity in dilated cardiomyopathy. Basic Res Cardiol. 2010;105:9–18. doi: 10.1007/s00395-009-0061-z. [DOI] [PubMed] [Google Scholar]
- 76.Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klovekorn WP, Schaper J. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 77.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 78.Powell SR, Wang P, Katzeff H, Shringarpure R, Teoh C, Khaliulin I, Das DK, Davies KJ, Schwalb H. Oxidized and ubiquitinated proteins may predict recovery of postischemic cardiac function: essential role of the proteasome. Antioxid Redox Signal. 2005;7:538–546. doi: 10.1089/ars.2005.7.538. [DOI] [PubMed] [Google Scholar]
- 79.Powell SR, Divald A. The ubiquitin-proteasome system in myocardial ischaemia and preconditioning. Cardiovasc Res. 2010;85:303–311. doi: 10.1093/cvr/cvp321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farout L, Mary J, Vinh J, Szweda LI, Friguet B. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Arch Biochem Biophys. 2006;453:135–142. doi: 10.1016/j.abb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 81.Ishii T, Sakurai T, Usami H, Uchida K. Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26 S proteasome. Biochemistry. 2005;44:13893–13901. doi: 10.1021/bi051336u. [DOI] [PubMed] [Google Scholar]
- 82.Amici M, Lupidi G, Angeletti M, Fioretti E, Eleuteri AM. Peroxynitrite-induced oxidation and its effects on isolated proteasomal systems. Free Radic Biol Med. 2003;34:987–996. doi: 10.1016/s0891-5849(02)01369-2. [DOI] [PubMed] [Google Scholar]
- 83.Taegtmeyer H, King LM, Jones BE. Energy substrate metabolism, myocardial ischemia, and targets for pharmacotherapy. Am J Cardiol. 1998;82:54K–60K. doi: 10.1016/s0002-9149(98)00538-4. [DOI] [PubMed] [Google Scholar]
- 84.Sohns W, van Veen TA, van der Heyden MA. Regulatory roles of the ubiquitin-proteasome system in cardiomyocyte apoptosis. Curr Mol Med. 2010;10:1–13. doi: 10.2174/156652410791065426. [DOI] [PubMed] [Google Scholar]
- 85.Naito AT, Okada S, Minamino T, Iwanaga K, Liu ML, Sumida T, Nomura S, Sahara N, Mizoroki T, Takashima A, Akazawa H, Nagai T, Shiojima I, Komuro I. Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury. Circ Res. 2010;106:1692–1702. doi: 10.1161/CIRCRESAHA.109.214346. [DOI] [PubMed] [Google Scholar]
- 86.Churchill EN, Mochly-Rosen D. The roles of PKCdelta and epsilon isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem Soc Trans. 2007;35:1040–1042. doi: 10.1042/BST0351040. [DOI] [PubMed] [Google Scholar]
- 87.Powell SR, Gurzenda EM, Teichberg S, Mantell LL, Maulik D. Association of increased ubiquitinated proteins with cardiac apoptosis. Antioxid Redox Signal. 2000;2:103–112. doi: 10.1089/ars.2000.2.1-103. [DOI] [PubMed] [Google Scholar]
- 88.Divald A, Kivity S, Wang P, Hochhauser E, Roberts B, Teichberg S, Gomes AV, Powell SR. Myocardial ischemic preconditioning preserves postischemic function of the 26S proteasome through diminished oxidative damage to 19S regulatory particle subunits. Circ Res. 2010;106:1829–1838. doi: 10.1161/CIRCRESAHA.110.219485. [DOI] [PubMed] [Google Scholar]
- 89.Churchill EN, Ferreira JC, Brum PC, Szweda LI, Mochly-Rosen D. Ischaemic preconditioning improves proteasomal activity and increases the degradation of deltaPKC during reperfusion. Cardiovasc Res. 2010;85:385–394. doi: 10.1093/cvr/cvp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Campbell B, Adams J, Shin YK, Lefer AM. Cardioprotective effects of a novel proteasome inhibitor following ischemia and reperfusion in the isolated perfused rat heart. J Mol Cell Cardiol. 1999;31:467–476. doi: 10.1006/jmcc.1998.0880. [DOI] [PubMed] [Google Scholar]
