

Abstract
The Escherichia coli UmuD′ protein is a subunit of the recently described error-prone DNA polymerase, pol V. UmuD′ is initially synthesized as an unstable and mutagenically inactive pro-protein, UmuD. Upon processing, UmuD′ assumes a relatively stable conformation and becomes mutagenically active. While UmuD and UmuD′ by themselves exist in vivo as homodimers, when together they preferentially interact to form heterodimers. Quite strikingly, it is in this context that UmuD′ becomes susceptible to ClpXP-mediated proteolysis. Here we report a novel targeting mechanism designed for degrading the mutagenically active UmuD′ subunit of the UmuD/D′ heterodimer complex, while leaving the UmuD protein intact. Surprisingly, a signal that is essential and sufficient for targeting UmuD′ for degradation was found to reside on UmuD not UmuD′. UmuD was also shown to be capable of channeling an excess of UmuD′ to ClpXP for degradation, thereby providing a mechanism whereby cells can limit error-prone DNA replication.
Keywords: ATP-dependent protease/degradation signal/SOS mutagenesis/UmuD
Introduction
The degradation of proteins is known to be an essential regulatory mechanism vital for biological processes ranging from apoptosis (Cohen, 1997; Grandgirard et al., 1998) and cell-cycle progression (King et al., 1996) to DNA repair (Kumar et al., 1999; Tinker-Kulberg and Morgan, 1999). A recurring theme in the degradation of labile proteins is the involvement of regions near the N- or C-terminus in protease recognition. For example, one of the better characterized degradation signals is the peptide tagging system encoded by Escherichia coli ssrA. This system functions by identifying stalled ribosomes at the 3′ end of truncated mRNA and attaching an 11 amino acid peptide (AANDENYALAA) to the C-terminus of the respective truncated protein (Tu et al., 1995; Keiler et al., 1996). Addition of this SsrA RNA-dependent peptide tag allows for the rapid removal of the resulting protein fusion (Keiler et al., 1996). The ClpXP and ClpAP proteases were shown to be responsible for the degradation of SsrA-tagged proteins (Gottesman et al., 1998); moreover, in a separate study the FtsH protease was also found to be capable of degrading SsrA-tagged proteins (Herman et al., 1998). The non-polar nature of the SsrA-tag is essential for protease recognition (Gottesman et al., 1998), and its extension from the C-terminus of targeted proteins is believed to make the SsrA-tag readily accessible to the respective proteases.
Another well characterized degradation signal is a component of the bacterial N-end rule degradation pathway (Tobias et al., 1991). This pathway also illustrates the importance of tail recognition in targeted proteolysis. The N-end rule relates the metabolic stability of a protein to the identity of the N-terminal amino acid. The pathway was identified by following the stability of engineered β-galactosidase fusions each exposing a different N-terminal amino acid. The bacterial N-end rule is hierarchically organized: primary destabilizing residues (leucine, phenylalanine, tryptophan and tyrosine) impart instability while the secondary destabilizing residues, arginine and lysine, require the activity of the Leu,Phe-tRNA–protein transferase for the conjugation of the primary destabilizing residues leucine or phenylalanine to the N-terminus before degradation occurs. As with the SsrA-tag, the ClpAP protease is responsible for degradation of bacterial N-end rule substrates (Tobias et al., 1991).
As with many temporally expressed systems, the response to DNA damage involves the rapid induction of proteins necessary for continued survival, followed by inactivation of those response proteins no longer required post-DNA repair. DNA damage in E.coli results in the expression of a set of unlinked genes necessary for DNA repair, cell division and damage tolerance that is often referred to as the ‘SOS-response’ (Radman, 1974). The prevailing hypothesis is that the SOS response evolved to ensure that repair of damaged DNA is accomplished with high fidelity (Friedberg et al., 1995). However, extensive DNA damage can result in the saturation of the available error-free repair systems, and cell survival becomes dependent upon translesion DNA synthesis. Unlike normal replication, which is highly accurate, synthesis of nascent DNA opposite normally replication-blocking lesions catalyzed by the UmuD′2C complex (DNA pol V) (Tang et al., 1998, 1999, 2000; Woodgate, 1999) is highly error-prone, resulting in a dramatic increase in the mutation rate (Kato and Shinoura, 1977). While the advantage of such error-prone replication is that it ultimately increases the ‘fitness’ of the organism (Radman, 1999), the levels of both transcriptional and translational regulation employed to keep the activity of the Umu proteins to a minimum are evidence that the cell uses the error-prone pathway only as a last resort (Koffel-Schwartz et al., 1996; Woodgate and Levine, 1996). Post-translational regulation of UmuD and UmuC is primarily achieved via their degradation by the Lon protease (Frank et al., 1996a; Gonzalez et al., 1998a). However, in cells that have incurred DNA damage, some of the UmuD molecules escape Lon-mediated degradation and instead are converted by a RecA-mediated self-cleavage reaction to the mutagenically active UmuD′ protein. Unlike UmuD, UmuD′ is relatively insensitive to Lon-mediated proteolysis and has a much greater half-life in vivo (Frank et al., 1996a). Survival of UmuD′ could be problematic for the cell as it would result in an increased likelihood that error-prone pol V (UmuD′2C) will replicate undamaged DNA (Fijalkowska et al., 1997). As a consequence, E.coli appears to have evolved a novel mechanism for targeting the mutagenically active UmuD′ protein for proteolysis. UmuD′ is unstable only in the presence of unprocessed UmuD, and is stabilized in E.coli strains carrying either a clpX or clpP mutation. We proposed earlier that UmuD′ becomes a substrate of the ATP-dependent ClpXP protease when in a heterodimer with UmuD (Frank et al., 1996a).
