

Abstract
Transforming growth factor-β (TGF-β) arrests growth of epithelial cells by inducing the transcription of p15Ink4B, a cyclin-dependent kinase inhibitor. In this study, we demonstrate that p15Ink4B induction was mediated by a TGF-β-induced complex of Smad2, Smad3, Smad4 and Sp1. Mutations in the Sp1- or Smad-binding sequences decreased or abolished the TGF-β responsiveness of the p15Ink4B promoter. Interference with, or deficiency in, Smad2, Smad3 or Smad4 functions also reduced or abolished the TGF-β-dependent p15Ink4B induction, whereas the absence of Sp1 reduced the basal and TGF-β-induced p15Ink4B transcription. In the nucleoprotein complex, Smad2 interacted through its C-domain with Sp1 and enhanced the DNA binding and transcriptional activity of Sp1. Smad3 interacted indirectly with Sp1 through its association with Smad2 and/or Smad4, and bound directly to the p15Ink4B promoter. Finally, Smad4 interacted through its N-domain with Sp1. Our data demonstrate the physical interactions and functional cooperativity of Sp1 with a complex of Smad2, Smad3 and Smad4 in the induction of the p15Ink4B gene. These findings explain the tumor suppressor roles of Smad2 and Smad4 in growth arrest signaling by TGF-β.
Keywords: Cdk inhibitor p15Ink4B/Smad/Sp1/TGF-β/transcription
Introduction
Transforming growth factor-β (TGF-β) is a secreted multifunctional protein that exhibits a diverse set of cellular responses, including cell proliferation and differentiation. TGF-β induces the expression of a variety of genes for extracellular matrix proteins, and functions as a potent growth inhibitor, best described in epithelial, endothelial and hematopoietic cells (Derynck and Feng, 1997; Massagué, 1998; Roberts, 1998). During development, TGF-β and related factors regulate cell and tissue differentiation, morphogenetic processes and embryonic organization (Whitman, 1998). TGF-β expression and responsiveness also regulate tumor development. Whereas TGF-β-induced extracellular matrix production provides an advantage to tumor development in vivo, the growth inhibitory response to TGF-β suppresses tumor formation. The TGF-β signaling pathway leading to growth inhibition is considered as a tumor suppressor pathway, and resistance to the antiproliferative effect of TGF-β is often observed in carcinomas (White, 1998). Restoration of TGF-β responsiveness in certain carcinoma cell lines has been shown to repress the tumorigenic behavior.
TGF-β exerts its function by interacting with a heteromeric complex of transmembrane serine/threonine kinase receptors, the type II and type I receptors (Wrana et al., 1994; Derynck and Feng, 1997; Hu et al., 1998; Massagué, 1998). Following TGF-β-induced phosphorylation of the type I by the type II receptors, Smads act as intracellular effectors of ligand-induced signaling (Heldin et al., 1997; Attisano and Wrana, 1998; Derynck et al., 1998; Hu et al., 1998; Massagué, 1998). Smads form a family of structurally related intracellular proteins that consists of eight mammalian members with homologs in Drosophila melanogaster and Caenorhabditis elegans (Heldin et al., 1997; Massagué, 1998). Upon activin or TGF-β stimulation, Smad2 and/or Smad3 are phosphorylated by the activated type I receptors (Abdollah et al., 1997; Souchelnytskyi et al., 1997). These C-terminally phosphorylated Smads then undergo a change in conformation, which results in dissociation from the receptors, and form complexes with Smad4 (Lagna et al., 1996; Macias-Silva et al., 1996; Nakao et al., 1997; Zhang et al., 1997). The heteromeric complexes of receptor-activated Smads and Smad4 are then translocated into the nucleus, where they exert ligand-induced changes in transcription of a variety of genes (Liu et al., 1996; Macias-Silva et al., 1996; Zhang et al., 1996). The heteromeric Smad complex activates transcription through its ability to cooperate functionally with several promoter-specific transcription factors and/or to bind specific DNA sequences (Derynck et al., 1998).
The mechanism of how Smads activate transcription has only been characterized for a few genes. To activate the activin-responsive genes Mix.2 and goosecoid, the activated heteromeric Smad 2/4 complex associates with a winged helix transcription factor, FAST-1 or -2, at the activin-responsive promoter elements (Chen et al., 1996, 1997; Liu et al., 1997, 1999; Labbé et al., 1998; Zhou et al., 1998). For TGF-β-responsive promoters, Smad3 has been shown to interact physically and functionally with the c-Jun–c-Fos complex at the collagenase I promoter (Zhang et al., 1998), or to cooperate with TFE3 at the PAI-1 promoter (Hua et al., 1998). Functional cross-talk of Smad3 with the vitamin D receptor has also been shown (Yanagisawa et al., 1999). Even though Smad2 can be activated in response to TGF-β, no examples of a natural involvement of Smad2 in TGF-β-induced transcription, or of Smad3 in activin responses, have been described. Neither has Smad2 nor Smad3 been shown to cooperate naturally to induce transcription of a specific gene.
While the potent growth inhibitory activity of TGF-β has been well documented, very little is known about how TGF-β-induced Smad activation is functionally connected with growth arrest. In HaCaT and Mv1Lu epithelial cells, TGF-β has been shown to induce the expression of the cyclin-dependent kinase (CDK) inhibitor p15Ink4B (Hannon and Beach, 1994; Reynisdóttir et al., 1995; Reynisdóttir and Massagué, 1997), which inhibits the activities of cyclin D-dependent CDK4 and CDK6. Recently, TGF-β-mediated induction of p15Ink4B was shown to be Smad3 dependent in astrocytes (Rich et al., 1999). Considering the inhibition of G1 phase progression by p15Ink4B, the induction of p15Ink4B expression by TGF-β most likely represents the key event that initiates TGF-β-induced growth arrest in cells. The p15Ink4B gene is frequently inactivated in carcinomas, often in conjunction with deletion of the gene for p16Ink4A, another CDK inhibitor, but this deletion rarely occurs in tumors that have escaped TGF-β-controlled growth inhibition. Although Smad3 and Smad4 have been shown to be involved in TGF-β-induced inhibition of DNA synthesis (Zhang et al., 1996), it is not clear whether or how Smad3/4 activate p15Ink4B expression. In several tumor types, inactivation of the gene for Smad4/DPC4 has been linked to abrogation of TGF-β-induced growth arrest and to tumorigenesis (Barrett et al., 1996; Hahn et al., 1996a,b; Schutte et al., 1996; Riggins et al., 1997). In fact, these studies led to the identification of Smad4/DPC4 as a candidate tumor suppressor and its role in TGF-β signaling (Hahn et al., 1996b). Similarly, the gene for Smad2 is also inactivated in some tumors (Eppert et al., 1996; Riggins et al., 1997), suggesting that Smad2 may also act as a tumor suppressor and its function may be somehow linked to TGF-β-induced growth arrest.
A previous study has shown that the proximal 113 bp promoter sequence of the p15Ink4B gene confers TGF-β inducible expression and has suggested that one of the two putative Sp1 binding sites may be required for the TGF-β response (J.-M.Li et al., 1995). In addition, Smad3 has been shown to enhance the transcriptional activity of Sp1 fused to a Gal4 DNA-binding domain (Moustakas and Kardassis, 1998), whereas TGF-β-mediated induction of p15Ink4B was recently shown to be Smad3 dependent in astrocytes (Rich et al., 1999). However, no mechanism has been proposed to explain the TGF-β-induced transcriptional activation of the p15Ink4B promoter, or to link mechanistically Smad activity with the function of Sp1 at the p15Ink4B promoter. In this report, we have shown that a functional cooperation and physical interaction of Smad2, Smad3 and Smad4 with Sp1 at the promoter of the p15Ink4B gene provides a mechanism underlying the TGF-β-induced growth arrest.
Results
Induction of CDK inhibitor p15Ink4B expression is an immediate early response to TGF-β
TGF-β induces growth arrest in the late G1 phase of the cell cycle in various cell types, including epithelial cells (Derynck and Feng, 1997; Massagué, 1998). This growth arrest is initiated by the ability of TGF-β to induce expression of the CDK inhibitor p15Ink4B, an inhibitor of G1 phase cyclin-dependent protein kinases such as CDK4 and CDK6 (Hannon and Beach, 1994; Reynisdóttir et al., 1995; Reynisdóttir and Massagué, 1997). Therefore, we evaluated the mechanism through which TGF-β activates the expression of p15Ink4B. Consistent with previous results (Hannon and Beach, 1994; Reynisdóttir et al., 1995; Reynisdóttir and Massagué, 1997), the levels of p15Ink4B mRNA were strongly increased upon TGF-β stimulation in human HaCaT keratinocytes (Figure 1A, lanes 1 and 2) and mink Mv1Lu epithelial cells (data not shown). To investigate whether the induction of p15Ink4B by TGF-β requires new protein synthesis, we assessed the effect of the protein synthesis inhibitor cycloheximide. As shown in Figure 1 (lanes 3 and 4), addition of cycloheximide did not alter the p15Ink4B mRNA levels in the absence or presence of TGF-β, indicating that de novo protein synthesis is not required for TGF-β-induced p15Ink4B expression.