- 91.Yu X, Patterson E, Kem DC. Targeting proteasomes for cardioprotection. Curr Opin Pharmacol. 2009;9:167–172. doi: 10.1016/j.coph.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 92.Bao J, Sato K, Li M, Gao Y, Abid R, Aird W, Simons M, Post MJ. PR-39 and PR-11 peptides inhibit ischemia-reperfusion injury by blocking proteasome-mediated I kappa B alpha degradation. Am J Physiol Heart Circ Physiol. 2001;281:H2612–2618. doi: 10.1152/ajpheart.2001.281.6.H2612. [DOI] [PubMed] [Google Scholar]
- 93.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 94.Wang J, Maldonado MA. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol. 2006;3:255–261. [PubMed] [Google Scholar]
- 95.Osna NA, Haorah J, Krutik VM, Donohue TM., Jr Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology. 2004;40:574–582. doi: 10.1002/hep.20352. [DOI] [PubMed] [Google Scholar]
- 96.Das S, Powell SR, Wang P, Divald A, Nesaretnam K, Tosaki A, Cordis GA, Maulik N, Das DK. Cardioprotection with palm tocotrienol: antioxidant activity of tocotrienol is linked with its ability to stabilize proteasomes. Am J Physiol Heart Circ Physiol. 2005;289:H361–367. doi: 10.1152/ajpheart.01285.2004. [DOI] [PubMed] [Google Scholar]
- 97.Selvetella G, Hirsch E, Notte A, Tarone G, Lembo G. Adaptive and maladaptive hypertrophic pathways: points of convergence and divergence. Cardiovasc Res. 2004;63:373–380. doi: 10.1016/j.cardiores.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 98.Morgan HE, Gordon EE, Kira Y, Chua HL, Russo LA, Peterson CJ, McDermott PJ, Watson PA. Biochemical mechanisms of cardiac hypertrophy. Annu Rev Physiol. 1987;49:533–543. doi: 10.1146/annurev.ph.49.030187.002533. [DOI] [PubMed] [Google Scholar]
- 99.Kaab S, Barth AS, Margerie D, Dugas M, Gebauer M, Zwermann L, Merk S, Pfeufer A, Steinmeyer K, Bleich M, Kreuzer E, Steinbeck G, Nabauer M. Global gene expression in human myocardium-oligonucleotide microarray analysis of regional diversity and transcriptional regulation in heart failure. J Mol Med. 2004;82:308–316. doi: 10.1007/s00109-004-0527-2. [DOI] [PubMed] [Google Scholar]
- 100.Hwang JJ, Allen PD, Tseng GC, Lam CW, Fananapazir L, Dzau VJ, Liew CC. Microarray gene expression profiles in dilated and hypertrophic cardiomyopathic end-stage heart failure. Physiol Genomics. 2002;10:31–44. [Google Scholar]
- 101.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120:S191–197. doi: 10.1161/CIRCULATIONAHA.108.842252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wohlschlaeger J, Sixt SU, Stoeppler T, Schmitz KJ, Levkau B, Tsagakis K, Vahlhaus C, Schmid C, Peters J, Schmid KW, Milting H, Baba HA. Ventricular Unloading Is Associated With Increased 20S Proteasome Protein Expression in the Myocardium. J Heart Lung Transplant. 2009 doi: 10.1016/j.healun.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 103.Wohlschlaeger J, Sixt SU, Stoeppler T, Schmitz KJ, Levkau B, Tsagakis K, Vahlhaus C, Schmid C, Peters J, Schmid KW, Milting H, Baba HA. Ventricular unloading is associated with increased 20s proteasome protein expression in the myocardium. J Heart Lung Transplant. 2010;29:125–132. doi: 10.1016/j.healun.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 104.Carrard G, Bulteau AL, Petropoulos I, Friguet B. Impairment of proteasome structure and function in aging. Int J Biochem Cell Biol. 2002;34:1461–1474. doi: 10.1016/s1357-2725(02)00085-7. [DOI] [PubMed] [Google Scholar]