In order to elucidate the mechanism of UmuD′ degradation, we first established that the previously described in vivo activity of ClpXP on the heterodimer is a direct interaction of the protease with substrate. We then identified the N-terminal region of UmuD, not UmuD′, as a region essential for ClpXP-mediated degradation of UmuD′ in a heterodimer complex. Furthermore, a 24 amino acid peptide corresponding to the N-terminus of UmuD was shown to be all that is necessary to target UmuD′ for degradation by ClpXP. Finally, we demonstrated that UmuD is capable of shuttling excess UmuD′ towards degradation by ClpXP, offering an explanation as to how cells return to a high fidelity mode of DNA replication after repair of DNA damage.
Results
Specificity of UmuD/D′ degradation by the ClpXP protease
Although both UmuD and UmuD′ can exist as homodimers, when both are present, the heterodimer is formed (Battista et al., 1990). We previously reported that in vivo the heterodimer was specifically recognized by ClpXP, yet ClpXP only degraded UmuD′ (Frank et al., 1996a). We have now reconstituted this system in vitro, and have shown that when highly purified UmuD/D′ heterodimer is incubated with ATP, ClpP and ClpX, UmuD′ is degraded (Figure 1A). No degradation of UmuD′ was seen when ClpX was omitted from the reaction (Figure 1A). Under the conditions used in Figure 1B in which UmuD/D′ was subsaturating, the degradation rate for UmuD′ was ∼0.4 min–1 although the maximum degradation rate is probably several times higher. Remarkably, degradation of UmuD′ was subunit-specific, with ClpXP having no effect on the UmuD component of the heterodimer.

Fig. 1. In vitro degradation of UmuD′ in a UmuD/D′ heterodimer complex by ClpXP. (A) Highly purified UmuD/D′ and ClpP proteins were incubated at 37°C in the presence or absence of ClpX as described. An aliquot was removed at the indicated time, electrophoresed on a 16% SDS–polyacrylamide gel, and visualized after staining with Coomassie Brilliant Blue. (B) The data were generated by quantifying the gels described in (A) on a ChemiImager 4000 low-light imaging system. The following decay curves are for the individual subunits of the heterodimer: (open squares) UmuD′ + ClpX; (filled squares) UmuD′ – ClpX; (open triangles) UmuD + ClpX; (filled triangles) UmuD – ClpX.
Although the role of ClpXP in selective degradation is well documented, the identification and subsequent binding of substrate amongst the vast number of stable proteins in a cell remains unclear. Degradation signals recognized by ClpXP vary from the non-polar nature of the SsrA-tag (Gottesman et al., 1998) to the positively charged MuA C-terminus (Levchenko et al., 1995, 1997). The heterodimer requirement for degradation therefore enabled us to assess the specificity of the ClpXP protease in vitro. When UmuD or UmuD′ homodimers were incubated separately with ClpXP no degradation was observed (Figure 2). Thus, ClpXP specifically degrades UmuD′ only when it is part of the heterodimeric complex. These experiments demonstrate the specificity of ClpXP and show that an otherwise stable protein can be targeted for degradation through an association with a protein that is itself insensitive to proteolysis.

Fig. 2. Specificity of ClpXP-mediated degradation of UmuD′ in vitro. (A) Purified UmuD, UmuD′ and UmuD/D′ were individually incubated with ClpP in the presence or absence of ClpX at 37°C for 1 h. Reaction mixtures were processed and proteins visualized as described in Figure 1. (B) Quantitation of ClpXP-mediated degradation of homodimeric UmuD and UmuD′ and heterodimeric UmuD/D′: closed bars, –ClpX; open bars, +ClpX. The data were generated by quantifying the gels described in (A) on a ChemiImager 4000 low-light imaging system. Heterodimer stability was determined for both UmuD and UmuD′ subunits.
Identification of a region within UmuD that is necessary to impart ClpXP-mediated sensitivity to UmuD′
In addressing the question of how UmuD imparts ClpXP sensitivity to UmuD′, we reasoned that a region unique to the heterodimer might be essential for efficient degradation of UmuD′. The only difference in the amino acid sequence between UmuD and UmuD′ is that UmuD retains the N-terminal 24 amino acids lacking in UmuD′. This N-terminal tail is important for both Lon-mediated degradation of UmuD and its RecA-mediated conversion to UmuD′ (Gonzalez et al., 1998a). We were therefore interested in determining whether it might also be important for UmuD′ degradation. To investigate this possibility, we used plasmids expressing multiple alanine mutants within the first 30 amino acids of UmuD (Figure 3A) (Gonzalez et al., 1998a) and measured the in vivo accumulation of UmuD′ after RecA-mediated cleavage of the mutant UmuD protein (Figure 3B). To ensure that our assay principally monitors ClpXP- mediated proteolysis, we eliminated two additional levels of regulation normally imposed on the UmuD protein by utilizing isogenic ΔumuDC lexA51(Def) recA730 strains that differ only in their clpX allele. The lexA51(Def) mutation allows constitutive expression of LexA-regulated genes, including umuD, in the absence of DNA damage, while recA730 is an allele of recA that mediates constitutive conversion of wild-type UmuD to UmuD′ (Shinagawa et al., 1988). In these strains, the wild-type UmuD protein was converted to UmuD′ with >95% efficiency, as was the UmuD9-4 mutant (Figure 3B). Under these conditions, cleavage of UmuD to UmuD′ is so efficient that little or no UmuD/D′ heterodimers form, and as a consequence, there is no in vivo (UmuD/D′) substrate upon which ClpXP can act. In comparison, conversion of UmuD26-4 to UmuD′ was obvious only in the clpX background and not the clpX+ cells (Figure 3B). We believe that these observations can be explained by the fact that conversion of UmuD26-4 to UmuD′ is greatly reduced (even in the recA730 background) with the resultant UmuD′ being ‘trapped’ in a heterodimeric complex with unconverted UmuD26-4. As the heterodimer is degraded rapidly by ClpXP, UmuD′ does not accumulate in the clpX+ cells but survives in clpX cells. Interestingly, when the double mutant UmuD26/9 was expressed, UmuD′ accumulated in both the clp+ and clpX background and was similar to that observed with UmuD26-4 in a clpX background. Based on these observations, we hypothesize that the multiple alanine stretch found in UmuD9-4 affects the ability of ClpXP to recognize the UmuD26-4/D′ heterodimer.