Fig. 1. TGF-β-induced p15Ink4B transcription requires functional TβRI and TβRII and is independent of de novo protein synthesis. (A) p15Ink4B expression is an immediate early response gene to TGF-β. Exponentially growing HaCaT cells were treated with or without 400 pM TGF-β or 10 µM cycloheximide (CHX). p15Ink4B mRNA was detected by northern hybridization. Equal levels of RNA were loaded per lane, as illustrated by equal levels of 28S and 18S RNA. (B) Transcriptional activation from the p15Ink4B promoter is induced by TGF-β in HaCaT and Mv1Lu cells. Cells were transfected with the p15P113luc luciferase reporter plasmid and, 40–45 h after transfection, treated with TGF-β for 4 h, and luciferase values were measured. (C) Luciferase mRNA expression from the p15Ink4B promoter in response to TGF-β does not require new protein synthesis. HaCaT cells were transfected with the p15P113luc luciferase reporter plasmid and a control β-galactosidase expression plasmid pSVβgal, incubated with TGF-β and/or cycloheximide for 4 h as shown, and RNA was isolated. The levels of luciferase and β-galactosidase mRNA were assessed by PCR cDNA amplification. While the 150 bp β-galactosidase cDNA band was constant in all lanes, the 700 bp luciferase cDNA band was induced by TGF-β, both in the absence and presence of cycloheximide (CHX). M, DNA fragment length markers (ΦX174 DNA/HaeIII). (D) Dominant-negative inhibition of TGF-β-induced p15Ink4B transcription by kinase-inactive (KR) TβRI and TβRII. HaCaT cells were cotransfected with p15P113luc and the indicated receptor expression plasmids. (E) TGF-β-induced p15Ink4B transcription requires both TβRII and TβRI. The reporter plasmid p15P113luc was transfected into wild-type Mv1Lu cells, which express both receptors, or the derivative DR26 and R1B cells, which lack functional TβRII and TβRI, respectively. Expression plasmids for TβRII or TβRI were cotransfected, as marked. All assays were done in triplicate and all values were normalized for transfection efficiency against the β-galactosidase expression directed from the cotransfected pSV-β-Gal control plasmid.
The TGF-β-induced increase of p15Ink4B mRNA levels was apparently regulated at the transcriptional level. An upstream regulatory sequence (nucleotides –113 to +75) of the human p15Ink4B gene promoter has previously been shown to mediate the induction of p15Ink4B expression in response to TGF-β (J.-M.Li et al., 1995). Using this promoter sequence to drive expression of a luciferase reporter gene, we observed that TGF-β induced luciferase expression in both HaCaT and Mv1Lu cells (Figure 1B) as well as in HepG2 cells (data not shown). The TGF-β-induced transcription of luciferase mRNA from this promoter segment also occurred in the presence of cycloheximide, similarly to the endogenous TGF-β-induced p15Ink4B mRNA expression (Figure 1C). Therefore, these assays indicate that this promoter sequence contains the necessary information to mediate the transcriptional activation of the p15Ink4B gene in response to TGF-β in these cells.
TGF-β-induced p15Ink4B expression is mediated by type I and II TGF-β receptors
TGF-β signals are transduced by a heteromeric complex of type I and type II receptors, TβRI and TβRII. Both receptors mediate the expression of several TGF-β-responsive genes in response to TGF-β (Derynck and Feng, 1997). To investigate the role of the two receptors in TGF-β-induced transcription from the p15Ink4B promoter, we overexpressed kinase-inactive mutants of TβRI and TβRII, which have been shown to act as specific dominant-negative inhibitors of the endogenous receptors (Feng et al., 1995). As shown in Figure 1D, overexpression of a dominant-negative mutant of TβRI or TβRII in HaCaT cells inhibited the TGF-β-induced transcriptional response from the p15Ink4B promoter. In contrast, the similarly truncated Tsk7L/ActRI type I receptor, which has been implicated in BMP, activin and TGF-β responses, did not interfere with this TGF-β-induced response. Consistent with these results, expression of TβRI(T202D), a constitutively active mutant of TβRI that confers TGF-β-independent signaling, induced transcription from the p15Ink4B promoter and this response was further enhanced by TGF-β (Figure 1D). In accordance with these findings, DR26 and R1B cells, two cell lines that are derived from Mv1Lu cells and lack functional TβRII and TβRI, respectively (Laiho et al., 1990), did not show a TGF-β-induced transcriptional activation from the p15Ink4B promoter. The responsiveness to TGF-β could be rescued by cotransfection of wild-type TβRII and TβRI in DR26 and R1B cells, respectively, similarly to the response of wild-type Mv1Lu cells (Figure 1E). Therefore, our data indicate that TβRI and TβRII mediate the transcriptional activation of the p15Ink4B gene in response to TGF-β.
TGF-β-induced expression of p15Ink4B requires Smad2, Smad3 and Smad4
Next, we investigated whether TGF-β induces p15Ink4B transcription through Smads. As shown in Figure 2A, overexpression of Smad2 or Smad3 induced p15Ink4B transcription, and Smad4 co-expression increased the effect of Smad2 or Smad3, whereas the highest induction was observed when Smad2, Smad3 and Smad4 were coexpressed. In addition, overexpression of C-terminally truncated forms of Smad2, Smad3 or Smad4, but not Smad1, inhibited TGF-β-induced transcription from the p15Ink4B promoter in these cells (Figure 2B). These truncated Smads are known to act as specific dominant-negative inhibitors of Smad signaling, since truncated versions of Smad3 or Smad4, but not Smad1 or Smad2, interfere with TGF-β-induced and Smad3-mediated transcription from the PAI-1 promoter (Zhang et al., 1996). These data suggest that both Smad2 and Smad3, in conjunction with Smad4, function as effectors of the TGF-β-induced transcription of the p15Ink4B gene.

Fig. 2. TGF-β-induced p15Ink4B transcription requires Smad2, Smad3 and Smad4. (A) Transcriptional activation of the p15Ink4B promoter by Smads. HaCaT cells were transfected with the p15P113luc reporter plasmid and indicated combinations of expression plasmids for Smad2, Smad3 and Smad4, and reporter gene expression were measured. (B) C-terminally truncated Smads inhibit TGF-β-induced transcription from the p15Ink4B promoter. HaCaT cells were cotransfected with the p15P113luc reporter plasmid, and indicated expression plasmids for Smad mutants. (C) Smad3 is required for TGF-β-induced transcription from the p15Ink4B promoter. Smad3–/– mouse embryonic fibroblasts (MEFs) were cotransfected with p15P113luc, and indicated expression plasmids for Smad2, Smad3 and Smad3 mutants. Smad3N, Smad3NL, Smad3LC and Smad3C contain the Smad3 regions of amino acids 2–144, 2–231, 144–425 and 232–425, respectively. (D) Smad4 is required for TGF-β-induced p15 transcription. Smad4-defective MDA-MB-468 cells were cotransfected with p15P113luc without or with an expression plasmid for Smad4.
The role of Smads in the transcriptional induction of the p15Ink4B promoter was also examined in cells lacking a specific Smad. Since cells that lack Smad2 are not available, we examined the ability of TGF-β to induce p15Ink4B transcription in cells that lack endogenous Smad3 or Smad4. As shown in Figure 2C, TGF-β did not induce transcription from the p15Ink4B promoter in Smad3–/– mouse embryonic fibroblasts, but exogenous Smad3 expression in these cells could rescue the induction of p15Ink4B expression by TGF-β. Increased Smad2 expression in these cells did not compensate for the Smad3 deficiency. These results indicate that Smad3 is required for TGF-β-dependent p15Ink4B induction, and that Smad2 and Smad3 are not functionally equivalent in activating transcription from the p15Ink4B promoter. In addition, Smad3 mutants with N- or C-terminal deletions were unable to rescue the p15Ink4B transcription in response to TGF-β (Figure 2C). Since the N- and C-terminal domains of Smads possess the DNA-binding and transactivation activities, respectively, our results suggest that both functions of Smad3 are required for TGF-β-induced expression of p15Ink4B.
To evaluate the essential functions of Smad4 in the p15Ink4B induction by TGF-β, we carried out luciferase reporter assays in MDA-MB-468 breast carcinoma cells, which lack endogenous Smad4 (Schutte et al., 1996). As shown in Figure 2D, TGF-β did not induce p15–luciferase expression in these cells, but re-introduction of Smad4 restored the induction of p15Ink4B transcription in response to TGF-β. These results, in agreement with Figure 2B, support the notion that transcription from the p15Ink4B promoter requires functional expression of Smad4.
