

Abstract
Regulated covalent modifications of lipid A are implicated in virulence of pathogenic Gram-negative bacteria. The Salmonella typhimurium PhoP/PhoQ-activated gene pagP is required both for biosynthesis of hepta-acylated lipid A species containing palmitate and for resistance to cationic anti-microbial peptides. Palmitoylated lipid A can also function as an endotoxin antagonist. We now show that pagP and its Escherichia coli homolog (crcA) encode an unusual enzyme of lipid A biosynthesis localized in the outer membrane. PagP transfers a palmitate residue from the sn-1 position of a phospholipid to the N-linked hydroxymyristate on the proximal unit of lipid A (or its precursors). PagP bearing a C-terminal His6-tag accumulated in outer membranes during overproduction, was purified with full activity and was shown by cross-linking to behave as a homodimer. PagP is the first example of an outer membrane enzyme involved in lipid A biosynthesis. Additional pagP homologs are encoded in the genomes of Yersinia and Bordetella species. PagP may provide an adaptive response toward both Mg2+ limitation and host innate immune defenses.
Keywords: endotoxin/lipid A/outer membranes/pagP/palmitate
Introduction
Lipid A is the hydrophobic anchor of lipopolysaccharide (LPS), a major component of the outer membrane of Gram-negative bacteria (Raetz, 1996). The Gram-negative outer membrane has unique permeability properties because of its asymmetric bilayer architecture, with LPS lining the outer leaflet and phospholipids lining the inner leaflet (Nikaido, 1996). Lipid A is the active component of LPS endotoxin, which can promote septic shock when shed from bacteria during severe infection. Lipid A is also an essential structural component unique to the Gram-negative cell envelope and, consequently, provides a target for the design of novel anti-bacterial and anti-inflammatory agents. The major lipid A species of enteric bacteria, such as Escherichia coli and Salmonella typhimurium, is a β(1′-6)-linked disaccharide of glucosamine, acylated with R-3-hydroxymyristate at the 2, 3, 2′ and 3′ positions, and phosphorylated at the 1 and 4′ positions (Figure 1A; Raetz, 1996). The characteristic acyloxyacyl linkages of lipid A result from the addition of laurate and myristate to the hydroxyl groups of the R-3-hydroxymyristate moieties at the 2′ and 3′ positions, respectively (Figure 1A). Over the past 15 years, the biosynthetic enzymes that generate the constitutive lipid A species shown in Figure 1A have been identified and their corresponding structural genes cloned, primarily from E.coli (Raetz, 1996).

Fig. 1. Structures of lipid A species and key precursors found in E.coli K-12 and S.typhimurium. (A) About two-thirds of lipid A is normally recovered from E.coli K-12 as a hexa-acylated 1,4′-bis-phosphate with the remaining one-third containing a 1-pyrophosphate group (dashed bond; Zhou et al., 1999). (B) Regulated covalent lipid A modifications found in Salmonella enterica serovars include the l-4-aminoarabinose and phosphoethanolamine substituents (blue), and the S-2-OH and palmitoyl groups (red; Guo et al., 1997). With the exception of the S-2-OH group, latent enzymes for these modifications also are present in E.coli K-12 (Zhou et al., 1999), and are induced by treatment with ammonium metavanadate, or accumulate in polymyxin-resistant mutants. (C) Substrates utilized in this investigation include the disaccharide lipid A precursors lipid IVA and Kdo2–lipid IVA (dashed bond), which can be acylated (red) by PagP to produce lipid IVB and Kdo2–lipid IVB, respectively. The monosaccharide lipid X can also be acylated (red) by PagP to produce lipid Y (Brozek et al., 1987).
Recently, several regulated covalent modifications of lipid A have been implicated in virulence of pathogenic Gram-negative bacteria, such as S.typhimurium (Guo et al., 1997). Substitution of the phosphate groups at the 1 and 4′ positions with phosphoethanolamine and/or l-4-amino arabinose (Figure 1B) provides resistance against poly myxin B, a lipid A-binding cationic cyclic peptide antibiotic (Vaara et al., 1981; Helander et al., 1994). In addition, modification of lipid A with palmitate in acyloxyacyl linkage at the 2 position (Figure 1B) provides resistance against cationic anti-microbial peptides (CAMPs) that are induced by the innate immune response to bacterial infections (Guo et al., 1998). Lipid A modified with palmitate also antagonizes LPS-mediated signal transduction in human cell lines (Tanamoto and Azumi, 2000). Lipid A modifications in S.typhimurium depend largely on the phoP/phoQ two-component signal transduction pathway (Groisman et al., 1989; Miller et al., 1989), which responds to the Mg2+-limited growth conditions encountered during infection (Garcia-Vescovi et al., 1996). Furthermore, phosphoethanolamine and l-4-amino arabinose addition to lipid A depends on the phoP/phoQ-activated genes pmrA/pmrB (Gunn et al., 1998), which encode a different two-component signal transduction pathway. The latter is coordinated with phoP/phoQ by a mediator known as pmrD (Kox et al., 2000). Recent studies have shown that the incorporation of palmitate into S.typhimurium lipid A additionally depends upon a specific phoP/phoQ-activated gene, designated pagP (Guo et al., 1998).
Modifications of lipid A with l-4-aminoarabinose, phosphoethanolamine and/or palmitate are seen in E.coli K-12 only when cells are treated with ammonium metavanadate (Zhou et al., 1999), or in polymyxin resistant mutants (Helander et al., 1994). These findings demonstrate that the modification enzymes are more latent (or regulated differently) in E.coli K-12 than in S.typhimurium. Lipid A modifications are not restricted to enteric Gram-negative bacteria. In the opportunistic pathogen Pseudomonas aeruginosa, phoP/phoQ-dependent lipid A modifications are enhanced in bacteria isolated from cystic fibrosis patients with chronic airway infections (Ernst et al., 1999).
The enzymology of lipid A modification has not been explored. In 1987, an enzyme was identified in E.coli membranes that catalyzes the transfer of a palmitate chain from a phospholipid to a monosaccharide lipid A precursor, termed lipid X, to produce lipid Y (Brozek et al., 1987) (Figure 1C). The enzyme was neither purified nor the corresponding gene cloned. It was found to be very specific for palmitate at the sn-1 position of the phospholipid donor substrate, but non-specific for the polar head group (Brozek et al., 1987). Unlike other enzymes that generate acyloxyacyl moieties on lipid A (Brozek and Raetz, 1990), the palmitoyl transferase does not require thioester-containing substrates. We now demonstrate that the Salmonella pagP gene codes for the palmitoyl transferase first described by Brozek et al. (1987). Its homolog, known as crcA in E.coli, had been discovered without knowledge of the relevant biochemical function because it is one of three genes that in multiple copies confer resistance to camphor vapors (Hu et al., 1996). The products of the pagP and crcA genes are hereafter referred to as PagP. PagP is the first example of an enzyme of lipid A biosynthesis that is localized in the outer membrane. Homologs of pagP are encoded in the genomes of Yersinia pestis, Bordetella pertussis, Bordetella bronchiseptica and various strains of Salmonella.