Fig. 3. Effects of the UmuD multiple alanine mutants on ClpXP-mediated proteolysis of UmuD′. (A) Areas boxed indicate the multiple alanine substitutions made within the UmuD N-terminus. The RecA-mediated posttranslational cleavage site within UmuD is located between Cys24 and Gly25. (B) Western blot analysis of the ability of the alanine stretch mutants to undergo RecA-mediated conversion to UmuD′ cleavage was assayed in isogenic ΔumuDClexA51(Def) recA730 strains that differed only in their clpX allele (RW244-clpX+; RW582-clpX). These strains constitutively express all LexA-regulated genes, including the UmuD mutants, and promote the constitutive cleavage of UmuD in the absence of an inducing signal. Strains were processed as described in Materials and methods.
To investigate further the role of the UmuD9-4 mutation in targeting UmuD′ for degradation by ClpXP, we constructed low-copy-number vectors that co-express each individual UmuD multiple alanine mutant in cis with UmuD′ and measured degradation rates of the expressed UmuD proteins in a ΔumuDC lexA51(Def) recA+ strain. Unlike recA730 strains, which constitutively mediate conversion of UmuD to UmuD′, significant UmuD cleavage only occurs in recA+ cells exposed to DNA damaging agents. As these experiments are performed in the absence of DNA damage, there is no spontaneous conversion of UmuD to UmuD′ in this background. As found previously (Frank et al., 1996a), both subunits of the wild-type heterodimer were labile (Figure 4A). The stability of the UmuD26-4/D′ complex mirrored that of the wild-type heterodimer, indicating that the UmuD26-4 mutant is competent for heterodimer formation and efficient recognition by the ClpXP protease (Figure 4A and B). In contrast, both subunits of the UmuD9-4/D′ heterodimer displayed a significant increase in stability (Figure 4A). The half-life of UmuD′ increased from ∼10 min in the wild-type complex to >60 min in the UmuD9-4/D′ complex (Figure 4B). ClpXP was also unable to degrade the UmuD26/9/D′ heterodimer (Figure 4A and B). These findings confirm that the UmuD9-4 region of UmuD is required for targeting UmuD′ for degradation by ClpXP. The stability of the full-length UmuD species also reflects the preferential formation of the heterodimeric UmuD9-4/UmuD′ complex in vivo, and the consequent protection of UmuD9-4 from degradation by the Lon protease (Frank et al., 1996a; Gonzalez et al., 1998a) when UmuD′ is not degraded by ClpXP.

Fig. 4. In vivo stability of UmuD′ when expressed in cis with individual multiple alanine mutants of UmuD. The cultures were grown at 37°C until early logarithmic growth when 100 µg/ml chloramphenicol was added to the medium to block protein synthesis. Aliquots were removed at the times indicated, and whole cell protein extracts of an equivalent number of cells were electrophoresed on 16% SDS–polyacrylamide gels and visualized as described in Materials and methods. (A) The stability of UmuD′ in the presence of the various UmuD multiple alanine mutants was analyzed by individually introducing each respective plasmid into the strain EC10 [recA+ lexA51(Def) Δ(umuDC)-596::ermGT clp+]. (B) Kinetics of UmuD′ degradation when expressed in cis with the following UmuD species: squares, wild type; circles, UmuD9-4; triangles, UmuD26-4; diamonds, UmuD26/9. The data were generated by quantifying the gels described in (A) on a ChemiImager 4000 low-light imaging system.
Characterization of the UmuD9-4/D′ heterodimer in vitro
To verify the role of the N-terminus of UmuD in heterodimer formation and stability, we purified the UmuD9-4 mutant and evaluated its ability to interact with UmuD′ in vitro. To rule out the possibility that UmuD9-4 (or UmuD26/9) is simply unable to form heterodimers and, as a consequence, is unable to target UmuD′ to ClpXP for proteolysis, we ran samples of UmuD9-4 alone or pre-incubated with UmuD′ on a native polyacrylamide gel. The sample of UmuD9-4 migrated as a dimer much like wild-type UmuD (Figure 5A). When pre-incubated with UmuD′, UmuD9-4 preferentially interacted with UmuD′, with most of the protein migrating at a position equivalent to that of the wild-type heterodimer, and ∼5% as a UmuD9-4 homodimer (Figure 5A). UmuD9-4 appears, therefore, to be as efficient as wild-type UmuD in forming heterodimers with UmuD′, and thus we can rule out that the lack of ClpXP-mediated proteolysis is due to inefficient heterodimer formation.