- 105.Cecarini V, Ding Q, Keller JN. Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic Res. 2007;41:673–680. doi: 10.1080/10715760701286159. [DOI] [PubMed] [Google Scholar]
- 106.Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121:997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, Hong C, Hittinger L, Ghaleh B, Sadoshima J, Vatner DE, Vatner SF, Madura K. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114:1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 108.Hedhli N, Lizano P, Hong C, Fritzky LF, Dhar SK, Liu H, Tian Y, Gao S, Madura K, Vatner SF, Depre C. Proteasome inhibition decreases cardiac remodeling after initiation of pressure overload. Am J Physiol Heart Circ Physiol. 2008;295:H1385–1393. doi: 10.1152/ajpheart.00532.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stansfield WE, Tang RH, Moss NC, Baldwin AS, Willis MS, Selzman CH. Proteasome inhibition promotes regression of left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2008;294:H645–650. doi: 10.1152/ajpheart.00196.2007. [DOI] [PubMed] [Google Scholar]
- 110.Meiners S, Dreger H, Fechner M, Bieler S, Rother W, Gunther C, Baumann G, Stangl V, Stangl K. Suppression of cardiomyocyte hypertrophy by inhibition of the ubiquitin-proteasome system. Hypertension. 2008;51:302–308. doi: 10.1161/HYPERTENSIONAHA.107.097816. [DOI] [PubMed] [Google Scholar]
- 111.Luss H, Schmitz W, Neumann J. A proteasome inhibitor confers cardioprotection. Cardiovasc Res. 2002;54:140–151. doi: 10.1016/s0008-6363(02)00232-8. [DOI] [PubMed] [Google Scholar]
- 112.Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, Richardson JA, Bassel-Duby R, Olson EN. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest. 2007;117:2486–2495. doi: 10.1172/JCI32827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol. 2003;4:192–201. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
- 114.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. Faseb J. 2006;20:1918–1920. doi: 10.1096/fj.05-5133fje. [DOI] [PubMed] [Google Scholar]
- 115.Kawamura N, Kubota T, Kawano S, Monden Y, Feldman AM, Tsutsui H, Takeshita A, Sunagawa K. Blockade of NF-kappaB improves cardiac function and survival without affecting inflammation in TNF-alpha-induced cardiomyopathy. Cardiovasc Res. 2005;66:520–529. doi: 10.1016/j.cardiores.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 116.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 117.Gianni L, Salvatorelli E, Minotti G. Anthracycline cardiotoxicity in breast cancer patients: synergism with trastuzumab and taxanes. Cardiovasc Toxicol. 2007;7:67–71. doi: 10.1007/s12012-007-0013-5. [DOI] [PubMed] [Google Scholar]
- 118.Maradia K, Guglin M. Pharmacologic prevention of anthracycline-induced cardiomyopathy. Cardiol Rev. 2009;17:243–252. doi: 10.1097/CRD.0b013e3181b8e4c8. [DOI] [PubMed] [Google Scholar]
- 119.Ranek MJ, Wang X. Activation of the ubiquitin-proteasome system in Doxorubicin cardiomyopathy. Curr Hypertens Rep. 2009;11:389–395. doi: 10.1007/s11906-009-0068-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu J, Zheng H, Tang M, Ryu YC, Wang X. A therapeutic dose of doxorubicin activates ubiquitin-proteasome system-mediated proteolysis by acting on both the ubiquitination apparatus and proteasome. Am J Physiol Heart Circ Physiol. 2008;295:H2541–2550. doi: 10.1152/ajpheart.01052.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ito T, Fujio Y, Takahashi K, Azuma J. Degradation of NFAT5, a transcriptional regulator of osmotic stress-related genes, is a critical event for doxorubicin-induced cytotoxicity in cardiac myocytes. J Biol Chem. 2007;282:1152–1160. doi: 10.1074/jbc.M609547200. [DOI] [PubMed] [Google Scholar]