Results
Palmitoyl transferase assays with Salmonella membranes
To determine whether or not pagP is required for palmitoyl transferase activity in vitro, we prepared membranes from Salmonella enterica serovar typhimurium strains ATCC 14028 (wild type), its PhoP-constitutive (PhoPc) mutant CS022, and a pagP mutant (LG069) derived from CS022. Membranes were assayed for palmitoyl transferase (PagP) activity, initially using the precursors lipid X, lipid IVA or 3-deoxy-d-manno-octulosonic acid (Kdo)2–lipid IVA as acyl acceptors (Figure 1C). The acylated products of the reactions were resolved by thin-layer chromatography (TLC) followed by PhosphorImager analysis (Figure 2). Wild-type Salmonella membranes convert lipid X to a more hydrophobic metabolite (Figure 2A), which migrates with lipid Y, as previously characterized (Brozek et al., 1987). Lipid IVB (Figure 1C) and Kdo2–lipid IVB are also generated under these conditions (Figure 2B and C). Acylated product formation was ∼5-fold higher in PhoPc membranes, and was largely eliminated in the pagP mutant membranes. Residual acylation detected in vitro with the pagP mutant (Figure 2) suggests that an additional lipid A-modification enzyme may be present in these extracts. However, the results of Guo et al. (1998) indicate that this residual activity does not contribute significantly to lipid A acylation in vivo. These findings are consistent with the idea that pagP is the structural gene for the palmitoyl transferase first described by Brozek and coworkers (Brozek et al., 1987). Unexpectedly, the PhoPc membranes also revealed the presence of an additional PhoP regulated enzyme that generates a more hydrophilic metabolite with each of the substrates (Figure 2). A separate investigation has shown that these hydrophilic products are made by a novel Salmonella phoP/phoQ-regulated lipid A 3-O-deacylase (M.S.Trent, W.Pabich, C.R.H.Raetz and S.I.Miller, unpublished data).

Fig. 2. Products of PagP reactions using various Salmonella membranes with lipid X, lipid IVA or Kdo2–lipid IVA as acceptors. PagP was assayed at 30°C for 40 min using 0.5 mg/ml membranes from S.enterica serovar typhimurium ATCC 14028 (Wild-type), CS022 (phoPc) and LG069 (pagP, phoPc). Assays contain 100 mM Tris–HCl pH 8, 10 mM EDTA, 0.25% Triton X-100, 1 mM sn-1-(16:0)-2-(18:1cΔ9)-PtdCho as acyl donor and 200 c.p.m./µl of the 32P-labeled lipid X, lipid IVA or Kdo2–lipid IVA acyl acceptors at 10 µM. Metabolites were separated by TLC and visualized by overnight exposure to a PhosphorImager screen. The solvent systems for the indicated acyl acceptors are followed by the Rf values listed in ascending order for each metabolite (identified with arrowheads). (A) Lipid X, chloroform:methanol:water:acetic acid (25:15:4:2 v/v). Rf1 = 0.42, Rf2 = 0.55, Rf3 = 0.71. (B) Lipid IVA, chloroform: pyridine:88% formic acid:water (50:50:16:5 v/v). Rf1 = 0.36, Rf2 = 0.43, Rf3 = 0.52, Rf4 = 0.55. (C) Kdo2–lipid IVA, chloroform:pyridine:88% formic acid/water (30:70:16:10 v/v). Rf1 = 0.38, Rf2 = 0.40, Rf3 = 0.43, Rf4 = 0.45.
Outer membrane localization of PagP
In contrast to the acyl carrier protein-dependent lauroyl- and myristoyl-transferases (LpxL and LpxM, respectively) (Brozek and Raetz, 1990; Carty et al., 1999), PagP is dependent on acyl groups derived from phospholipids (Brozek et al., 1987). The unique substrate requirements of PagP do not restrict it to an inner membrane localization, which is the case for all other known membrane-bound enzymes of lipid A biosynthesis (Raetz, 1996). Therefore, protein profiles of highly purified S.typhimurium outer membranes were compared by 2D-PAGE as described previously (Guina et al., 2000). One of the PhoP-activated outer membrane protein species that was missing in the isogenic pagP mutant migrated at the molecular weight and pI predicted for mature PagP (18 kDa and pI 6.5; Figure 3). Corresponding protein was digested in-gel with trypsin and the resulting peptide masses were recorded by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (MS) for peptide mass fingerprinting using the MS-Fit program of Protein Prospector (http://prospector.ucsf.edu) that was linked to the NCBI genome database (ftp://ncbi.nlm.nih.gov/blast/db/). Allowing a mass accuracy of ±0.5 Da, seven peptides matched the masses of predicted PagP tryptic fragments. Therefore, PagP is localized in the outer membrane of S.typhimurium.

Fig. 3. Salmonella PagP is an outer membrane protein. Purified outer membrane proteins (350 µg) from the S.typhimurium PhoP-constitutive strain (CS022) were prepared and separated by 15% 2D-PAGE as described previously (Guina et al., 2000). An ∼18-kDa protein species migrating at pI 6.5 was identified as PagP by MALDI-TOF MS tryptic peptide fingerprinting. Corresponding protein was absent from the outer membranes of the pagP mutant strain (LG069). The locations of PagP and the major outer membrane proteins OmpF/C and OmpA are indicated with arrowheads. The positions of molecular weight standards are indicated to the left of the gel and the linear pH gradient generated during the isoelectric focusing step is indicated above the gel.
This observation predicts that PagP activity should co-localize with outer membranes. In order to avoid interference from the Salmonella lipid A 3-O-deacylase and to exploit the established lipid A enzymology of E.coli, we elected to focus our investigation on E.coli PagP. We separated inner and outer membranes of wild-type E.coli using isopycnic sucrose density gradient centrifugation (Figure 4). Appropriate marker enzymes were used to identify the inner membrane (NADH oxidase) and outer membrane (phospholipase A) fractions. PagP activity was clearly localized to the outer membrane.

Fig. 4. Outer membrane localization of PagP activity in wild-type E.coli membranes. The inner and outer membranes from E.coli MC1061 were separated by isopycnic sucrose density gradient centrifugation, and ∼0.7 ml fractions were collected. Light scattering material was detected by measuring the A550. The inner and outer membranes were located by measuring NADH oxidase and phospho lipase A activities, respectively. PagP activity was measured as described in the legend to Figure 2, but with 12.5 µl portions from each fraction using 32P-labeled lipid IVA (200 c.p.m./µl) at 100 µM for 220 min. Measurements are expressed as a percentage of the total activity units across the entire gradient. NADH oxidase (squares), phospholipase A (triangles), light scattering (crosses) and PagP (circles).
Expression and in vitro activity of PagP from E.coli
Under our assay conditions, the PagP specific activity of wild-type Salmonella membranes using disaccharide acceptors as substrates at 10 µM (150 pmol/min/mg) is ∼5- to 10-fold greater than that observed with wild-type E.coli membranes (15–30 pmol/min/mg). Therefore, the pagP homolog of E.coli (crcA) (Hu et al., 1996) was cloned with its endogenous ribosome-binding site placed behind an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible tac-promoter in the expression vector pMS119HE (Strack et al., 1992) to create plasmid pCrcHD. Outer membranes were prepared from induced cells transformed with the vector or with pCrcHD, and assayed for palmitate transfer to lipid X, lipid IVA or Kdo2–lipid IVA as acyl acceptors. Expression of crcA yielded ∼500-fold overproduction of PagP above the background activity. The enzyme utilizes the disaccharides lipid IVA and Kdo2–lipid IVA nearly 3-fold more rapidly under standard assay conditions than the monosaccharide lipid X (Figure 5). Unlike LpxL and LpxM (Brozek and Raetz, 1990), PagP clearly does not depend upon the presence of the two Kdo sugars.

Fig. 5. Relative rates of product formation by E.coli PagP-overproducing outer membranes with lipid X, lipid IVA or Kdo2–lipid IVA as acyl acceptors. PagP was assayed as described in the legend to Figure 2 using outer membranes at 10 µg/ml. Each substrate was present at 10 µM in 25 µl reaction volumes. Induced outer membranes were prepared from E.coli MC1061 transformed with pMS119HE (open symbols) or pCrcHD (solid symbols). Acyl acceptors are lipid X (circles), lipid IVA (squares) or Kdo2–lipid IVA (triangles).