Fig. 5. In vitro characterization of the UmuD9-4/D′ heterodimer. (A) Heterodimerization of UmuD9-4 with UmuD′. UmuD and UmuD9-4 were each individually incubated with UmuD′ at 25°C for 30 min and electrophoresed through a 12% native polyacrylamide gel. UmuD, UmuD9-4 and UmuD′ were also electrophoresed to determine mobility of the homodimer species. Visualization of the reactions was accomplished by staining with Coomassie Brilliant Blue. The UmuD/D′ sample was purified from E.coli as a heterodimer complex (Woodgate et al., 1989). Migration of the homodimer species and UmuD/D′ is indicated. Concentrations of the various components are described in Materials and methods. (B) In vitro stability of UmuD9-4/D′ in the presence of ClpXP. Purified UmuD or UmuD9-4 was incubated in the presence of UmuD′ at 25°C for 30 min, followed by incubation with ClpP ± ClpX at 37°C for the designated times. Reactions were stopped with the addition of 4× SDS sample buffer, followed by freezing in dry ice. Samples were electrophoresed, visualized, and quantitated as described in Figure 1. Kinetics of degradation are for UmuD′ in each respective heterodimer. UmuD is unaffected by ClpXP. Open squares, UmuD/D′ + ClpX; filled squares, UmuD/D′ – ClpX; open triangles, UmuD9-4/D′ + ClpX; filled triangles, UmuD9-4/D′ – ClpX.
We next compared the in vitro degradation kinetics of UmuD′ when complexed to either UmuD or UmuD9-4. There was little difference in UmuD′ degradation between heterodimers purified from cells and those formed in vitro (Figures 1B and 5B). Furthermore, the in vitro degradation kinetics of the UmuD9-4/D′ heterodimer showed a similar stabilization of UmuD′ to that observed in vivo (Figures 4B and 5B).
The N-terminal 24 amino acids of UmuD is all that is necessary for targeting UmuD′ for degradation
The N-terminus of UmuD plays an important role in the regulation of error-prone translesion synthesis. It functions in targeting UmuD for degradation by the Lon protease (Gonzalez et al., 1998a), regulates the levels of RecA-mediated self cleavage of UmuD (McDonald et al., 1998), and as we demonstrate in this report, it depletes the levels of UmuD′ during non-inducing conditions. The importance of this region directed us to consider that the N-terminal 24 amino acids of UmuD might be all that is necessary to impart ClpXP-mediated degradation on UmuD′. To test this hypothesis, a 24 amino acid peptide corresponding to the N-terminus of UmuD was synthesized and pre-incubated with UmuD′ prior to the addition of ClpXP and ATP to initiate degradation. At stoichiometric levels (10 µM), the peptide was capable of promoting UmuD′ degradation by ClpXP (Figure 6). Approximately 40% of the UmuD′ protein is degraded within 1 h; no increase in degradation was seen at higher peptide concentrations (Figure 6). In contrast, the peptide did not stimulate ClpXP activity towards λO or a cI-SsrA fusion protein (M.Maurizi, unpublished results), indicating that the peptide-stimulated proteolysis is specific to UmuD′. Furthermore, the peptide does not appear to result in the monomerization of UmuD′ (M.Gonzalez and R.Woodgate, unpublished results), suggesting that it binds to a UmuD′ homodimer rather than to a UmuD′ monomer. Experiments are currently in progress to test this hypothesis as well as to determine the fate of the peptide after ClpXP proteolysis of UmuD′. In complete agreement with our expectations, incubation with a variant peptide corresponding to the UmuD9-4 mutant had no stimulating effect on UmuD′ degradation (Figure 6). These peptide studies, together with the in vitro and in vivo results described above, establish that residues 9–12 of UmuD are required to generate the degradation signal that imparts susceptibility to ClpXP-mediated proteolysis upon UmuD′.

Fig. 6. The role of the N-terminus in ClpXP-mediated degradation of UmuD′. UmuD (squares) and UmuD9-4 (triangles) peptides, at the concentrations indicated, were incubated with UmuD′ at 25°C for 30 min, followed by the addition of ClpXP. After addition of ClpXP the reactions were incubated at 37°C for 1 h and the samples were electrophoresed on a 16% polyacrylamide gel. Samples were processed, visualized and quantitated as described in Figure 1. These experiments reveal that the N-terminal 24 amino acids of UmuD are sufficient to target UmuD′ for ClpXP-mediated proteolysis.
UmuD is capable of channeling excess UmuD′ for degradation by ClpXP in vitro
Degradation of UmuD′ is important for E.coli to minimize mutagenesis both during normal growth and after repair of DNA damage. We have shown that ClpXP plays a role in removal of UmuD′ generated by spontaneous RecA-mediated cleavage of UmuD under non-inducing conditions (Gonzalez et al., 1998b). Because heterodimers were readily formed from mixtures of homodimers, we reasoned that following repair of DNA damage when the levels of UmuD′ are relatively high in relation to UmuD, UmuD might act in multiple cycles to promote degradation of the excess of mutagenically active UmuD′ homodimers. Indeed, as seen in Figure 7, UmuD was able to promote degradation of a 3-fold molar excess of UmuD′. This result provides strong support for a mechanism whereby, in vivo, mutagenic UmuD′ homodimers, which are not directly susceptible to degradation, can be efficiently eliminated if small amounts of UmuD are present (Gonzalez et al., 1998b).