Direct binding of Smad3 and Sp1 to the p15Ink4B promoter is essential for TGF-β-induced p15Ink4B transcription
As illustrated in Figures 1 and 2, the 113 bp promoter sequence of the p15 gene was sufficient to direct transcription in response to TGF-β and to Smads. Examination of this sequence (Figure 3A) revealed two GC-rich putative Sp1 binding sites (Gidoni et al., 1984), Sp1a and Sp1b, and four sequences resembling the proposed Smad-binding element (SBE) (Dennler et al., 1998; Shi et al., 1998). Two of these putative SBEs were located between positions –113 and –95 (relative to the transcription initiation site), a promoter segment that is not required for TGF-β-inducible transcription of the p15Ink4B gene (J.-M.Li et al., 1995; data not shown). In addition, this dispensable DNA sequence did not bind Sp1 (data not shown). Therefore, we focused on the two putative Sp1 binding sites, Sp1a and Sp1b, and the two SBEs, SBE1 and SBE2, which are located downstream from the nt –95 position (Figure 3A). Two oligonucleotide DNA probes were synthesized in order to determine the abilities of Sp1 and Smads to interact with these sequence elements using gel shift assays. As shown in Figure 3A, probe A contained the Sp1a and SBE1, while probe B contained the Sp1b and SBE2 sites.


Fig. 3. Sp1 binding sites and Smad3-binding elements are required for the activation of the p15Ink4B gene. (A) Nucleotide sequence of the –113 to +1 segment of the p15Ink4B promoter. Predicted Sp1 binding sites (Sp1a and Sp1b) and SBEs 1 and 2 are indicated. The oligonucleotide probes A and B, used in the gel shift experiments, with their inactivating mutations are also shown. (B) Sp1 and Smad3 bind to the p15Ink4B promoter. Purified Sp1 (0.5 or 5 U) or GST–Smad fusion proteins (1 µg) were incubated with the 32P-labeled probe A or B, or the mutant probes A1 or B1, in which the Sp1a or Sp1b sites are mutated, or the mutant A2 or B2 probes, in which the SBE1 or SBE2 sites are inactivated. Gel-shifted DNA–protein complexes are marked. (C) Sp1 binding to the Sp1 binding sites in oligonucleotides A or B can be competed with unlabeled ‘wild-type’ oligonucleotides, but not by oligonucleotides with mutated Sp1 binding sites. Purified Sp1 was incubated with the 32P-labeled probe A or B in the presence of indicated unlabeled DNA. The DNA–Sp1 complex is marked. (D) Mutational analysis of the p15Ink4B promoter. Sp1 sites and SBEs were individually, or in combination, mutated in the p15Ink4B promoter, as shown in Figure 3A. Transcription was measured by luciferase activity. Transfection, TGF-β treatment and luciferase assay in HaCaT cells were performed as in Figure 1.
Both putative Sp1 sequences, Sp1a in probe A and Sp1b in probe B, were able to bind purified Sp1 protein (Figure 3B). The Sp1a site, which has been proposed to be required for TGF-β responsiveness (J.-M.Li et al., 1995), bound Sp1 with an efficiency of at least 20-fold higher than the Sp1b sequence. Consequently, a much higher amount of Sp1 protein was required to demonstrate Sp1 binding to probe B (Figure 3B). Point mutations in the Sp1a or Sp1b sequences abolished their ability to interact with Sp1 (Figure 3A and B), indicating the authenticity of these two Sp1 binding sites. Unlabeled Sp1a DNA competed with the radiolabeled Sp1a or Sp1b sequences for Sp1 binding, but point mutations in the Sp1a sequence abolished its ability to compete (Figure 3C). In contrast, the unlabeled Sp1b sequence, with its weaker Sp1 binding ability, only competed with radiolabeled Sp1b but not with the Sp1a site (Figure 3C). Mutations in the SBEs did not affect the ability of probes A and B to bind Sp1 protein (data not shown).
We used the same approach to evaluate whether Smad3 and Smad4 bound to the potential SBEs in the p15Ink4B promoter. Besides full-length Smad3 and Smad4, we also tested their NL segments, i.e. the N (MH1) domain with the adjacent linker (L) segment, since removal of the C (MH2) domain is known to enhance the DNA binding ability of the NL segment (Hata et al., 1997; Kim et al., 1997). Smad2 was not tested, because it lacks the ability to bind DNA (Dennler et al., 1998; Zawel et al., 1998). Smad3NL bound to probes A and B, whereas full-length Smad3 did not detectably associate with probe B, yet displayed binding to probe A (Figure 3B). As also shown in Figure 3B, Smad3NL was unable to bind these oligonucleotides when the SBEs were point-mutated (marked in Figure 3A). Smad4 or Smad4NL did not bind to these oligonucleotides (Figure 3B), even though Smad4NL bound as efficiently as Smad3NL to the optimal Smad binding sequence GTCTAGAC (data not shown).
Next, we defined the contributions of the Smad and Sp1 binding elements to the basal and TGF-β-induced transcription from the 113 bp promoter. Mutations were introduced to abolish the Smad3 or Sp1 binding (Figure 3A and D). Mutations of either Sp1 binding site decreased the basal transcription level as well as the TGF-β-induced transcription, whereas mutation of both sites further decreased the basal transcription and totally abolished the TGF-β inducibility (Figure 3D). Mutations of individual SBEs also decreased the TGF-β-inducible transcription level, but did not affect the basal transcription level. When both SBEs were mutated, only a minimal degree of TGF-β inducibility was apparent (Figure 3D). We conclude that all SBEs and Sp1 binding sites contribute to the TGF-β responsiveness of the p15Ink4B promoter, and that inactivation of the SBEs primarily affects the TGF-β inducibility, whereas inactivation of the Sp1 sites has a drastic effect on both the basal and TGF-β-inducible transcription.
Smad2 and Smad4 interact with Sp1 in vivo and in vitro
Our findings demonstrated that Smad2, Smad3 and Smad4 are all required for TGF-β-induced transcription from the p15Ink4B promoter (Figure 2), and that the Sp1 binding sites in the promoter are essential for this response (Figure 3D). Therefore, we evaluated the ability of these three Smads to interact with Sp1 in vivo. Interaction of endogenous Sp1 with individually expressed Smad2, Smad3 or Smad4 was determined in the absence or presence of TGF-β using co-immunoprecipitation analysis. As shown in Figure 4A, TGF-β-dependent interaction between Smad2 and Sp1 was detected in vivo. Similarly, TGF-β induced an interaction of Smad4 with Sp1, although a weak interaction was already apparent in the absence of TGF-β. In control experiments, CBP did not co-immunoprecipitate with Sp1 (Figure 4A). Unlike Smad2 or Smad4, Smad3 did not detectably interact with Sp1, either in the absence or presence of TGF-β. However, Smad3 was able to form a complex efficiently with Sp1, when Smad2 or Smad4 expression was increased (Figure 4B). These results strongly suggest that, following TGF-β stimulation, Smad3 interacts with Sp1 through Smad2 and/or Smad4.

Fig. 4. Physical association of Smad2, Smad3 and Smad4 with Sp1. (A) TGF-β-dependent association of Smads and Sp1 in vivo. HaCaT cells were transfected with HA-tagged Smads or CBP, with (+) or without (–) an expression plasmid for activated TβRI. Cell lysates were immunoprecipitated (IP) with an anti-HA antibody, followed by immunoblotting (IB) with an anti-Sp1 antibody to detect Smad-bound Sp1 (upper panel). Cell lysates were also directly immunoblotted with anti-Sp1 or anti-HA antibodies to demonstrate expression of endogenous Sp1 (middle panel) or transfected Smads (lower panel). (B) Smad3 co-immunoprecipitates with endogenous Sp1 in the presence of Smad2 or Smad4. HaCaT cells were transfected with expression plasmids for Flag-tagged Smad3 and HA-tagged Smad2 or 4. Immunoprecipitation with anti-Flag antibody was followed by anti-Sp1 immunoblotting to detect Smad3-bound Sp1. Expression levels of Flag-tagged Smads (middle panel) or HA-tagged Smads (lower panel) were shown by anti-Flag or anti-HA immunoblotting. (C) Direct interaction of GST–Sp1(1–621) with 35S-labeled Smad2 and its segments. Smad2N, Smad2NL, Smad2LC and Smad2C cover the Smad2 regions of amino acids 2–183, 2–273, 181–467 and 270–467, respectively. (D) Direct interaction of GST–Sp1(1–621) with 35S-labeled Smad4 and its segments. Smad4N, Smad4NL, Smad4LC and Smad4C cover the Smad4 regions of amino acids 2–154, 2–300, 141–552 and 294–552, respectively.