Lipid A is conjugated to the core oligosaccharide and the O-antigen polymer in the inner membrane before it is targeted to the outer membrane (Raetz, 1996). Therefore, an outer membrane enzyme involved in lipid A biosynthesis must utilize fully assembled LPS as a substrate. We examined whether PagP in outer membranes could acylate Kdo2–lipid A (Re endotoxin), which is the simplest chemotype of LPS capable of supporting the growth of E.coli K-12. Kdo2–[32P]lipid A (hexa-acylated) was prepared from the heptose-deficient E.coli strain WBB06 (Brabetz et al., 1997). Using induced outer membranes from E.coli MC1061 transformed with either pMS119HE or pCrcHD, hepta-acylated Kdo2–[32P]lipid A was rapidly detected only when PagP was overproduced (Figure 6). This result indicates that PagP is capable of transferring palmitate to hexa-acylated lipid A, as would be present as the hydrophobic anchor of fully assembled LPS in the outer membrane.
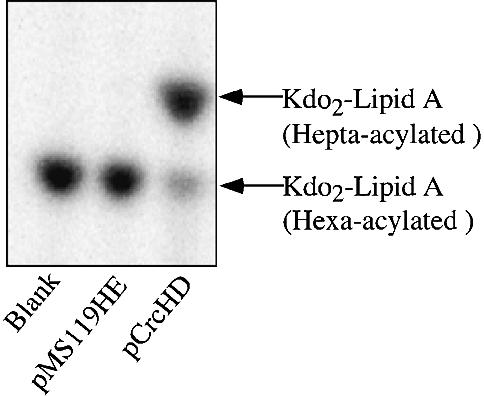
Fig. 6. Kdo2–lipid A is a substrate for acylation by PagP. Kdo2–[32P]lipid A (100 c.p.m./µl) at ∼10 µM was assayed for PagP activity for 5 min as described in the legend to Figure 2 using induced outer membranes (10 µg/ml) from E.coli MC1061 transformed with either pMS119HE or pCrcHD. The TLC plate was developed in the solvent system of chloroform:pyridine:88% formic acid:water (30:70:16:10 v/v) and the separated products were visualized by overnight exposure to a PhosphorImager screen. The Rf values for Kdo2–lipid A are 0.52 (hexa-acylated) and 0.60 (hepta-acylated). The positions of the two Kdo2–lipid A derivatives are indicated to the right of the figure.
Expression and activity of His6-PagP
We have purified the crcA gene product in order to establish unequivocally its identity with the palmitoyl transferase enzyme. To achieve massive overproduction of the protein, we used PCR to amplify the E.coli crcA gene for cloning behind the T7-RNA polymerase promoter and ribosome-binding site in plasmid pET21a+. Two PCR products were constructed to clone the wild-type crcA open reading frame (pETCrcA) and to include a C-terminal His6 tag (pETCrcAH). Induced membranes from E.coli BL21(DE3)/pLysE transformed with pET21a+, pETCrcA or pETCrcAH were prepared and separated by isopycnic sucrose density gradient centrifugation. Whole membranes, inner membranes and outer membranes were analyzed by SDS–PAGE (Figure 7). The majority of both overproduced proteins were recovered in the outer membranes, although a small amount of the overproduced His6-tagged protein is present in the inner membranes. This appears to result from outer membrane contamination since several porins and OmpA are clearly present in this fraction. The specific activities in outer membranes for both proteins using 10 µM lipid IVA as the acceptor substrate were identical (3.3 µmol/min/mg) and represent ∼105-fold overproduction of PagP activity. This finding demonstrates that the C-terminal His6-tag does not interfere with the expression, outer membrane assembly, or activity of PagP.

Fig. 7. Outer membrane localization of the PagP and His6-PagP proteins in induced membranes of E.coli BL21(DE3)pLysE transformed with pET21a+, pETCrcA or pETCrcAH. The inner and outer membranes were separated by isopycnic sucrose density gradient centrifugation. Next, 40 µg samples of protein from the whole membranes, the inner membranes, or the outer membranes were solubilized, boiled for 10 min, analyzed by 15% SDS–PAGE and stained with Coomassie Blue dye. The positions of molecular weight standards, PagP and His6-PagP, are indicated to the right of the gel.
In order to establish that the overproduced protein was encoded by crcA, we transferred the outer membrane proteins from the gel to a polyvinylidene fluoride membrane for protein micro-sequencing. We focused on the His6-tagged protein band for this purpose, because it did not migrate with any other proteins in the gel. The first 10 amino acid residues were identified in the order NADEWMTTFR, corresponding to the sequence following a probable 25 amino acid signal peptide at the N-terminus of the E.coli crcA gene. Signal peptides similar to that of E.coli PagP are apparent in the homologs currently identified in the genomes of Y.pestis (53% identical), Bordetella species pertussis and bronchiseptica (38% identical), and Salmonella enterica serovars typhi, paratyphi and typhimurium (75% identical). These can be accessed through DDBJ/EMBL/GenBank and the NCBI microbial genomes database (http://www.ncbi.nlm.nih.gov/Microb_blast/unfinishedgenome.html).
Purification and dimeric structure of His6-PagP
In order to purify the His6-tagged outer membrane protein by metal chelate affinity chromatography, it was necessary first to solubilize it from the membranes. Two successive solubilization steps with 0.01 and 0.1% lauroyl dimethylamine N-oxide (LDAO) in the presence of MgSO4 served to solubilize most cytoplasmic membrane proteins, whereas the PagP activity remained with the insoluble outer membrane proteins. PagP was then solubilized quantitatively (Table I) by increasing the LDAO concentration to 0.5%. The majority of other outer membrane proteins remained insoluble under these conditions. The His6-tagged protein was observed in the 0.5% LDAO extract by SDS–PAGE and appeared to be >90% homogenous (not shown).
Table I. Solubilization and purification of His6-PagP.
| Fraction | Total protein (mg) | Total volume (ml) | Specific activitya (µmol/min/mg) | Total units (µmol/min) | % yield |
|---|---|---|---|---|---|
| Membranes | 34.6 | 8 | 2.9 | 100 | 100 |
| 0.5% LDAO | 10.6 | 8 | 10.6 | 113 | 100+ |
| Ni2+-eluate | 0.85 | 2.5 | 80.2 | 68 | 68 |
aAssayed with 10 µM lipid IVA and 1 mM sn-1-(16:0)-2-(18:1cΔ9)-PtdCho.
The solubilized His6-tagged protein was bound to a Ni2+-affinity matrix, washed and then eluted with steps of increasing imidazole concentrations. After the protein peak was pooled and dialyzed, palmitoyl transferase activity was recovered in 68% yield (Table I). The migration of the purified His6-tagged protein in SDS–PAGE (Figure 8) was found to be heat-modifiable, which is diagnostic of many integral outer membrane proteins (Schnaitman, 1973). The migration in SDS–PAGE of the heat-treated sample corresponds to the predicted molecular weight of the mature His6-tagged protein (20.0 kDa). The unheated sample migrated further, suggesting that it maintains some secondary structure in the gel. A possible contaminant of roughly twice the molecular weight was also apparent in the unheated sample (Figure 8). We tested whether the purified His6-tagged protein might actually be a dimer by chemically cross-linking it with glutaraldehyde. Indeed, the glutaraldehyde-treated sample produced a major band that migrated with the apparent contaminant in the untreated His6-tagged protein. The migration of the heat-modified cross-linked material corresponds exactly to the predicted molecular weight (40.0 kDa) of a dimer of the His6-tagged protein. The association of the palmitoyl transferase activity with the purified His6-tagged protein establishes its identity as the crcA gene product.