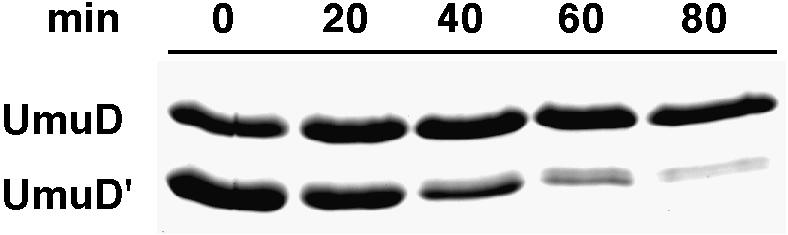
Fig. 7. The role of UmuD in removal of excess UmuD′ by ClpXP-mediated degradation. Purified UmuD/D′ was mixed with UmuD′ and incubated in the presence of ClpXP at 37°C for the indicated times. Aliquots were removed, processed and visualized as described in Materials and methods. In this experiment, UmuD′ was targeted for ClpXP-mediated proteolysis despite the fact that it is in a 3-fold molar excess over UmuD. Similar results were also obtained when UmuD′ was in 6-fold excess over UmuD (not shown).
Such a mechanism provides the cell with an opportunity to temporarily postpone error-prone translesion replication in favor of error-free DNA repair. If damage persists in vivo however, the increased rate of RecA-mediated conversion of UmuD to UmuD′ results in fewer UmuD/D′ heterodimers and a concomitant reduction in ClpXP-mediated degradation of UmuD′. As a result, intracellular levels of homodimeric UmuD′ increase, allowing it to interact with UmuC and form pol V, where it promotes error-prone translesion DNA synthesis (Tang et al., 1999). Once DNA repair has occurred, the inducing signal wanes and so does RecA-mediated cleavage of UmuD to UmuD′. UmuD/D′ heterodimers prevail once more and the intracellular concentration of UmuD′ is returned to a basal level by the action of ClpXP (Gonzalez et al., 1998b).
Discussion
Understanding the mechanisms by which proteases selectively recognize and degrade substrates is important in determining how they act in a temporal fashion to regulate biological processes. We have discovered a novel mechanism for destabilizing an otherwise stable protein through a protein–protein interaction that provides protease recognition in trans. How might ClpXP recognize the heterodimer and not the respective homodimers? One model is that each subunit of the heterodimer provides a portion of the degradation signal recognized by ClpXP. For example, neither region alone is capable of directing ClpX recognition, but together they create a protein environment conducive for ClpX recognition. A second possible model is based on the assumption that the binding of the UmuD N-terminus to UmuD′ promotes a conformational change that exposes a degradation signal normally masked in the UmuD′ homodimer. Either mechanism described above presents the challenge to identify the cognate region(s) of UmuD′ necessary for ClpXP-mediated degradation.
As with many labile proteins, the stability of the UmuD/D′ heterodimer is also contingent on the presence of a destabilizing motif at the N- or C-terminus. The UmuD/D′ heterodimer deviates from other examples of destabilizing termini in that it is the N-terminus of UmuD, not the substrate UmuD′, that is required for degradation by ClpXP. Our work implicates amino acids 9–12 of UmuD as necessary for UmuD′ instability. It is of particular interest to note that a region of the N-terminus of UmuD (amino acids 15–19) is also vital for the Lon-mediated degradation of UmuD (Gonzalez et al., 1998a). Despite the close proximity of these putative degradation signals, the UmuD9-4 homodimer is degraded by Lon with the same efficiency as the wild-type UmuD homodimer, suggesting that the Lon degradation signal does not overlap the ClpXP component located at amino acids 9–12 (Gonzalez et al., 1998a). We are currently assessing the role, if any, of amino acids 15–19 of UmuD in ClpXP-mediated degradation of the UmuD/D′ heterodimer. The proximity of these degradation signals could allow regulation of the sequence of proteolytic attack by the respective proteases on the UmuD/D′ heterodimer. We show that stabilization of UmuD′ in the UmuD9-4/D′ heterodimer (Figure 4A), or the wild-type heterodimer in a clpX background (Frank et al., 1996a), also results in an increase in stability of the UmuD species of the heterodimer. Because ClpXP has no significant effect on the stability of the UmuD homodimer (Figure 2), stabilization of UmuD suggests that in a heterodimer UmuD′ must first be degraded by ClpXP before Lon can effectively act upon the UmuD species. By ordering the accessibility of protease recognition motifs the cell ensures that the mutagenically active UmuD′ is removed before any response is generated against mutagenically inactive UmuD.
Protein–protein interactions have long been known to influence selective degradation. In many cases, interactions involving a labile protein lead to its stabilization (Gottesman and Maurizi, 1992). For example, the Saccharomyces cerevisiae MAT transcription factors, α2 and a1, are rapidly degraded in haploid cells (Johnson et al., 1998). In a/α diploids, a1 and α2 form heterodimers that are more resistant to degradation by the ubiquitin–proteasome pathway (Johnson et al., 1998). Other examples of this phenomenom found in E.coli include the stabilization of the antidote protein of the ccd post-segregation killing system, CcdA, by CcdB (Van Melderen et al., 1996), the stabilization of the positive regulator of capsular synthesis, RcsA, by RcsB (Stout et al., 1991) and the stabilization of the phage Mu replication protein, MuA, by MuB (Levchenko et al., 1997). Examples of targeting proteins for degradation by trans recognition are much rarer. The example for which trans recognition was defined is the targeting of β-galactosidase subunits carrying a ubiquitylation site but lacking an N-end degradation signal by associated subunits carrying the N-end degradation signal but lacking the ubiquitylation site (Johnson et al., 1990). Another instance occurs in human cells upon infection with HIV-1. The virus-encoded protein Vpu interacts with β-TrCP, but it is the associated receptor protein, CD4, that is ubiquitylated and degraded rather than the Vpu (Margottin et al., 1998). Trans recognition in the case of UmuD/D′ is mediated directly through the protease and, in this way, is more reminiscent of ubiquitin–conjugate recognition by the proteasome. Ubiquitin binding to a component of the regulatory complex positions the tagged protein for unfolding and degradation by the base complex and proteasome; ubiquitin itself is released and not degraded (Hershko and Ciechanover, 1998). Interest ingly, two examples of trans-targeting in E.coli involve the ClpXP protease: degradation of MuC repressor in hetero-complexes with mutant MuC subunits (Welty et al., 1997) and degradation of the sigma factor, RpoS, complexed with RssB (Bearson et al., 1996; Muffler et al., 1996; Pratt and Silhavy, 1996; S.Wickner, Y.Zhou, M.R.Maurizi and S.Gottesman, unpublished results). Trans recognition by ClpXP implies that there are subsites on the protease that recognize different elements in protein complexes, and that translocation and degradation proceed from some binding sites but not others. Elucidation of the specificity of these sites and their location in the ClpXP complex will help us understand the complex process of selective protein degradation in vivo.