Since Smad2 and Smad4 interacted with Sp1 in vivo, we determined whether the interactions were direct using glutathione S-transferase (GST) adsorption assays. Purified GST–Sp1 protein was tested for its ability to interact with in vitro-translated 35S-labeled Smad2 or Smad2 segments. Because the zinc finger domain (amino acids 622–788) of Sp1 did not interact with Smad2 or Smad4 (data not shown) and it is difficult to prepare GST-fused full-length Sp1, we used GST-fused Sp1(1–621) to detect in vitro binding of Sp1 to Smad2 or Smad4. As shown in Figure 4C, Smad2, Smad2LC and Smad2C bound to GST–Sp1(1–621), while Smad2N and Smad2NL did not. This suggests that Smad2 interacts directly through its C-domain with Sp1. The higher level interaction of Smad2LC and Smad2C, when compared with Smad2, is consistent with the notion that ligand-induced activation of a full-length Smad exposes the N- and C-domains with a higher affinity for interacting proteins (Hata et al., 1997). In reciprocal experiments, in vitro-translated 35S-labeled Sp1 interacted with GST-fused Smad2LC, but not GST–Smad2NL (data not shown). Smad4 also interacted directly with GST–Sp1 (1–621). In contrast to Smad2, Smad4 interacted through its N-domain. Thus, both Smad4N and Smad4NL interacted with GST–Sp1, whereas the LC or C segments of Smad4 did not associate (Figure 4D). Finally, whereas efficient interaction of Smad3 with Sp1 in vivo depended on the presence of Smad2 or Smad4, Sp1 was able to associate with GST–Smad3 in vitro (data not shown). We speculate that the absence of a direct Smad3–Sp1 interaction in vivo may be due to a lower affinity of Sp1 for Smad3 than for Smad2 or Smad4, and to a concomitant steric hindrance when Smad2 and Smad4 interact with Sp1 in the transcription complex.
Smads and Sp1 form a nucleoprotein complex on the p15Ink4B promoter
Since Sp1 was able to interact directly with Smad2 and Smad4 in vivo, we analyzed their ability to form a complex with the Sp1 binding sites in the p15Ink4B promoter. Purified Sp1 and GST–Smad proteins were incubated individually or together with probe A or B (Figure 3A), which contain the Sp1a or Sp1b binding sites, respectively. Using lower concentrations of GST–Smad proteins than in Figure 3B, we were unable to detect GST–Smad3 binding to the DNA, and, consistent with the results in Figure 3B, GST–Smad2 and GST–Smad4 were also unable to interact by themselves with either oligonucleotide (Figure 5A, lanes 2–4). As in Figure 3B, Sp1 interacted efficiently with the Sp1a sequence (Figure 5A, lane 1). However, even though Smad2 did not interact with DNA, addition of Smad2 together with Sp1 resulted in a more intense and slower migrating band when compared with Sp1 alone (Figure 5A, compare lane 5 with lane 1), presumably representing a Smad2–Sp1 complex at the Sp1a site. Consistent with its ability to interact with Sp1, Smad4 was also able to synergize with Sp1 for DNA binding similarly to Smad2, albeit with a lower efficiency. (Figure 5A, lane 7). In contrast to the Sp1a sequence, Sp1 had only a low affinity for the Sp1b element (Figures 3B and 5B, lane 1). However, combined Sp1 and Smad2 formed a DNA binding complex efficiently (Figure 5B, lane 5). Unlike at the Sp1a site, the presence of Smad4 did not result in detectable complex formation with Sp1 at the Sp1b site (Figure 5, lane 5) and appeared to decrease the intensity of the Smad2–Sp1 complex (Figure 5B, compare lane 8 with lane 5). Smad3 did not enhance Sp1 binding at the Sp1a or the Sp1b sequence (Figure 5A and B, lane 3), indicating that Smad3 can not functionally replace Smad2. Under similar conditions, no Smad2–Sp1 or Smad4–Sp1 binding was observed when the Sp1 binding sites were mutated (data not shown).


Fig. 5. Interaction of Smad2 or Smad4 with Sp1 at the Sp1a and Sp1b binding sites. Purified Sp1 (0.5 U) and GST–Smad fusion proteins (0.2 µg) were incubated with the 32P-labeled probe A, containing the Sp1a binding site (A), or probe B, containing the Sp1b binding site (B) of the p15Ink4B promoter. DNA-bound Sp1 and Sp1–Smad complexes are marked. (C) Gel shift analyses using oligonucleotide A and purified proteins were carried out as in (A). Antibodies, shown above the gel, were added to the gel shift reactions and incubated for 90 min, prior to gel analysis. Sp1–DNA, Sp1–Smad–DNA complex, and the supershifted (SS) complexes are marked. (D) Sp1-binding ability of p15 promoter elements. Nuclear extracts from HaCaT cells were incubated with probe A or B in gel shift reactions, with or without anti-Sp1 antibody. The Sp1–DNA complex, which is displaced in the presence of anti-Sp1 antibody, is indicated. (E) TGF-β-induced formation of Smad–DNA complex in cell lysates. Expression plasmids for Flag–Smad2, HA–Smad3 and Myc–Smad4 were transfected. Forty-eight hours after transfection, cells were lysed and incubated with streptavidin paramagnetic beads (Dynal) with immobilized biotinylated p15 promoter DNA oligonucleotide (nt –84 to –46). After extensive washing, DNA-bound proteins were detected by SDS–PAGE followed by western blotting using the indicated antibodies (lanes 4–6). Immunoblotting of the cell lysates in parallel demonstrates the expression level of endogenous Sp1 and transfected Smads. (F) The TGF-β-induced formation of Smad–DNA complex in cell lysates is largely dependent on intact Sp1 binding sites. Binding of Sp1 and Smad2, Smad3, Smad4 and Sp1 to the wild-type oligonucleotide, as was also done in (E), was compared with their binding to the corresponding mutant oligonucleotides, in which the SBEs and Sp1 binding sequences were mutated, as shown in Figure 3A.
The participation of Sp1, Smad2, Smad3 and Smad4 in the same complex was also evaluated by co-incubation of the four proteins with oligonucleotide A (Figure 5C). While Sp1 interacted with the DNA as expected, co-incubation of all three Smads resulted in the formation of a single complex with slower mobility and a higher binding efficiency than the Sp1–DNA complex. These findings are consistent with those in Figure 5A. Addition of antibodies against Sp1 or Smad3, which interfere with their DNA binding, totally abolished the formation of this DNA–multiprotein complex, while anti-Smad2 and anti-Smad4 antibodies reduced the mobility of this complex. These results strongly suggest that Smad2, Smad3 and Smad4 participate in the formation of a multiprotein complex with Sp1 at the promoter DNA.
We also evaluated the formation of gel shift complexes with the p15Ink4B promoter in HaCaT cell nuclear extracts. As shown in Figure 5D, both oligonucleotides A and B were able to form complexes, and incubation of these reaction mixtures with the same anti-Sp1 antibody as in Figure 5C resulted in the disappearance of the most predominant complexes. These results strongly suggest that endogenous Sp1 protein in the nuclear extract bound to the Sp1 binding sites of probes A and B. The interaction of endogenous Smads with these oligonucleotides could not be detected under these conditions, probably due to the lack of sensitivity of the antibodies and to the interaction of the nucleoprotein complexes of Smads and Sp1 with other nuclear factors. Because of these technical limitations, we used an oligonucleotide-DNA precipitation method to assess the interaction of tagged Smads and endogenous Sp1 in the cell lysates with the p15Ink4B promoter sequence. The p15Ink4B promoter oligonucleotide corresponded to the –84 to –46 sequence, shown in Figure 3A, and contained both Sp1 binding sites and both SBEs. As shown in Figure 5E, endogenous Sp1 interacted in the presence or absence of TGF-β receptor activation. In the absence of receptor activation, Smad2 did not interact, whereas a low level of Smad3 and Smad4 interaction with the DNA was observed, presumably a result of overexpression of these Smads. TGF-β receptor activation resulted in efficient interaction of Smad2, Smad3 and Smad4 with the p15Ink4B promoter sequence. These results strongly suggest that Sp1 constitutively interacts with the p15Ink4B promoter, and that TGF-β receptor activation induces the formation of the heteromeric Smad complex with Sp1 at the promoter. Consistent with the physical interactions of Smad2, Smad3 and Smad4 with Sp1, and the gel shift analyses in Figure 5A–C, mutation of both Sp1 binding sites, to abolish Sp1 binding, strongly decreased or abolished the interaction of Smad2, Smad3 and Smad4 with the p15Ink4B promoter sequence (Figure 5F). Mutation of the SBEs strongly decreased Smad3 and Smad4 binding, but did not decrease Smad2 binding (Figure 5F), which is consistent with the inability of Smad2 to bind DNA and its association with Sp1.