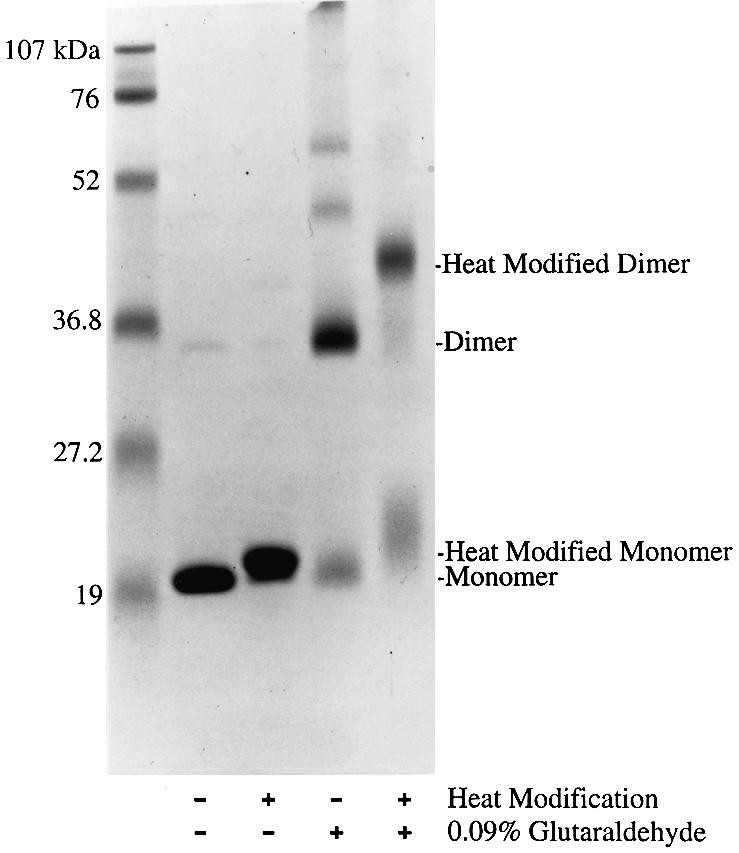
Fig. 8. Heat modification and chemical crosslinking of purified His6-PagP. Two 17 µg samples of purified His6-PagP were treated with or without prior incubation in 0.09% glutaraldehyde for 100 min at 25°C. Each sample was then split into two 8.5 µg fractions and solubilized, with or without boiling for 10 min, for analysis by 15% SDS–PAGE. The gel was stained with Coomassie Blue dye. The positions of monomeric and dimeric bands, and their heat-modified derivatives, are indicated to the right of the gel. The positions of molecular weight standards are indicated to the left of the gel.
Phospholipid substrate requirements of purified His6-PagP
Brozek et al. (1987) previously established that the in vitro acylation of lipid X catalyzed by ethanol-washed E.coli membranes exhibited a strong preference for a phospholipid cosubstrate that contained palmitate at the sn-1 position, but little preference for the polar head group. In order to confirm that purified His6-PagP behaved similarly, we examined its specific activity using various phospholipids of defined acyl-chain composition (Table II). Trace activity above background was observed in the absence of added co-substrate, indicating that some phospholipids may remain associated with the purified enzyme. Using a set of phosphatidylcholine (PtdCho) substrates, we observed the expected strong preference for palmitate at the sn-1 position. Alterations in acyl-chain length as little as one carbon atom reduced the acylation of lipid IVA by up to 6-fold. The presence of oleate at the sn-2 position was beneficial, provided that palmitate was present at the sn-1 position. This observation may reflect the unique physical properties imparted by the presence of an unsaturated acyl-chain. Purified His6-PagP is completely unable to utilize either sn-1-(16:0)-2-lyso PtdCho or the sn-2/sn-3 isomer of dipalmitoyl PtdCho (Table II). Since E.coli does not make PtdCho, we examined whether common E.coli phospholipids containing palmitate at the sn-1 position could be utilized as substrates. Phosphatidylglycerol (PtdGro), phosphatidylethanolamine (PtdEtn), phosphatidylserine (PtdSer) and phosphatidic acid (Ptd-OH) were all effective acyl donors to varying extents (Table II). However, dipalmitoylglycerol (16:0/16:0 Gro) was completely inactive (Table II). We conclude that purified His6-PagP possesses the same phospholipid substrate specificity as displayed by the enzyme reported by Brozek et al. (1987).
Table II. Phospholipid requirements of purified His6-PagPa.
| Phospholipid cosubstrateb | Specific activity (µmol/min/mg) |
|---|---|
| None | 1.07 |
| 15:0/15:0 PtdCho | 5.71 |
| 17:0/17:0 PtdCho | 3.77 |
| 18:1cΔ9/18:1cΔ9 PtdCho | 4.14 |
| 16:0/18:1cΔ9 PtdCho | 29.59 |
| 18:1cΔ9/16:0 PtdCho | 9.68 |
| 16:0/16:0 PtdCho | 23.29 |
| 16:0/lyso PtdCho | 0.51 |
| 16:0/16:0 PtdCho (sn-2/sn-3) | 0.73 |
| 16:0/16:0 Gro | 0.83 |
| 16:0/16:0 Ptd-OH | 9.82 |
| 16:0/16:0 PtdSer | 18.20 |
| 16:0/16:0 PtdEtn | 3.93 |
| 16:0/18:1cΔ9 PtdEtn | 7.22 |
| 16:0/18:1cΔ9 PtdGro | 9.18 |
aAssayed with 10 µM lipid IVA and 1 mM of the specified phospholipid, and using purified His6-PagP at 10 ng/ml.
bStereochemistry is sn-1/sn-2 unless indicated otherwise. 15:0, pentadecanoyl; 16:0, palmitoyl; 17:0, heptadecanoyl; 18:1cΔ9, oleoyl.
Discussion
An important role for lipid A modifications is to provide resistance against CAMPs. A large body of evidence indicates that CAMPs associate with the cell surface of Gram-negative bacteria by electrostatic interactions with negatively charged groups on LPS (Schindler and Osborn, 1979; Rana et al., 1991; Bruch et al., 1999). Partial neutralization of the negatively charged phosphate groups at the 1 and 4′ positions of lipid A by increased covalent modification with l-4-aminoarabinose and phosphoethanolamine moieties proabably explains the phenomenon of polymyxin B and CAMP resistance in S.typhimurium and E.coli (Vaara et al., 1981; Helander et al., 1994). CAMP resistance in Salmonellae depends in part on the PhoP/PhoQ-activated gene pagP, which is required for the palmitoylation of lipid A (Guo et al., 1998). The transfer of an additional (seventh) acyl-chain to the normally hexa-acylated lipid A may prevent the self-promoted uptake of CAMPs across the hydrophobic bilayer of the outer membrane before they proceed to damage the inner membrane (Guo et al., 1998). Our results demonstrate that pagP and its homolog in E.coli (crcA) encode the enzyme (PagP) that is responsible for lipid A palmitoylation.
Escherichia coli PagP is an outer membrane protein that is synthesized with a cleavable type-I signal peptide. We also detected the PagP protein in outer membranes of a Salmonella PhoPc mutant. The outer membrane localization of PagP casts doubt on the physiological significance of its role in the conversion of lipid X to lipid Y (Brozek et al., 1987), both of which accumulate in mutants conditionally defective in the gene for the lipid A disaccharide synthase lpxB(pgsB) (Nishijima and Raetz, 1979; Ray et al., 1984). While lipid X is a key cytoplasmic monosaccharide precursor in the lipid A biosynthetic pathway (Raetz, 1996), accumulation of lipid Y in lpxB mutants may be secondary to the excessive accumulation of lipid X under non-permissive conditions and/or to outer membrane reorganization under conditions of limited lipid A biosynthesis. Although a regulatory function for palmitoylated precursors of lipid A cannot be ruled out, the most plausible physiological substrate for PagP would appear to be the lipid A moiety of fully assembled LPS in the outer membrane. The simplest chemotype of LPS known to support growth, Kdo2–lipid A (Re endotoxin), was found to be an acceptor for PagP catalyzed acylation in vitro.