Materials and methods
Bacterial strains and plasmids
All strains carry a lexA51(Def) mutation, which results in constitutive expression of LexA-regulated genes (including the umu operon) in the absence of exogenous DNA damage. They also carry a deletion of the chromosomal umu operon (either ΔumuDC595::cat or ΔumuDC596:: ermGT). Strain EC10 [relevant genotype: recA+ lexA51(Def) Δ(umuDC)::ermGT] (Frank et al., 1996a) was used to characterize the stability of the heterodimer mutants. To determine the ability of the various UmuD multiple alanine mutants to undergo RecA-mediated cleavage to UmuD′, we utilized strains RW244 [recA730 srlC300::Tn10 lexA51(Def) ΔumuDC::cat] (McDonald et al., 1998) and RW582 [recA730 srlC300::Tn10 lexA51(Def) ΔumuDC::cat clpX::Kan].
Plasmid pET-D9 expresses the UmuD9-4 protein from an isopropyl-β-d-thiogalactopyranoside-inducible T7 promoter and was constructed by digesting pKSD9-4 with NdeI and HindIII and cloning the resulting 613 bp fragment into the similarly digested plasmid pET22b. All of the remaining plasmid constructs described below express their respective proteins from the native umu operon operator-promoter. Construction of all heterodimer expressing plasmids was accomplished by digesting the respective pBR322-derived UmuD multiple alanine expressing plasmids (Gonzalez et al., 1998a) with AgeI, blunt ending with PolI (Kf), and subsequently digesting with EcoRI. The resulting ∼545 bp fragment was then subcloned into the PmlI and EcoRI digested low-copy-number plasmid, pRW66, which expresses UmuD′. pKSD16 expresses wild-type UmuD in cis with UmuD′; pKSD17 expresses UmuD9-4 in cis with UmuD′; pKSD20 expresses UmuD26-4 in cis with UmuD′; and pKSD21 expresses UmuD26/9 in cis with UmuD′.
Proteins and peptides
ClpX (Grimaud et al., 1998), ClpP (Maurizi et al., 1994), UmuD (McDonald et al., 1998), UmuD9-4 (McDonald et al., 1998), UmuD′ (McDonald et al., 1998) and UmuD/D′ (Woodgate et al., 1989) were all purified as previously described. The UmuD wild-type peptide was synthesized by Covance Laboratories (Vienna, VA). The UmuD9-4 peptide was synthesized on an Applied Biosystems 431A synthesizer, cleaved, and deprotected prior to reverse-phase HPLC purification.
In vitro protein degradation
Purified UmuD, UmuD′ and UmuD/D′ (9 µM) were each individually incubated with ClpXP (0.35 µM) in buffer containing 50 mM Tris–HCl pH 8.0, 100 mM KCl, 4 mM ATP, 10 mM MgCl2, 1 mM dithiothreitol (DTT). The reaction mixture was incubated at 37°C and a 25 µl aliquot was removed at the specified times, added to 4× SDS buffer, and frozen in dry ice. After heating at 100°C for 5 min, proteins were run on a 16% SDS–polyacrylamide gel and visualized after staining with Coomassie Blue R-250. Identical reaction conditions (UmuD, UmuD′ or UmuD/D′ at 9 µM, ±0.35 µM ClpXP) were employed when assaying ClpXP specificity except that only a 1 h time point was assayed. Degradation of heterodimer in the presence of excess UmuD′ was performed as follows. The reaction mixture (40 µl) contained UmuD/D′ (15 µM), additional UmuD′ (16 µM), ClpXP (1.0 µM), 50 mM Tris–HCl pH 8.0, 100 mM KCl, 8 mM ATP, 15 mM MgCl2, 1 mM DTT, 50 mM creatine phosphate and 3 µg of creatine kinase. Reactions were incubated at 37°C, and at the indicated times 7 µl aliquots were removed, processed and visualized as described above. Peptide-mediated degradation of UmuD′ was similar to that described above, except that each peptide was pre-incubated with UmuD′ (10 µM) for 30 min at 25°C at the indicated peptide concentrations before ClpXP (0.70 µM) was added.
Heterodimerization of UmuD9-4 with UmuD′
UmuD (6 µM) and UmuD9-4 (6 µM) were each individually incubated with UmuD′ (6 µM) at 25°C for 30 min and electrophoresed through a 12% native polyacrylamide gel (Novex, San Diego, CA) as recommended by the manufacturer and visualized after staining with Coomassie Blue R-250. UmuD (6 µM), UmuD9-4 (6 µM) and UmuD′ (6 µM) were also electrophoresed to determine mobility of the homodimer species.