Smads enhance the transcriptional activity of Sp1 through its glutamine-rich domain
To characterize the effect of Smad2, Smad3 and Smad4 on the transcriptional function of Sp1, we tested whether TGF-β and Smads can enhance the ability of Sp1, linked to the Gal4 DNA binding domain, to transactivate a Gal4-binding promoter. Consistent with previous findings (J.-M.Li et al., 1995; Moustakas and Kardassis, 1998), TGF-β increased the transactivation function of Gal4–Sp1 ∼8-fold in HaCaT cells (Figure 6A). In addition, increased expression of the TGF-β-responsive Smad2 or Smad3 enhanced the Gal4–Sp1 activity in these cells (Figure 6A and B), which is consistent with their ability to interact directly or indirectly with Sp1. However, mutation of the two distal serines in the SSXS motif of either Smad2 or Smad3 inactivated their ability to enhance transcription of Gal4–Sp1 (Figure 6B), which is consistent with their decreased ability to interact with the coactivator CBP/p300 (Feng et al., 1998). In contrast, Smad1 did not regulate the activity of Gal4–Sp1 (Figure 6A).

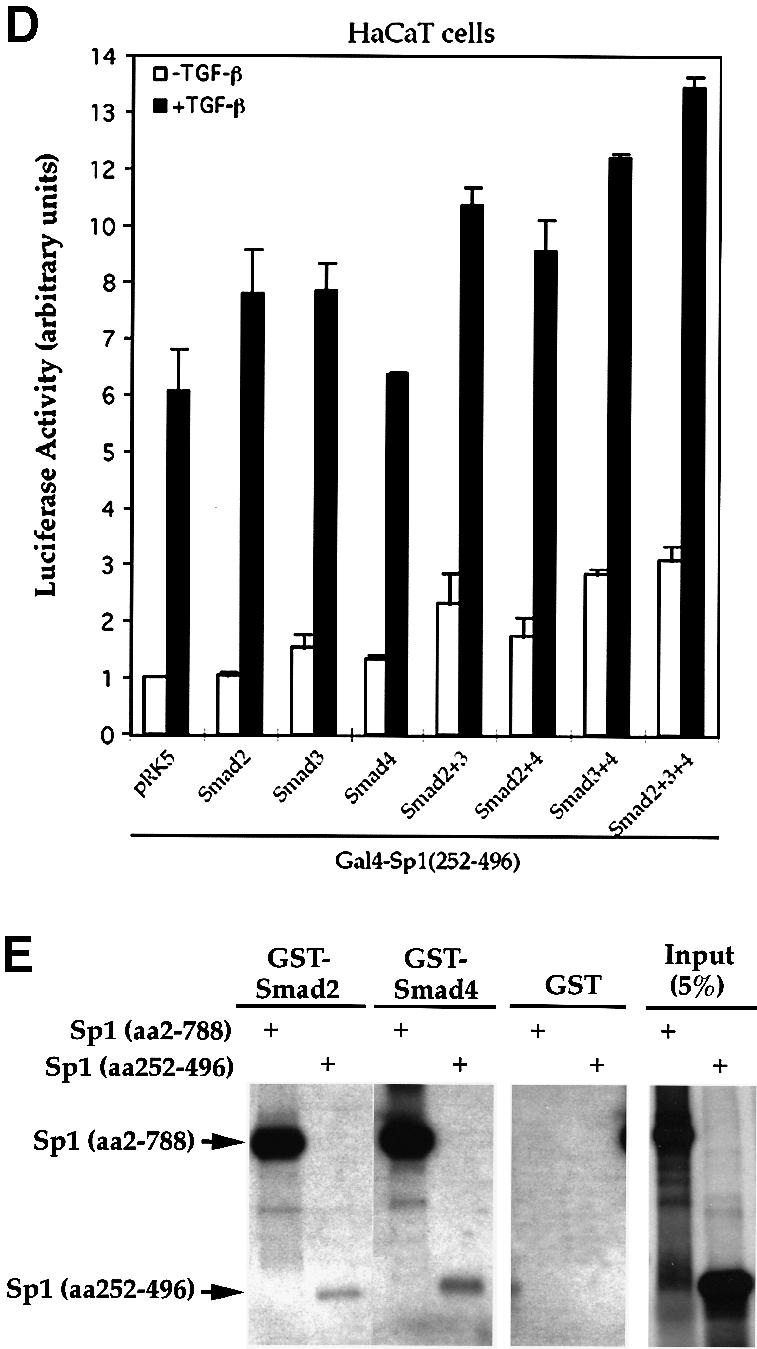
Fig. 6. Smads increase the transcriptional activity of Sp1 through its second S/T-rich and Q-rich domains. (A) Smad2 and 3 stimulate the transactivation activity of Gal4–Sp1. HaCaT cells were cotransfected with Gal4–Sp1(WT) and the luciferase reporter plasmid pFR-Luc, and expression plasmids for the indicated Smads. (B) Smad-dependent stimulation of Gal4–Sp1 transactivation activity requires the C-terminal SSXS motif of Smad2 or Smad3. HaCaT cells were cotransfected with Gal4–Sp1(WT) and pFR-Luc, and expression plasmids for the indicated Smads or mutants. (C) Localization of the TGF-β-responsive domain of Gal4–Sp1. HaCaT cells were transfected with expression plasmids for Gal4–Sp1 or its derivatives, as shown, and the pFR-Luc reporter plasmid. pXF1Gal4 is the control plasmid containing only the GAL4 DNA binding domain. (D) Effects of Smads on the activity of Gal4–Sp1(252–496). The Gal4–Sp1(252–496) plasmid, in combination with the indicated expression plasmids for Smads, was transfected into HaCaT cells, together with the pFR-Luc reporter plasmid. (E) Direct interaction of GST–Smads with Sp1 (amino acids 252–496). Equal amounts of GST–Smads or control GST were used to adsorb 35S-labeled full-length Sp1 or its aa 252–496 fragment.
Several deletion mutants of Sp1, linked to the Gal4 DNA binding domain, were generated to identify the segment that mediates TGF-β- and Smad-responsiveness. Sp1 contains two serine/threonine (S/T)-rich domains, two Gln (Q)-rich domains and a DNA binding zinc finger domain (Figure 6C). Deletion of the first S/T- and Q-rich domains did not affect the ability of TGF-β to enhance the transcription activity. Further deletion of the zinc finger domain of Sp1 (amino acids 497–788) did not affect the TGF-β inducible transcription either. Thus, Gal4–Sp1 (252–496) was transcriptionally as active and TGF-β inducible as full-length Gal4–Sp1 (Figure 6C). The transcriptional activity of Gal4–Sp1(252–496) could be further enhanced by increasing the levels of Smad2 or Smad3 (Figure 6D). Combination of any two Smads, including Smad2 or Smad3 with Smad4, further enhanced the activity, and the highest level activity of Gal4–Sp1 (252–496) was achieved by co-expressing all three Smads (Figure 6D). This enhancement of transcription by overexpression of Smads was consistently observed in HaCaT cells, but was more pronounced in HepG2 cells (data not shown), presumably due to differences in the endogenous Smad levels. Thus, the segment from amino acid 252 to 496, which contained the second S/T-rich and Q-rich domains, provided full responsiveness to TGF-β and Smads (Figure 6C and D). In accordance, this 252–496 segment still possessed the ability to interact directly with both Smad2 and Smad4 in GST adsorption assays, albeit more weakly than Sp1(2–788) (Figure 6E).
Sp1 and Smads functionally cooperate in TGF-β-induced p15Ink4B gene transcription
Since Sp1 binds to the Sp1a and Sp1b sites in the p15Ink4B promoter, we further explored the role of Sp1 in p15Ink4B transcription. These experiments were carried out in Drosophila Schneider (S2) cells, which are known to lack endogenous Sp1 or functional homologs (Courey and Tjian, 1988). As shown in Figure 7A, the p15Ink4B promoter could only drive a minimal level of transcription in the absence of Sp1, but increasing the level of Sp1 strongly increased the constitutive transcription of p15Ink4B–luciferase reporter gene. No additional induction of transcription was observed when the cells were cotransfected with an activated mutant of the type I TGF-β receptor, TβRI(T202D). These results indicate that Sp1 confers a high constitutive level of transcription from the p15Ink4B promoter but does not confer TGF-β responsiveness.