The activation of integral outer membrane enzymes in E.coli by physical perturbations to the outer membrane is now emerging as a potential mechanism for the maintenance of outer membrane lipid asymmetry. Integral outer membrane enzymes of E.coli include the duplicated OmpT and OmpP proteases, which are 87% identical and hydrolyze peptide bonds between paired basic residues (Sugimura and Nishihara, 1988; Kaufmann et al., 1994), as well as the Ca2+-dependent phospholipase OMPLA (Snijder et al., 1999; Dekker, 2000). OmpT provides resistance against protamine, a polycationic anti-microbial peptide known to disorganize outer membrane lipids (Stumpe et al., 1998). In addition, a Salmonella homolog of OmpT is required for resistance to α-helical CAMPs and is regulated in a PhoP/PhoQ-dependent manner (Guina et al., 2000). The recently determined crystal structure of the outer membrane phospholipase OMPLA of E.coli shows how it hydrolyzes phospholipids in the outer membrane (Snijder et al., 1999). Events that disturb the outer membrane can activate OMPLA, which may function to restore outer membrane lipid asymmetry by hydrolyzing phospholipids that migrate into the outer leaflet (Dekker, 2000). PagP is only the third enzyme shown to be an integral outer membrane protein in E.coli and is the first example of an outer membrane enzyme of lipid A biosynthesis.
The Gram-negative outer membrane is a major reservoir for Mg2+, which serves to bridge the negative charges between individual LPS molecules (Coughlin et al., 1983a,b). The strong Mg2+-dependent lateral interactions between LPS molecules help to maintain outer membrane lipid asymmetry (Nikaido, 1996). Treatment of E.coli or Salmonellae with the Mg2+ chelating agent ethylenediaminetetraacetic acid (EDTA) disrupts outer membrane lipid asymmetry by selectively stripping 30–50% of LPS molecules from the cell surface, presumably as a result of electrostatic repulsion (Leive et al., 1968). Consequently, phospholipids are thought to migrate into the outer leaflet to replace the lost LPS and create patches of phospholipid bilayers within the outer membrane (Nikaido, 1996). Cells treated with EDTA remain viable, but become permeable to lipophilic solutes that cannot normally penetrate the outer membrane (Leive, 1968). OMPLA is dependent on Ca2+ for both activity and dimerization, and is unlikely to be fully active at micromolar divalent cation concentrations where large numbers of phospholipid molecules populate the outer leaflet of the outer membrane (Nikaido, 1996). It is conceivable that total hydrolysis of phospholipids under these conditions might be deleterious. PagP activity is independent of divalent cations and may thereby provide a specific response to those perturbations in outer membrane lipid asymmetry induced by Mg2+ limitation. The resulting hepta-acylated lipid A should be more firmly anchored to the cell surface, while the lysophospholipid byproduct might be degraded and recycled (Hsu et al., 1991). The regulation of pagP by the Mg2+-sensing PhoP/PhoQ virulence regulator in pathogenic Salmonellae is consistent with a role for PagP in providing an adaptive response toward Mg2+ limitation.
Lipid A is a distinguishing feature of Gram-negative bacteria, and the genomes of ∼30 species have revealed key enzymes in the lipid A biosynthetic pathway. However, PagP is narrowly distributed within a subgroup of primarily pathogenic Gram-negative organisms, which include Y.pestis, B.pertussis, B.bronchiseptica and various strains of Salmonella. Effective PagP inhibitors may have value in the treatment of infectious diseases by sensitizing pathogenic Gram-negative bacteria to the innate immune response (Guo et al., 1998). While such inhibitors would probably not be useful in the treatment of acute infections, they might have value in preventing recurrence. The specificity of PagP for palmitate at the sn-1 position of phospholipids provides a rational basis for the design of transition state analog inhibitors. Further investigation of PagP structure and function will be needed for the development of anti-infective agents.
Materials and methods
Materials
32Pi and [γ-32P]ATP were purchased from NEN LifeScience Products Inc. Phospholipids, antibiotics, NADH, 25% glutaraldehyde and Ponceau S were obtained from Sigma. LDAO was obtained from Fluka. Pyridine, methanol and 88% formic acid were obtained from Mallinckrodt. Chloroform and glacial acetic acid were purchased from EM Science. Glass-backed Silica Gel 60 TLC and HPTLC plates (0.25 mm) were from E.Merck, Germany. Bicinchoninic acid protein assay reagents and Triton X-100 were obtained from Pierce. The His-bind resin and buffer kit were obtained from Novagen. QIAprep spin miniprep, Qiaquick PCR purification, and QIAEX II gel extraction kits were obtained from Qiagen. Gibco-BRL custom primers were manufactured by Life Technologies, Inc. Pfu polymerase and supercompetent E.coli XL1-Blue were obtained from Stratagene. Restriction endonucleases, deoxyribonucleotide triphosphates and dodecylmaltoside were obtained from Boehringer Mannheim.
Bacterial strains, plasmids and growth conditions
The bacterial strains and plasmids used in this study are described in Table III. Plasmid pKH1 was kindly provided by Dr Nancy J.Trun, National Institutes of Health, Baltimore, MD. Cells were generally grown at 37°C in LB broth, consisting of 10 g of NaCl, 5 g of yeast extract and 10 g of Tryptone per liter. Antibiotics were added when necessary at final concentrations of 12 µg/ml for tetracycline, 10 µg/ml for chloramphenicol, 100 µg/ml for ampicillin, 100 µg/ml for streptomycin and 50 µg/ml for kanamycin.
Table III. Bacterial plasmids and strains used in this work.
| Plasmid/strain | Description/genotype | Sourcea |
|---|---|---|
| Plasmid | ||
| pMS119HE | IPTG-inducible tac-promoter expression vector (ApR) | Strack et al. (1992) |
| pET21a+ | IPTG-inducible T7 RNA polymerase-promoter expression vector (ApR) | Novagen |
| pKH1 | pBR322-derived plasmid carrying the 2200 bp HindIII–EcoRI fragment of E.coli crcAcspEcrcB locus | Hu et al. (1996) |
| pCrcHD | 1100 bp HindIII–DraI fragment of pKH1 carrying crcA cloned into HindIII–SmaI-digested pMS119HE | this study |
| pETCrcA | 600 bp NdeI–EcoRI PCR product carrying crcA cloned into pET21a+ | this study |
| pETCrcAH | 600 bp NdeI–XhoI PCR product carrying crcA cloned into pET21a+ with C-terminal His6 tag | this study |
| E.coli | ||
| BL21(DE3)pLysE | F–, ompT, hsdSB (rB–mB–), gal, dcm, λ(DE3), pLysE (CmR) | Novagen |
| XL1-Blue | F–, recA1, endA1, gyrA96, thi-1, hsdR17, supE44, relA1, lac, [F′ proAB, lacIqZΔM15, Tn10(Tetr)]c | Stratagene |
| MC1061 | F–, araD139, Δ(ara-leu)7697, Δ(lac)X74, galU, galK, hsdR2 (rK–mK+), mcrB1, rpsL | Casadaban and Cohen (1980) |
| MN7 | F–, rpsL136, his-4, pgsA444, nalA, pgsB1(lpxB1) | Nishijima et al. (1981) |
| WBB06 | W3110 mtl, Δ(rfaC-rfaF)::tet6 | Brabetz et al. (1997) |
| S.typhimurium | ||
| ATCC 14028 | wild-type Salmonella enterica serovar. typhimurium | ATCC |
| CS022 | ATCC 14028, pho-24(phoPc, PhoP constitutive) | Miller and Mekalanos (1990) |
| LG069 | ATCC 14028, pho-24, pagP2::Tn10d-tet, phoN2, zxx6251::Tn10d-cam | Guo et al. (1998) |
aATCC, American Type Culture Collection.