In vivo measurement of heterodimer stability and UmuD cleavage
The stability of the heterodimers (both wild type and mutant) and the cleavage of UmuD were assayed as previously described (Frank et al., 1996a). Briefly, cells were grown in Luria-Bertani media at 37°C until they reached early exponential phase. At time zero, 100 µg/ml chloramphenicol was added to the medium to block protein synthesis and a 1.5 ml aliquot was removed at the time indicated. To monitor the RecA-mediated conversion of UmuD, chloramphenicol was omitted and only a single sample processed as described here. Cells were harvested by centrifugation and the resulting cell pellet resuspended in 4× SDS sample buffer. Aliquots representing equal cell numbers were electrophoresed in 16% SDS–PAGE gels. Proteins were transferred to an Immobilon P membrane (Millipore) and subsequently probed with a mixture of polyclonal antibodies raised against the N-terminal 24 amino acids of UmuD (Covance, Vienna, VA) and the full-length UmuD′ protein (Frank et al., 1996b). The transferred proteins were visualized on Kodak Bio-MaxMR film using the CPSD-Western light chemiluminescent assay (Tropix, Bedford, MA). Decay curves were generated by quantitating multiple exposures of the blots to assure that the relative intensity of UmuD and UmuD′ was within a linear range. All time points were normalized to the zero time point (100% protein) of each respective decay curve.
Quantitation for each experiment was performed on a ChemiImager 4000, low-light imaging system (Alpha Innotech Corporation, San Leandro, CA). All experiments were performed two to three times with no significant variation in results.
Acknowledgments
Acknowledgements
We thank Ekaterina Frank for providing purified UmuD/D′ complex and Jan Rozycki for synthesis of the UmuD9 peptide. We also thank David Dean for critical reading of the manuscript. This work was supported in part by the NIH Intramural research program and by a National Research Council fellowship to M.G.
References
- Battista J.R., Ohta,T., Nohmi,T., Sun,W. and Walker,G.C. (1990) Dominant negative umuD mutations decreasing RecA-mediated cleavage suggest roles for intact UmuD in modulation of SOS mutagenesis. Proc. Natl Acad. Sci. USA, 87, 7190–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearson S.M., Benjamin,W.H.,Jr, Swords,W.E. and Foster,J.W. (1996) Acid shock induction of RpoS is mediated by the mouse virulence gene mviA of Salmonella typhimurium. J. Bacteriol., 178, 2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen G.M. (1997) Caspases: the executioners of apoptosis. Biochem. J., 326, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska I.J., Dunn,R.L. and Schaaper,R.M. (1997) Genetic requirements and mutational specificity of the Escherichia coli SOS mutator activity. J. Bacteriol., 179, 7435–7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E.G., Ennis,D.G., Gonzalez,M., Levine,A.S. and Woodgate,R. (1996a) Regulation of SOS mutagenesis by proteolysis. Proc. Natl Acad. Sci. USA, 93, 10291–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E.G., Gonzalez,M., Ennis,D.G., Levine,A.S. and Woodgate,R. (1996b) In vivo stability of the Umu mutagenesis proteins: a major role for RecA. J. Bacteriol., 178, 3550–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. American Society for Microbiology, Washington, DC. [Google Scholar]
- Gonzalez M., Frank,E.G., Levine,A.S. and Woodgate,R. (1998a) Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: in vitro degradation and identification of residues required for proteolysis. Genes Dev., 12, 3889–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M., Frank,E.G., McDonald,J.P., Levine,A.S. and Woodgate,R. (1998b) Structural insights into the regulation of SOS mutagenesis. Acta Biochemica Polonica, 45, 163–172. [PubMed] [Google Scholar]
- Gottesman S. and Maurizi,M.R. (1992) Regulation by proteolysis: energy-dependent proteases and their targets. Microbiol. Rev., 56, 592–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S., Roche,E., Zhou,Y. and Sauer,R.T. (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev., 12, 1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandgirard D., Studer,E., Monney,L., Belser,T., Fellay,I., Borner,C. and Michel,M.R. (1998) Alphaviruses induce apoptosis in Bcl-2-overexpressing cells: evidence for a caspase-mediated, proteolytic inactivation of Bcl-2. EMBO J., 17, 1268–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaud R., Kessel,M., Beuron,F., Steven,A.C. and Maurizi,M.R. (1998) Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. J. Biol. Chem., 273, 12476–12481. [DOI] [PubMed] [Google Scholar]
- Herman C., Thevenet,D., Bouloc,P., Walker,G.C. and D’Ari,R. (1998) Degradation of carboxy-terminal-tagged cytoplasmic proteins by the Escherichia coli protease HflB (FtsH). Genes Dev., 12, 1348–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Johnson E.S., Gonda,D.K. and Varshavsky,A. (1990) Cis–trans recognition and subunit-specific degradation of short-lived proteins. Nature, 346, 287–291. [DOI] [PubMed] [Google Scholar]
- Johnson P.R., Swanson,R., Rakhilina,L. and Hochstrasser,M. (1998) Degradation signal masking by heterodimerization of MATα2 and MATa1 blocks their mutual destruction by the ubiquitin–proteasome pathway. Cell, 94, 217–227. [DOI] [PubMed] [Google Scholar]