Fig. 7. Requirement of Sp1 and its functional cooperation with Smads in the transcription of p15Ink4B in Drosophila S2 cells. (A) Sp1 strongly transactivates the p15 promoter. S2 cells were transfected with the p15P113luc reporter plasmid and increasing amounts of the Sp1 expression plasmid Pac-Sp1, in the presence or absence of an activated TβRI. Luciferase assays were done as in HaCaT cells. (B) Inability of Smads to transactivate the p15 promoter in the absence of Sp1. S2 cells were transfected with the p15P113luc reporter and expression plasmids for Sp1 or for Smads, in the presence or absence of an activated TβRI, as shown. (C) Sp1 and Smads cooperate to activate the p15 promoter in response to TGF-β receptor activation. S2 cells were transfected with the p15P113luc reporter, the Sp1 expression plasmid and expression plasmids for Smads, as marked.
We also examined the role of individual Smads in the transcription from the p15Ink4B promoter in S2 cells. Transfection of expression plasmids for Smad2, Smad3 or Smad4, either individually or in combination, did not affect the basal transcription level from this promoter in S2 cells, even in the presence of activated TβRI (Figure 7B). However, coexpression of Sp1 with Smad2 or Smad3 enhanced transcriptional activation in the presence of TβRI (T202D) (Figure 7C). The transcriptional induction by the activated TβRI, when not all three Smads are introduced, is most likely due to the endogenous expression of Medea and/or dSmad2. Medea is the Drosophila homolog of Smad4 (Das et al., 1998; Hudson et al., 1998; Inoue et al., 1998; Wisotzkey et al., 1998) and dSmad2 is a Drosophila homolog with characteristics of Smad2 and Smad3 (Brummel et al., 1999; Das et al., 1999). Our results indicate that cooperation of Sp1 and Smads is required for transcriptional activation in response to TGF-β.
Discussion
Cell cycle progression is controlled by a set of CDKs and their regulators, e.g. cyclins and CDK inhibitors (CKIs) (for review see Sherr and Roberts, 1999). There are two families of CKIs, the Cip/Kip family, which includes p21Cip1, p27Kip1 and p57Kip2, and the Ink4 family, which includes p15Ink4B, p16Ink4A, p18Ink4C and p19Ink4D. Numerous reports illustrate how environmental and developmental cues induce Cip family members during growth and differentiation. In contrast, our knowledge of the regulation of Ink4 family members is limited. p15Ink4B expression is strongly induced in response to TGF-β and this event initiates the TGF-β-induced growth arrest (Hannon and Beach, 1994; Reynisdóttir et al., 1995). In some cell lines, however, the induction of p21Cip1 is thought to initiate TGF-β-induced growth arrest as well (Datto et al., 1995; C.Y.Li et al., 1995; Reynisdóttir et al., 1995). The recent characterization of Smads as effectors of TGF-β signaling raises the question as to how Smads induce growth arrest. In this report, we have described the mechanism by which Smads induce p15Ink4B expression and growth arrest in response to TGF-β.
In agreement with the model that Smads activate transcription through cooperativity with a set of other transcription factors (Derynck et al., 1998), our study demonstrates that Smads cooperate with Sp1 to activate transcription of the p15Ink4B gene in response to TGF-β. Sp1 is the primary DNA-binding component, i.e. to the Sp1 sites in the p15Ink4B promoter, and provides the basal transcription. In the nucleoprotein complex with Sp1, three different Smads (Smad2, Smad3 and Smad4) provide the ligand-induced coactivator function. TGF-β induces the formation and nuclear translocation of a trimeric Smad complex, which in this case is likely to consist of one monomer each of Smad2, Smad3 and Smad4. Smad2 and Smad4 associate directly with Sp1 and co-activate the transcriptional activity of Sp1. Smad3 transactivates the p15 promoter by binding directly to SBE(s) adjacent to the Sp1 binding sites and by enhancing the activity of Sp1 through indirect interactions. The physical interaction of the heterotrimeric complex of Smad2, Smad3 and Smad4 mediates the TGF-β-induced transcriptional enhancement, presumably through the direct interaction of Smad2 and/or Smad3 with the transcriptional coactivator CBP/p300 (Figure 8).

Fig. 8. Model for TGF-β-dependent transcriptional activation of the p15Ink4B gene. An oligomeric, likely trimeric, complex consisting of Smad2, Smad3 and Smad4 is translocated into the nucleus upon TGF-β stimulation. Smad3 contacts DNA at the SBE on the p15Ink4B promoter, to which Sp1 was already bound at the Sp1 site (only one Sp1 binding site and one SBE are shown). Additional protein–protein interactions occur between Sp1 and Smad2, Sp1 and Smad4, and Smad2 and/or 3 and CBP/p300. TAF/TBP represents TATA-binding protein (TBP) and its associated factors (TAF), the transcription factors for general transcription associated with RNA polymerase II.
Several features distinguish this described mechanism of Smad-mediated transcription from previous observations. First, our demonstration that Sp1 physically interacts and functionally cooperates with the heteromeric Smad complex identifies Sp1 as a new transcription partner for the Smads and characterizes a new function for Sp1. In response to TGF-β, Smad3 has been shown to cooperate with c-Jun or the vitamin D receptor through direct interaction (Zhang et al., 1998; Yanagisawa et al., 1999). In the case of activation of the p15 promoter, Sp1 is the transcription factor with which the Smads cooperate. Thus, Sp1 interacts constitutively with the promoter and provides a basal level of transcription, independent of external stimuli. Upon TGF-β treatment, the Smad complex undergoes nuclear translocation, interacts with DNA-bound Sp1 and increases the level of transcription. This increase has also been illustrated in Drosophila Schneider cells, which lack endogenous Sp1. In this system, the basal transcription level from the p15Ink4B promoter was increased with increasing levels of Sp1, and Sp1 was required for transcriptional induction by Smads. Finally, inactivation of the Sp1 binding sites in the p15Ink4B promoter decreased the basal transcription level and TGF-β responsiveness.
A second important feature of the mechanism described in this study is that Smad2, Smad3 and Smad4 are all required for TGF-β-induced transcription from the p15Ink4B promoter. This is in contrast to the binary combinations of Smad2 and Smad4, which mediate activation of activin-responsive Mix.2 and goosecoid promoters (Chen et al., 1996, 1997; Liu et al., 1997; Labbé et al., 1998; Zhou et al., 1998), and of Smad3 and Smad4, which mediate TGF-β-induced transcription from the few TGF-β-responsive promoters characterized so far (Zhang et al., 1996, 1998; Dennler et al., 1998; Jonk et al., 1998; Song et al., 1998; Vindevoghel et al., 1998; Stroschein et al., 1999). The requirement of all three Smads for TGF-β-induced activation of the p15Ink4B promoter was demonstrated using several approaches. Dominant- negative Smad mutants of Smad2, Smad3 and Smad4, but not Smad1, inhibit TGF-β-induced activation of the p15Ink4B promoter. In addition, Smad3- or Smad4-null cells do not allow TGF-β-induced transcription from the p15Ink4B promoter, unless this defect is rescued with exogenous Smad3 or Smad4 expression, respectively. Finally, the Sp1 interaction with Smads can be reconstituted in Schneider cells by expressing Sp1, Smad2, Smad3 and Smad4. Whereas Sp1 provides the basal expression, the Smads do not have any effect on p15Ink4B transcription in the absence of Sp1. The highest level of TGF-β-induced transcription, however, can be achieved by co-expressing all three Smads with Sp1. Nevertheless, expression of Sp1 with Smad2, Smad3 or Smad4, alone or in binary combination, also provides inducible expression, presumably due to the presence of Medea (Das et al., 1998; Hudson et al., 1998; Inoue et al., 1998; Wisotzkey et al., 1998) and dSmad2 (Brummel et al., 1999; Das et al., 1999).
We also determined the biochemical basis for the requirement for all three Smads in cooperation with Sp1. Smads form trimeric complexes (Shi et al., 1997; Kawabata et al., 1998) and the heteromeric combination most likely consists of one Smad4 with two receptor-activated Smads. In this complex, Smad2 interacts through its C-domain directly with Sp1, whereas Smad4 interacts with Sp1 through its N-domain. Smad3 does not interact directly with Sp1, but participates in the complex because of its direct interaction with Smad2 and Smad4, and furthermore is in direct contact with the SBE of the promoter DNA through its N-domain.
A third important feature of the mechanism of transcriptional activation is the synergy in binding between Sp1 and Smad2 (and to some extent Smad4). In gel shift analyses, Smad2 enhanced the binding of Sp1, for binding to both the Sp1a and Sp1b sites. This was most striking in the case of the Sp1b site. Sp1 has only a low affinity for the Sp1b site, but the binding efficiency was strongly increased in the presence of Smad2. Since Smad2 is unable to bind DNA, we conclude that Smad2 induces a conformational change in Sp1 that strongly enhances the DNA binding of Sp1. The participation of Smad3 in the complex provides a further interaction with DNA at the SBEs that may result in DNA bending required for the assembly of a larger activator complex.