DNA manipulations
Restriction enzyme digestions, ligations, transformations and DNA electrophoresis were performed according to Sambrook et al. (1989). Plasmid pCrcHD was constructed by cloning the 1100 bp HindIII–DraI fragment carrying E.coli crcA from plasmid pKH1 into the IPTG-inducible tac-promoter expression vector pMS119HE, which was opened by HindIII–SmaI digestion. PCR gene amplification was performed with 2.5 U of Pfu polymerase in a volume of 50 µl of the supplied buffer with 10 ng of pCrcHD as template, 0.2 µM of the appropriate primers (Table IV) and 10 µM dNTPs. After initial denaturation for 7 min at 94°C, 25 cycles of 30 s at 94°C, 30 s at 50°C and 30 s at 72°C were performed, followed by 5 min at 72°C. The NdeI–EcoRI-digested PCR product that was amplified from pCrcHD using the primers crcA5Nde and crcA3Eco was cloned into pET21a+ digested with the same enzymes to create plasmid pETCrcA. The C-terminal His6-tag fusion plasmid pETCrcAH was constructed similarly, except that PCR was performed using the primers crcA5Nde and crcA3Xho, and the product cloned into pET21a+ using NdeI–XhoI digestion. All cloned PCR products were subjected to double strand DNA sequencing at the Duke University Comprehensive Cancer Center DNA Analysis Facility to confirm the absence of any spurious mutations.
Table IV. Oligonucleotide primers used in this work.
| Name | Function | Sequence |
|---|---|---|
| T7 promoter | DNA sequencing | 5′-TAATACGACTCACTATAGGG-3′ |
| T7 terminator | DNA sequencing | 5′-GCTAGTTATTGCTCAGCGG-3′ |
| crcA5Nde | PCR | 5′-GGGAATTCCATATGAACGTGAGTAAATATGTC-3′ |
| crcA3Eco | PCR | 5′-CCGGAATTCTCAAAACTGAAAGCGCATC-3′ |
| crcA3Xho | PCR | 5′-TCCCTCGAGAAACTGAAAGCGCATCCA-3′ |
Protein analysis
Protein extracts (40 µg) were solubilized in an equal volume of 2× SDS buffer (Sambrook et al., 1989) with or without boiling for 10 min where indicated. SDS–PAGE on 1.5-mm thick 15% acrylamide slab gels was performed with a Bio-Rad Protean II XI apparatus and operated at 90–180 V. Gels were stained with Coomassie Blue dye and destained with 10% acetic acid. The Bio-Rad mini-Protean Transblot apparatus was used to electroblot protein to an Immobilon-P polyvinylidene fluoride membrane (Millipore) in 10 mM 3-(cyclohexylamino)-1-propanesulfonic acid pH 11 with 10% methanol at 100 V (∼200–340 mA) for 30 min. The membrane was stained with 0.1% Ponceau S in 1% acetic acid and destained with 1% acetic acid. The band of interest was excised with a razor blade, washed with water and subjected to high sensitivity protein micro-sequence analysis on an ABI model 492A Procise Sequencer at the University of Massachusetts Medical School Worcester Foundation Campus Core Laboratory for Protein Microsequencing and Mass Spectrometry. Protein was assayed with the bicinchoninic acid protocol (Smith et al., 1985) as described by the manufacturer (Pierce) and using bovine serum albumin as the standard.
Preparation and isolation of lipid substrates
All lipid substrates in aqueous buffer were dispersed prior to use by sonic irradiation for 1 min in a bath sonicator. [32P]lipid X was prepared and isolated from E.coli MN7 (Nishijima et al., 1981) as described by Radika and Raetz (1988). [4′-32P]lipid IVA was prepared using [γ-32P]ATP according to the procedure of Garrett et al. (1997), and isolated as described by Basu et al. (1999). Kdo2–[4′-32P]lipid IVA was prepared and isolated according to Basu et al. (1999). Laboratory stocks of non-radioactive substrates were prepared and isolated as described for lipid X (Radika and Raetz, 1988), lipid IVA (Raetz et al., 1985) and Kdo2–lipid IVA (Brozek et al., 1989). Kdo2–[32P]lipid A (hexa-acylated) was prepared using the heptose-deficient E.coli strain WBB06 (Brabetz et al., 1997). The cells were grown to an A600 of ∼1.0 in 100 ml of G56 minimal medium with 0.3 mM potassium phosphate (Ganong et al., 1980) containing 5 µCi/ml of 32Pi. The cells were harvested and lysed by re-suspension in 12 ml of a single phase Bligh/Dyer mixture, containing chloroform:methanol:water (1:2:0.8 v/v), and incubated for 60 min at room temperature. The insoluble material was removed by centrifugation, and the radiolabeled Kdo2–lipid A found in the supernatant was isolated by chromatography on DEAE cellulose, as described for E.coli lipid A (Odegaard et al., 1997). Approximately 3 µCi of the 32Pi was incorporated into the Kdo2–lipid A fraction.
Palmitoyl transferase reactions
Palmitoyl transferase assays were performed with varying amounts of membranes or purified His6-PagP in 25 µl vol. as described by Brozek et al. (1987), with minor modifications. The reaction contains 100 mM Tris–HCl pH 8, 10 mM EDTA and 0.25% Triton X-100 with 1 mM sn-1-(16:0)-2-(18:1cΔ9)-PtdCho as acyl donor and 100–200 c.p.m./µl of the 32P-labeled lipid X, lipid IVA, Kdo2–lipid IVA or Kdo2–lipid A acceptors at 10–100 µM. Reactions were stopped by spotting 5 µl to the origin of a Silica Gel 60 TLC plate. The various chromatographic solvent systems that were used include chloroform:methanol:water:acetic acid (25:15:4:2 v/v), chloroform:pyridine:88% formic acid/water (50:50:16:5 v/v and 30:70:16:10 v/v). Plates were dried and exposed to a Molecular Dynamics PhosphorImager screen overnight to identify and quantify both substrates and products.
Protein expression and membrane isolation
Salmonella membranes were prepared from 25 ml cultures grown to A600 = 1–3. Escherichia coli membranes were prepared from 100 ml cultures that were grown to mid-logarithmic phase (A600 = 0.5–0.6) before addition of 1 mM IPTG, when required, and then grown for an additional 3–4 h. Cells were harvested at 7740 g for 10 min at 4°C, washed in 10 ml of ice-cold phosphate buffered saline (PBS; Sambrook et al., 1989) and frozen at –80°C. Subsequent steps were performed at 4°C. Cells were thawed in 5 ml of ice-cold PBS and passed twice through a small French Pressure cell at 10 000 psi. Debris was removed by centrifugation at 7740 g for 10 min, and membranes were prepared from the supernatant by centrifugation at 318 000 g in a Sorval RC-M150GX micro-ultracentrifuge using the S80-AT3 rotor. Membrane pellets were resuspended in 250 µl of PBS by repeated passage through a syringe equipped with a 25 guage needle and stored at –80°C. To separate inner and outer membranes, 200 µl of membranes at 18–28 mg/ml were layered on top of a discontinuous sucrose gradient composed of 0.5 ml of 60%, 1 ml of 55%, 2.4 ml of 50%, 2.5 ml of 45%, 2.4 ml of 40%, 1.4 ml of 35% and 0.8 ml of 30% sucrose in PBS (w/v) and centrifuged for 16 h at 35 000 g with a Beckman SW41 rotor. Membrane fractions were assayed directly for the inner membrane NADH oxidase and the outer membrane phospholipase A as described by Zhou et al. (1998). Pooled fractions were diluted 2- to 5-fold with water and centrifuged at 318 000 g for 20 min to remove sucrose. The inner and outer membrane pellets were resuspended in PBS and stored at –80°C.