- Kato T. and Shinoura,Y. (1977) Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol. Gen. Genet., 156, 121–131. [DOI] [PubMed] [Google Scholar]
- Keiler K.C., Waller,P.R. and Sauer,R.T. (1996) Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science, 271, 990–993. [DOI] [PubMed] [Google Scholar]
- King R.W., Deshaies,R.J., Peters,J.M. and Kirschner,M.W. (1996) How proteolysis drives the cell cycle. Science, 274, 1652–1659. [DOI] [PubMed] [Google Scholar]
- Koffel-Schwartz N., Coin,F., Veaute,X. and Fuchs,R.P.P. (1996) Cellular strategies for accommodating replication-hindering adducts in DNA: control by the SOS response in Escherichia coli. Proc. Natl Acad. Sci. USA, 93, 7805–7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Talis,A.L. and Howley,P.M. (1999) Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem., 274, 18785–18792. [DOI] [PubMed] [Google Scholar]
- Levchenko I., Luo,L. and Baker,T.A. (1995) Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes Dev., 9, 2399–2408. [DOI] [PubMed] [Google Scholar]
- Levchenko I., Yamauchi,M. and Baker,T.A. (1997) ClpX and MuB interact with overlapping regions of Mu transposase: implications for control of the transposition pathway. Genes Dev., 11, 1561–1572. [DOI] [PubMed] [Google Scholar]
- Margottin F., Bour,S.P., Durand,H., Selig,L., Benichou,S., Richard,V., Thomas,D., Strebel,K. and Benarous,R. (1998) A novel human WD protein, h-β TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell, 1, 565–574. [DOI] [PubMed] [Google Scholar]
- Maurizi M.R., Thompson,M.W., Singh,S.K. and Kim,S.H. (1994) Endopeptidase Clp: ATP-dependent Clp protease from Escherichia coli. Methods Enzymol., 244, 314–331. [DOI] [PubMed] [Google Scholar]
- McDonald J.P., Frank,E.G., Levine,A.S. and Woodgate,R. (1998) Intermolecular cleavage of the UmuD-like mutagenesis proteins. Proc. Natl Acad. Sci. USA, 95, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffler A., Fischer,D., Altuvia,S., Storz,G. and Hengge-Aronis,R. (1996) The response regulator RssB controls stability of the σS subunit of RNA polymerase in Escherichia coli. EMBO J., 15, 1333–1339. [PMC free article] [PubMed] [Google Scholar]
- Pratt L.A. and Silhavy,T.J. (1996) The response regulator SprE controls the stability of RpoS. Proc. Natl Acad. Sci. USA., 93, 2488–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman M. (1974) Phenomenology of an inducible mutagenic DNA repair pathway in Escherichia coli: SOS repair hypothesis. In Prakash,L., Sherman,F., Miller,M., Lawrence,C.W. and Tabor,H.W. (eds), Molecular and Environmental Aspects of Mutagensis. Charles C.Thomas, Springfield, IL, pp. 128–142. [Google Scholar]
- Radman M. (1999) Enzymes of evolutionary change. Nature, 401, 866–867. [DOI] [PubMed] [Google Scholar]
- Shinagawa H., Iwasaki,H., Kato,T. and Nakata,A. (1988) RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc. Natl Acad. Sci. USA, 85, 1806–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout V., Torres-Cabassa,A., Maurizi,M.R., Gutnick,D. and Gottesman,S. (1991) RcsA, an unstable positive regulator of capsular polysaccharide synthesis. J. Bacteriol., 173, 1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Bruck,I., Eritja,R., Turner,J., Frank,E.G., Woodgate,R., O’Donnell,M. and Goodman,M.F. (1998) Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA. Proc. Natl Acad. Sci. USA, 95, 9755–9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Shen,X., Frank,E.G., O’Donnell,M., Woodgate,R. and Goodman,M.F. (1999) UmuD′2C is an error-prone DNA polymerase, Escherichia coli, DNA pol V. Proc. Natl Acad. Sci. USA, 96, 8919–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Pham,P., Shen,X., Taylor,J.-S., O’Donnell,M., Woodgate,R. and Goodman,M. (2000) Roles of E.coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature, 404, 1014–1018. [DOI] [PubMed] [Google Scholar]
- Tinker-Kulberg R.L. and Morgan,D.O. (1999) Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and after DNA damage. Genes Dev., 13, 1936–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias J.W., Shrader,T.E., Rocap,G. and Varshavsky,A. (1991) The N-end rule in bacteria. Science, 254, 1374–1377. [DOI] [PubMed] [Google Scholar]
- Tu G.F., Reid,G.E., Zhang,J.G., Moritz,R.L. and Simpson,R.J. (1995) C-terminal extension of truncated recombinant proteins in Escherichia coli with a 10Sa RNA decapeptide. J. Biol. Chem., 270, 9322–9326. [DOI] [PubMed] [Google Scholar]
- Van Melderen L., Thi,M.H.D., Lecchi,P., Gottesman,S., Couturier,M. and Maurizi,M.R. (1996) ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem., 271, 27730–27738. [DOI] [PubMed] [Google Scholar]
- Welty D.J., Jones,J.M. and Nakai,H. (1997) Communication of ClpXP protease hypersensitivity to bacteriophage Mu repressor isoforms. J. Mol. Biol., 272, 31–41. [DOI] [PubMed] [Google Scholar]
- Woodgate R. (1999) A plethora of lesion-replicating DNA polymerases. Genes Dev., 13, 2191–2195. [DOI] [PubMed] [Google Scholar]
- Woodgate R. and Levine,A.S. (1996) Damage inducible mutagenesis: recent insights into the activities of the Umu family of mutagenesis proteins. In Lindahl,T. (ed.), Cancer Surveys: Genetic Instability in Cancer. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 117–140. [PubMed] [Google Scholar]
- Woodgate R., Rajagopalan,M., Lu,C. and Echols,H. (1989) UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD′. Proc. Natl Acad. Sci. USA, 86, 7301–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]