A fourth important feature is that the interaction of Smads with Sp1 strongly enhances the transcriptional activity of Sp1 and provides linkage to another coactivator system. Consistent with the ability of TGF-β to enhance the transcriptional activity of Sp1 fused to the Gal4 DNA binding domain (J.-M.Li et al., 1995; Moustakas and Kardassis, 1998), Smad2 and Smad3 enhance the transcriptional activity of Gal4–Sp1. This ability of Smad2 and Smad3 to enhance the transcription is most likely related to the recruitment of CBP/p300 as coactivator. CBP/p300 has been shown to interact directly with the C-termini of Smad2 or Smad3, and to serve as coactivator for Smad-mediated transcription (Feng et al., 1998; Janknecht et al., 1998; Nishihara et al., 1998; Pouponnot et al., 1998; Shen et al., 1998; Topper et al., 1998). Efficient interaction with CBP/p300 requires the C-terminal phosphorylation of the two serines of Smad2 or Smad3 (Feng et al., 1998), and mutation of these serines to alanines inactivates their ability to enhance the transcriptional activity of Gal4–Sp1. Importantly, Sp1 does not interact with CBP/p300, but instead interacts with the CRSP coactivator complex, which contains several components shared by other transcriptional complexes such as the murine Mediator complex or ARC/DRIP complex (Ryu and Tjian, 1999; Ryu et al., 1999). The recruitment of CBP/p300 into the transcription complex of Smad2–Smad3–Smad4 with Sp1 is also supported by the observation that increased CBP/p300 expression enhances transcription from the p15Ink4B promoter (data not shown). In addition, E1A, which inhibits CBP/p300 function, blocks the TGF-β-induced, but not the basal transcription from the p15Ink4B promoter (Datto et al., 1997). This suggests that the recruitment of CBP/p300 by the Smads does not displace the interaction of Sp1 with the CRSP complex.
Our observation that the heteromeric Smad complex interacts with Sp1 to mediate TGF-β-induced transcription is most likely not restricted to the p15Ink4B promoter. Sp1 sites are also found in the promoter for another CDK inhibitor, p21Cip1, and have been shown to be required for TGF-β-induced activation of this promoter (Datto et al., 1995). In addition, overexpression of Smads increases transcription from the p21 promoter (Moustakas and Kardassis, 1998). Therefore, we expect that the mechanisms of TGF-β-induced transcriptional activation of the promoters for p15Ink4B and p21Cip1 are similar, and that the same mechanism underlies the TGF-β-induced growth arrest, irrespective of whether the primary event is induction of p15Ink4B or p21Cip1 expression. Multiple Sp1 sites are also present in the promoters of the genes for TGF-β1 (Kim et al., 1989; Geiser et al., 1993; Kim et al., 1998), TGF-β2 (Noma et al., 1991), TGF-β3 (Geiser et al., 1993), and the TGF-β receptors TβRI (Bloom et al., 1996; Ji et al., 1996; Kim et al., 1998) and TβRII (Humphries et al., 1994; Bae et al., 1995; Kim et al., 1998). TGF-β treatment induces its own expression (Van Obberghen-Schilling et al., 1988) and that of its receptors (Bloom et al., 1996), thus providing a positive feedback mechanism. Therefore, it is possible that the Sp1–Smad interaction is critical for the TGF-β-induced autoregulation. Finally, Sp1 sites are also required for TGF-β-induced transcriptional activation of the α2(I) collagen promoter (Greenwel et al., 1997).
The mechanism of transcriptional activation of the p15Ink4B promoter in TGF-β-induced growth arrest also provides insight into the mechanistic role of Smads as tumor suppressors. TGF-β is growth inhibitory for normal epithelial cells and the TGF-β signaling that leads to growth arrest, from TGF-β receptors to Smads to p15Ink4B, is considered as a tumor suppressor pathway. Inactivation of the functions of individual components in this pathway has been associated with tumor progression into carcinomas. For example, the p15Ink4B gene is deleted in many tumors and tumor cell lines, thus eliminating the TGF-β-induced CDK inhibitors (Derynck and Feng, 1997; Sherr and Roberts, 1999). Overexpression of c-myc could also downregulate the expression of the p15Ink4B gene and results in TGF-β resistance (Warner et al., 1999). Smad4 is often inactivated in carcinomas (Barrett et al., 1996; Hahn et al., 1996a,b; Schutte et al., 1996; Riggins et al., 1997) and this observation resulted in its initial identification in pancreatic carcinomas as the candidate tumor suppressor DPC4 (Hahn et al., 1996b). Our demonstration that Smad4 is required in the Smad–Sp1 complex for TGF-β-induced activation of p15Ink4B expression, further pinpoints the activity of Smad4 as a tumor suppressor. In the absence of Smad4, TGF-β is unable to induce expression of p15Ink4B (or p21Cip1) and, consequently, can not induce growth arrest. Interestingly, among the two Smads that can be activated by TGF-β (Smad2 and Smad3), only Smad2 has been inactivated in human tumors (Eppert et al., 1996; Riggins et al., 1997). Here we show that Smad2 interacts directly with Sp1 and is essential for p15Ink4B induction. In contrast, all other examples of TGF-β-induced gene expression that have been mechanistically characterized act through Smad3–Smad4 in epithelial cells. Thus, the inactivation of Smad2 and not Smad3 effectively eliminates the TGF-β-induced growth arrest, presumably without effect on the ability of TGF-β to induce expression of many other genes. Since TGF-β-induced extracellular matrix production is advantageous for tumor growth and development (Wakefield et al., 1995; Arrick and Derynck, 1996), these tumors may have maintained the advantageous aspects of the TGF-β response and eliminated the tumor suppressor role of TGF-β. Although Smad3 has not been found to be inactivated in human tumors, Smad3–/– mice have been shown to develop colon carcinomas (Zhu et al., 1998), which is consistent with the essential role of Smad3 in the induction of p15Ink4B expression and growth arrest in response to TGF-β.
Materials and methods
Expression plasmids
The sequences coding for N-terminally hemagglutanin- (HA) or Flag-tagged, or Gal4 DNA-binding domain fused Smad and Sp1 proteins (or defined regions of these proteins) were generated by PCR-based techniques. To obtain expression in mammalian cells, these coding sequences were inserted as EcoRI–SalI fragments into the EcoRI–SalI sites of the mammalian expression plasmid pRK5 (Graycar et al., 1989). Detailed information on the construction of the plasmids used in this report will be provided upon request. The pRK5-based expression plasmid for constitutively active TβRI was described (Feng and Derynck, 1996). The expression plasmid Pac-Sp1 was obtained from Robert Tjian (University of California, Berkeley), and GST–Sp1 plasmid was provided by Hans Rotheneder and Erhard Wintersberger (University of Vienna, Austria).
Cell culture, transfections and immunoprecipitations
293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS), and HaCaT, Mv1Lu and MDA-MB-468 cells were maintained in MEM, 10% FBS, supplemented with non-essential amino acids. Mouse embryonic Smad3–/– fibroblasts were grown in DMEM containing 20% FBS. RI14 cells are R1B/L17 (Wrana et al., 1994) that were stably transfected to express TβRI (data not shown). 293 cells and HaCaT cells were transfected using LipofectAMINE (Gibco-BRL) and DEAE-dextran, respectively. All mammalian cells were incubated at 37°C in a 5% CO2 incubator. Drosophila S2 cells were cultured in Drosophila Schneider cell medium (Gibco-BRL) with 10% FBS, at 25°C without CO2.
Immunoprecipitations using anti-Flag or anti-HA antibody were carried out as described (Feng et al., 1995). HaCaT cells were transfected with expression plasmids for Flag- or HA-tagged Smads. Anti-Flag (Sigma) or anti-HA antibodies (Roche) were used to immunoprecipitate Smad proteins from transfected cell lysates. For subsequent western blotting, the immunoprecipitated proteins were separated by SDS–PAGE, transferred onto Immobilon (Millipore), and detected with the primary antibody. Antibody-bound proteins were visualized by chemiluminescence (Pierce). To detect endogenous Sp1 bound to precipitated Smads, western analysis was performed with anti-Sp1 antibody (Santa Cruz Biotechnology).
Transcriptional reporter assays
Plasmid p15P113Luc, which contains the luciferase gene under control of the p15 promoter (J.-M.Li et al., 1995), was used to measure TGF-β- and Smad-induced transcription from the p15 promoter. pSVβgal (Promega), which expresses β-galactosidase under the control of the SV40 early promoter, was cotransfected to allow normalization of transfection efficiency. Transfections, TGF-β treatment and reporter assays were carried out as described (Feng et al., 1995). HaCaT and Mv1Lu cells were transfected using DEAE-dextran, and other cells were transfected using commercial reagents [LipofectAMINE for MDA-MB-468 cells, FuGene 6 (Roche) for mouse Smad3–/– fibroblasts, and Cytofectin GSV (Glen Research) for S2 cells]. Generally, exponentially grown cells at 25–30% confluency were transfected with expression plasmids for Smads and/or reporter plasmids, e.g. p15P113luc and pSV-β-Gal. The amounts of individual plasmid DNAs used for transfection depended on the transfection reagents used, but the total amount of transfected DNA was always the same due to the addition of vector DNA when necessary. Forty to forty-five hours after transfection, cells were treated with 400 pM TGF-β for 4 h. Cells were then harvested for measurement of luciferase and β-galactosidase activities. All assays were done in triplicate and all values were normalized for transfection efficiency against β-galactosidase activity.