Solubilization and purification of PagP
To express the C-terminal His6-tagged PagP, a single colony of E.coli BL21(DE3)pLysE transformed with pETCrcAH was inoculated into 10 ml of medium, grown to an A600 = 0.7, and then added directly to 1 l of fresh medium. The culture was then grown to A600 = 0.5 before induction with 1 mM IPTG for 4 h. Cells were harvested at 7740 g for 10 min at 4°C, washed in 40 ml of ice-cold PBS, and divided into two fractions. The cell pellets were frozen at –80°C. Upon thawing of the two cell pellets in 5 ml each of PBS, a viscous high cell-density suspension was formed due to the presence of the pLysE plasmid. Membranes were then prepared from French Press lysates as described in the preceding section. However, the two membrane fractions were each washed with 5 ml of PBS by resuspending them using syringes equipped serially with 18, 21 and 25 guage needles. Each washed membrane fraction was then resuspended in the same manner using 2 ml of 10 mM Tris–HCl pH 8, 1 mM MgSO4, 0.01% LDAO. The volume of each fraction was adjusted to 4 ml with the same buffer prior to centrifugation at 318 000 g. This procedure was repeated twice with the same buffer containing 0.1% LDAO, then 0.5% LDAO, followed by a final resuspension with 10 mM Tris–HCl pH 8 containing 10 mM EDTA and 1.0% LDAO. The supernatant from the 0.5% LDAO solubilization was adjusted to 250 mM NaCl and 5 mM imidazole. It was loaded onto a 1.4 ml bed of charged His-bind resin that had been prepared according to the manufacturer’s instructions (Novagen) in a 0.8 × 4 cm open Poly-Prep chromatography column (Bio-Rad), pre-equilibrated with three column volumes of buffer A (10 mM Tris–HCl pH 8, 250 mM NaCl, 0.1% LDAO) containing 5 mM imidazole. After the sample flowed through the column, the resin was washed with 10 column volumes of the pre-equilibration buffer, followed by five column volumes of buffer A containing 25 mM imidazole, and then eluted with serial 1 ml volumes of buffer A containing 50, 75, 100, 125 and 250 mM imidazole. The 75, 100 and 125 mM imidazole fractions were pooled and dialyzed against 1 l of 10 mM Tris–HCl pH 8 containing 0.1% LDAO.
Chemical cross-linking of His6-PagP was performed by incubating 17 µg of purified protein with 0.09% glutaraldehyde for 100 min at 25°C before the sample was solubilized and split into two fractions for analysis by SDS–PAGE. For most assays, purified His6-PagP was diluted to an appropriate concentration in 0.1% Triton X-100 prior to its addition to the reaction mixture.
Acknowledgments
Acknowledgements
The authors wish to thank Dr Nancy J.Trun for providing plasmid pKH1. This work was supported by National Institutes of Health grants GM-51310 to C.R.H.R. and AI-30479 to S.I.M. R.E.B. was supported by a fellowship from the Alberta Heritage Foundation for Medical Research.
References
- Basu S.S., York,J.D. and Raetz,C.R.H. (1999) A phosphotransferase that generates phosphatidylinositol 4-phosphate (PtdIns-4-P) from phosphatidylinositol and lipid A in Rhizobium leguminosarum. A membrane-bound enzyme linking lipid A and PtdIns-4-P biosynthesis. J. Biol. Chem., 274, 11139–11149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabetz W., Muller-Loennies,S., Holst,O. and Brade,H. (1997) Deletion of the heptosyltransferase genes rfaC and rfaF in Escherichia coli K-12 results in an Re-type lipopolysaccharide with a high degree of 2-aminoethanol phosphate substitution. Eur. J. Biochem., 247, 716–724. [DOI] [PubMed] [Google Scholar]
- Brozek K.A. and Raetz,C.R.H (1990) Biosynthesis of lipid A in Escherichia coli. Acyl carrier protein-dependent incorporation of laurate and myristate. J. Biol. Chem., 265, 15410–15417. [PubMed] [Google Scholar]
- Brozek K.A., Bulawa,C.E. and Raetz,C.R.H. (1987) Biosynthesis of lipid A precursors in Escherichia coli. A membrane-bound enzyme that transfers a palmitoyl residue from a glycerophospholipid to lipid X. J. Biol. Chem., 262, 5170–5179. [PubMed] [Google Scholar]
- Brozek K.A., Hosaka,K., Robertson,A.D. and Raetz,C.R.H. (1989) Biosynthesis of lipopolysaccharide in Escherichia coli. Cytoplasmic enzymes that attach 3-deoxy-d-manno-octulosonic acid to lipid A. J. Biol. Chem., 264, 6956–6966. [PubMed] [Google Scholar]
- Bruch M.D., Cajal,Y., Koh,J.T. and Jain,M.K. (1999) Higher-order structure of polymyxin B: the functional significance of topological specificity. J. Am. Chem. Soc., 121, 11993–12004. [Google Scholar]
- Carty S.M., Sreekumar,K.R. and Raetz,C.R.H. (1999) Effect of cold shock on lipid A biosynthesis in Escherichia coli: induction at 12°C of an acyltransferase specific for palmitoleoyl-acyl carrier protein. J. Biol. Chem., 274, 9677–9685. [DOI] [PubMed] [Google Scholar]
- Casadaban M.J. and Cohen,S.N. (1980) Analysis of gene control signals by DNA fusion and cloning in Escherichia coli.J. Mol. Biol., 138, 179–207. [DOI] [PubMed] [Google Scholar]
- Coughlin R.T., Tonsager,S. and McGroarty,E.J. (1983a) Quantitation of metal cations bound to membranes and extracted lipopolysaccharide of Escherichia coli. Biochemistry, 22, 2002–2007. [DOI] [PubMed] [Google Scholar]
- Coughlin R.T., Haug,A. and McGroarty,E.J. (1983b) Physical properties of defined lipopolysaccharide salts. Biochemistry, 22, 2007–2013. [DOI] [PubMed] [Google Scholar]
- Dekker N. (2000) Outer-membrane phospholipase A: known structure, unknown biological function. Mol. Microbiol., 35, 711–717. [DOI] [PubMed] [Google Scholar]
- Ernst R.K., Yi,E.C., Guo,L., Lim,K.B., Burns,J.L., Hackett,M. and Miller,S.I. (1999) Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science, 286, 1561–1565. [DOI] [PubMed] [Google Scholar]
- Ganong B.R., Leonard,J.M. and Raetz,C.R.H. (1980) Phosphatidic acid accumulation in the membranes of Escherichia coli mutants defective in CDP-diglyceride synthetase. J. Biol. Chem., 255, 1623–1629. [PubMed] [Google Scholar]
- Garcia-Vescovi E., Soncini,F.C. and Groisman,E.A. (1996) Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell, 84, 165–174. [DOI] [PubMed] [Google Scholar]
- Garrett T.A., Kadrmas,J.L. and Raetz,C.R.H. (1997) Identification of the gene encoding the Escherichia coli lipid A 4′-kinase. Facile phosphorylation of endotoxin analogs with recombinant LpxK. J. Biol. Chem., 272, 21855–21864. [DOI] [PubMed] [Google Scholar]
- Groisman E.A., Chiao,E., Lipps,C.J. and Heffron,F. (1989) Salmonella typhimurium phoP virulence gene is a transcriptional regulator. Proc. Natl Acad. Sci. USA, 86, 7077–7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guina T., Yi,E.C., Wang.H., Hackett,M. and Miller,S.I. (2000) A PhoP-regulated outer-membrane protease of Salmonella enterica serovar typhimurium promotes resistance to α-helical antimicrobial peptides. J. Bacteriol., 182, 4077–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn J.S., Lim,K.B., Krueger,J., Kim,K., Guo,L., Hackett,M. and Miller,S.I. (1998) PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol., 27, 1171–1182. [DOI] [PubMed] [Google Scholar]
- Guo L., Lim,K.B., Gunn,J.S., Bainbridge,B., Darveau,R.P., Hackett,M. and Miller,S.I. (1997) Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP–phoQ. Science, 276, 250–253. [DOI] [PubMed] [Google Scholar]