- 122.Tsimokha AS, Mittenberg AG, Kulichkova VA, Kozhukharova IV, Gause LN, Konstantinova IM. Changes in composition and activities of 26S proteasomes under the action of doxorubicin--apoptosis inductor of erythroleukemic K562 cells. Cell Biol Int. 2007;31:338–348. doi: 10.1016/j.cellbi.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 123.Luo H, Wong J, Wong B. Protein degradation systems in viral myocarditis leading to dilated cardiomyopathy. Cardiovasc Res. 2010;85:347–356. doi: 10.1093/cvr/cvp225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pauschinger M, Chandrasekharan K, Schultheiss HP. Myocardial remodeling in viral heart disease: possible interactions between inflammatory mediators and MMP-TIMP system. Heart Fail Rev. 2004;9:21–31. doi: 10.1023/B:HREV.0000011391.81676.3c. [DOI] [PubMed] [Google Scholar]
- 125.Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol. 2008;99:95–114. doi: 10.1016/S0065-2776(08)00604-4. [DOI] [PubMed] [Google Scholar]
- 126.Esfandiarei M, McManus BM. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol. 2008;3:127–155. doi: 10.1146/annurev.pathmechdis.3.121806.151534. [DOI] [PubMed] [Google Scholar]
- 127.Szalay G, Meiners S, Voigt A, Lauber J, Spieth C, Speer N, Sauter M, Kuckelkorn U, Zell A, Klingel K, Stangl K, Kandolf R. Ongoing coxsackievirus myocarditis is associated with increased formation and activity of myocardial immunoproteasomes. Am J Pathol. 2006;168:1542–1552. doi: 10.2353/ajpath.2006.050865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gao G, Zhang J, Si X, Wong J, Cheung C, McManus B, Luo H. Proteasome inhibition attenuates coxsackievirus-induced myocardial damage in mice. Am J Physiol Heart Circ Physiol. 2008;295:H401–408. doi: 10.1152/ajpheart.00292.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Retnakaran R, Zinman B. Type 1 diabetes, hyperglycaemia, and the heart. Lancet. 2008;371:1790–1799. doi: 10.1016/S0140-6736(08)60767-9. [DOI] [PubMed] [Google Scholar]
- 130.Bell DS. Heart failure: the frequent, forgotten, and often fatal complication of diabetes. Diabetes Care. 2003;26:2433–2441. doi: 10.2337/diacare.26.8.2433. [DOI] [PubMed] [Google Scholar]
- 131.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 132.Marfella R, DAM, Di Filippo C, Siniscalchi M, Sasso FC, Ferraraccio F, Rossi F, Paolisso G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc Diabetol. 2007;6:35. doi: 10.1186/1475-2840-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- 134.Bulteau AL, Szweda LI, Friguet B. Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys. 2002;397:298–304. doi: 10.1006/abbi.2001.2663. [DOI] [PubMed] [Google Scholar]
- 135.Li F, Zhang L, Craddock J, Bruce-Keller AJ, Dasuri K, Nguyen A, Keller JN. Aging and dietary restriction effects on ubiquitination, sumoylation, and the proteasome in the heart. Mech Ageing Dev. 2008;129:515–521. doi: 10.1016/j.mad.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Grune T. Oxidative stress, aging and the proteasomal system. Biogerontology. 2000;1:31–40. doi: 10.1023/a:1010037908060. [DOI] [PubMed] [Google Scholar]
- 137.Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- 138.Powell SR, Wang P, Divald A, Teichberg S, Haridas V, McCloskey TW, Davies KJ, Katzeff H. Aggregates of oxidized proteins (lipofuscin) induce apoptosis through proteasome inhibition and dysregulation of proapoptotic proteins. Free Radic Biol Med. 2005;38:1093–1101. doi: 10.1016/j.freeradbiomed.2005.01.003. [DOI] [PubMed] [Google Scholar]