- Guo L., Lim,K.B., Poduje,C.M., Daniel,M., Gunn,J.S., Hackett,M. and Miller,S.I. (1998) Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell, 95, 189–198. [DOI] [PubMed] [Google Scholar]
- Helander I.M., Kilpeläinen,I. and Vaara,M. (1994) Increased substitution of phosphate groups in lipopolysaccharides and lipid A of the polymyxin-resistant pmrA mutants of Salmonella typhimurium: a 31P-NMR study. Mol. Microbiol., 11, 481–487. [DOI] [PubMed] [Google Scholar]
- Hsu L., Jackowski,S. and Rock,C.O. (1991) Isolation and characterization of Escherichia coli K-12 mutants lacking both 2-acyl-glycerophosphoethanolamine acyltransferase and acyl–acyl carrier protein synthetase activity. J. Biol. Chem., 266, 13783–13788. [PubMed] [Google Scholar]
- Hu K.H., Liu,E., Dean,K., Gingras,M., DeGraff,W. and Trun,N.J. (1996) Overproduction of three genes leads to camphor resistance and chromosome condensation in Escherichia coli.Genetics, 143, 1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann A., Stierhof,Y.D. and Henning,U. (1994) New outer membrane-associated protease of Escherichia coli K-12. J. Bacteriol., 176, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kox L.F.F., Wösten,M.M.S.M. and Groisman,E.A. (2000) A small protein that mediates the activation of a two-component system by another two-component system. EMBO J., 19, 1861–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leive L. (1968) Studies on the permeability change produced in coliform bacteria by ethylenediaminetetraacetate. J. Biol. Chem., 243, 2373–2380. [PubMed] [Google Scholar]
- Leive L., Shovlin,V.K. and Mergenhagen,S.E. (1968) Physical, chemical and immunological properties of lipopolysaccharide released from Escherichia coli by ethylenediaminetetraacetate. J. Biol. Chem., 243, 6384–6391. [PubMed] [Google Scholar]
- Miller S.I. and Mekalanos,J.J. (1990) Constitutive expression of the phoP regulon attenuates Salmonella virulence and survival within macrophages. J. Bacteriol., 172, 2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.I., Kukral,A.M. and Mekalanos,J.J. (1989) A two-component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc. Natl Acad. Sci. USA, 86, 5054–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. (1996) Outer membrane. In Neidhardt,F.C. (ed.), Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd edn, Vol. 1. American Society for Microbiology, Washington, DC, pp. 29–47. [Google Scholar]
- Nishijima M. and Raetz,C.R.H. (1979) Membrane lipid biogenesis in Escherichia coli: identification of genetic loci for phosphatidyl glycerophosphate synthetase and construction of mutants lacking phosphatidylglycerol. J. Biol. Chem., 254, 7837–7844. [PubMed] [Google Scholar]
- Nishijima M., Bulawa,C.E. and Raetz,C.R.H. (1981) Two interacting mutations causing temperature-sensitive phosphatidylglycerol synthesis in Escherichia coli membranes. J. Bacteriol., 145, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard T.J., Kaltashov,I.A., Cotter,R.J., Steeghs,L., van der Ley,P., Khan,S., Maskell,D.J. and Raetz,C.R.H. (1997) Shortened hydroxyacyl chains on lipid A of Escherichia coli cells expressing a foreign UDP-N-acetylglucosamine O-acyltransferase. J. Biol. Chem., 272, 19688–19696. [DOI] [PubMed] [Google Scholar]
- Radika K. and Raetz,C.R.H. (1988) Purification and properties of lipid A disaccharide synthase of Escherichia coli.J. Biol. Chem., 263, 14859–14867. [PubMed] [Google Scholar]
- Raetz C.R.H. (1996) Bacterial lipopolysaccharides: a remarkable family of bioactive macroamphiphiles. In Neidhardt,F.C. (ed.), Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd edn, Vol. 1. American Society for Microbiology, Washington, DC, pp. 1035–1063. [Google Scholar]
- Raetz C.R.H., Purcell,S., Meyer,M.V., Qureshi,N. and Takayama,K. (1985) Isolation and characterization of eight lipid A precursors from a 3-deoxy-d-manno-octylosonic acid-deficient mutant of Salmonella typhimurium. J. Biol. Chem., 260, 16080–16088. [PubMed] [Google Scholar]
- Rana F.R., Macias,E.A., Sultany,C.M., Modzrakowski,M.C. and Blazyk,J. (1991) Interactions between magainin 2 and Salmonella typhimurium outer membranes: effect of lipopolysaccharide structure. Biochemistry, 130, 5858–5866. [DOI] [PubMed] [Google Scholar]
- Ray B.L., Painter,G. and Raetz,C.R.H. (1984) The biosynthesis of Gram-negative endotoxin. Formation of lipid A disaccharides from monosaccharide precursors in extracts of Escherichia coli. J. Biol. Chem., 259, 4852–4859. [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Schindler M. and Osborn,M.J. (1979) Interaction of divalent cations and polymyxin B with lipopolysaccharide. Biochemistry, 18, 4425–4430. [DOI] [PubMed] [Google Scholar]
- Schnaitman C.A. (1973) Outer membrane proteins of Escherichia coli. I. Effect of preparative conditions on the migration of protein in polyacrylamide gels. Arch. Biochem. Biophys., 157, 541–552. [DOI] [PubMed] [Google Scholar]
- Smith P.K. et al. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem., 150, 76–85. [DOI] [PubMed] [Google Scholar]
- Snijder H.J., Ubarretxena-Belandia,I., Blaauw,M., Kalk,K.H., Verheij,H.M., Egmond,M.R., Dekker,N. and Dijkstra,B.W. (1999) Structural evidence for dimerization-regulated activation of an integral membrane phospholipase. Nature, 401, 717–721. [DOI] [PubMed] [Google Scholar]
- Strack B., Lessl,M., Calendar,R. and Lanka,E. (1992) A common sequence motif, -E-G-Y-A-T-A-, identified within the primase domains of plasmid-encoded I- and P-type DNA primases and the α protein of the Escherichia coli satellite phage P4. J. Biol. Chem., 267, 13062–13072. [PubMed] [Google Scholar]
- Stumpe S., Schmid,R., Stephens,D.L., Georgiou,G. and Bakker,E.P. (1998) Identification of OmpT as the protease that hydrolyzes the antimicrobial peptide protamine before it enters growing cells of Escherichia coli.J. Bacteriol., 180, 4002–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura K. and Nishihara,T. (1988) Purification, characterization and primary structure of Escherichia coli protease VII with specificity for paired basic residues: identity of protease VII and OmpT. J. Bacteriol., 170, 5625–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanamoto K. and Azumi,S. (2000) Salmonella-type heptaacylated lipid A is inactive and acts as an antagonist of lipopolysaccharide action on human line cells. J. Immunol., 164, 3149–3156. [DOI] [PubMed] [Google Scholar]
- Vaara M., Vaara,T., Jensen,M., Helander,I., Nurminen,M., Rietschel,E.T. and Makela,P.H. (1981) Characterization of the lipopoly saccharide from the polymyxin-resistant pmrA mutants of Salmonella typhimurium. FEBS Lett., 129, 145–149. [DOI] [PubMed] [Google Scholar]
- Zhou Z., White,K.A., Polissi,A., Georgopoulos,C. and Raetz,C.R.H. (1998) Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem., 273, 12466–12475. [DOI] [PubMed] [Google Scholar]
- Zhou Z., Lin,S., Cotter,R.J. and Raetz,C.R.H. (1999) Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-l-arabinose, phosphoethanolamine and palmitate. J. Biol. Chem., 274, 18503–18514. [DOI] [PubMed] [Google Scholar]