Gal4 transactivation assays
Plasmids encoding Gal4–Sp1 (Sp1 fused to the Gal4 DNA-binding domain) and derivatives were cotransfected into cells with the Gal4–luciferase reporter plasmid pFR-Luc (Stratagene) and other expression plasmids, as specified in the figure legends. Transfected cells were treated for 24 h with or without 400 pM TGF-β. The ability of Gal4–Sp1 to transactivate the heterologous Gal4-binding promoter was quantitated by measuring the luciferase expression from the Gal4-binding promoter. Transfections, TGF-β treatment and luciferase assays were done essentially as for the p15–luc luciferase assays.
Gel shift assays
Gel shift assays were performed using a commercial kit (Promega). A 10 µl reaction, containing 10 µg of nuclear extract in 10 mM Tris–HCl pH 7.5, 0.5 mM EDTA, 1 mM MgCl2, 50 mM NaCl, 0.5 mM dithiothreitol, 4% glycerol and 0.05 mg/ml poly(dI–dC)·poly(dI–dC), was incubated at room temperature for 30 min with 50 000 d.p.m. of 32P-labeled probe. When using purified proteins for gel shift analyses, indicated amounts of purified Sp1 (Promega) or GST–Smads were used in place of nuclear extract in the binding reaction. Where indicated, a 25-fold molar excess of unlabeled competitor DNA was used in competition experiments. The probe and competitor oligonucleotides corresponded to segments of the p15Ink4B promoter (Figure 3A), as indicated in text and figures. For antibody supershift or interference experiments, anti-Sp1 or anti-Smad antibodies were added to the reaction mixture, which was then incubated at 4°C for 90 min. Anti-Sp1 (Santa Cruz Biotechnology), anti-Smad2 (Upstate Biotechnology), anti-Smad3 and anti-Smad4 (Santa Cruz Biotechnology) were used as indicated. DNA–protein complexes were separated in a 5% polyacrylamide gel in 0.5× TBE buffer and visualized by autoradiography.
Biotinylated oligonucleotide precipitation
Biotinylated p15 promoter DNA oligonucleotides, corresponding to nt –84 to –46 (Figure 3A), either wild type or with point mutations in the SBE or Sp1 sites as shown in Figure 3B, were synthesized. Immobilization of biotinylated p15 promoter oligonucleotides and adsorption of cellular proteins to the DNA were carried out using Dynabeads (Dynal) following the manufacturer’s instructions. Briefly, DNA was incubated with streptavidin paramagnetic beads, washed three times with BW buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 2 M NaCl) followed by one wash in S1 buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 150 NaCl, 0.1% NP-40). The DNA was then incubated with nuclear extracts from transfected 293 cells expressing Smad2, Smad3 and Smad4, in the presence or absence of activated TβRI, for 1 h at 4°C. After extensive washing with S1 buffer, DNA-bound protein was subjected to SDS–PAGE, followed by western blotting using antibodies, as shown in Figure 5E and F. In parallel, the cell lysates were immunoblotted with different antibodies to demonstrate the expression level of endogenous Sp1 and transfected Smads.
GST fusion proteins and in vitro protein binding assays
GST fusion proteins of Sp1 and Smads were prepared according to Ausubel et al. (1998). In Figure 4C and D, 35S-labeled Smad2 or Smad4 and their domains, obtained by in vitro transcription/translation (Promega) from the SP6 promoter, were incubated with 1 µg of GST or GST–Sp1 fragments bound to glutathione–Sepharose beads (Pharmacia). In Figure 6E, 1 µg of GST or GST–Smad bound to glutathione–Sepharose beads was used to adsorb in vitro-translated 35S-labeled Sp1(2–788) or Sp1(252–496). After extensive washing of GST fusions and bound proteins in 25 mM Tris–HCl pH 8.0, 300 mM NaCl and 1% Triton X-100, associated proteins were separated by SDS–PAGE and visualized by autoradiography.
Northern blot hybridization
Exponentially growing HaCaT cells were treated with 400 pM TGF-β and 10 µM cycloheximide for the time indicated in Figure 1 and total RNA was prepared from the cells using TriZol kit (Gibco-BRL) following the manufacturer’s instructions. Cycloheximide was able to inhibit new protein synthesis in control experiments (data not shown). Equal amounts of RNA were then separated in an agarose gel, and stained with ethidium bromide for visualization of ribosomal RNA. RNA was then transferred onto a nitrocellulose membrane. Hybridization to a 32P-labeled p15Ink4B gene probe (the 310 bp SpeI–KpnI fragment of human p15Ink4B cDNA coding region) was carried out at 65°C in a solution containing 6× SSC, 2× Denhardt’s reagent and 0.1% SDS. Hybridized bands were then visualized by autoradiography.
Detection of luciferase mRNA using RT–PCR
Reporter plasmids, 2 µg of p15P113luc and 2 µg of SVβGal were transfected using DEAE-dextran into HaCaT cells at 25% confluency in a 10 cm petri dish. Forty-five hours after transfection, cells were treated with 400 pM TGF-β in the presence or absence of 10 µM cycloheximide for 4 h. Total RNA was then extracted using Trizol (Gibco-BRL). RNA (1 µg) was reverse-transcribed to generate the first strand cDNA using Superscript reverse transcriptase (Gibco-BRL) following the manufacturer’s instructions. One-tenth of the cDNA was used as template in PCR (50 µl) using a pair of primers for luciferase coding region (forward: ATGGAAGACGCCAAAAACATAAAG; reverse: TATCCGGAATGA TTTGATTGCCAA) and another pair of primers for β-galactosidase coding region (forward: ACATTTAATGTTGATGAAAGCT; reverse: TGCGCTCAGGTCAAATTCAGAC). The reaction contained 25 pmol of each primer, 1 U of Taq polymerase (Gibco-BRL), and was incubated for 25 cycles of denaturation (94°C), annealing (58°C) and extension (72°C). PCR products were separated on a 2% agarose gel and stained with ethidium bromide.
Acknowledgments
Acknowledgements
We thank Xiao-Fan Wang (Duke University, Durham) for stimulating discussion, reporter plasmid p15P113luc and Smad3–/– mouse embryonic fibroblasts; Robert Tjian (University of California, Berkeley) for Sp1 expression plasmids and anti-Sp1 antibody; Joan Massagué (Memorial Sloan-Kettering Cancer Center, New York) for mink cell lines DR26 and R1B; Norbert Fusenig (German Cancer Center, Heidelberg) for HaCaT cells; Hans Rotheneder and Erhard Wintersberger (University of Vienna, Vienna, Austria) for GST–Sp1; and Yue Xiong (University of North Carolina, Chapel Hill) for p15 cDNA. This research was supported by NIH grants R01-CA63101 and P01-HL60231 (Project III) to R.D., Baylor College of Medicine start-up funds to X.-H.F. and X.L., and American Cancer Society Grant RPG-00-214-01-CCG to X.-H.F.
References
- Abdollah S., Macías-Silva,M., Tsukazaki,T., Hayashi,H., Attisano,L. and Wrana,J.L. (1997) TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2–Smad4 complex formation and signaling. J. Biol. Chem., 272, 27678–27685. [DOI] [PubMed] [Google Scholar]
- Arrick B.A. and Derynck,R. (1996) The biological role of transforming growth factor-β in cancer development. In Waxman,J. (ed.), Molecular Endocrinology of Cancer. Cambridge University Press, Cambridge, UK, pp. 51–78. [Google Scholar]
- Attisano L. and Wrana,J.L. (1998) Mads and Smads in TGFβ signalling. Curr. Opin. Cell Biol., 10, 188–194. [DOI] [PubMed] [Google Scholar]
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1998) Current Protocols in Molecular Biology. John Wiley and Sons, Inc., New York, NY. [Google Scholar]
- Bae H.W., Geiser,A.G., Kim,D.H., Chung,M.T., Burmester,J.K., Sporn,M.B., Roberts,A.B. and Kim,S.-J. (1995) Characterization of the promoter region of the human transforming growth factor-β type II receptor gene. J. Biol. Chem., 270, 29460–29468. [DOI] [PubMed] [Google Scholar]
- Barrett M.T., Schutte,M., Kern,S.E. and Reid,B.J. (1996) Allelic loss and mutational analysis of the DPC4 gene in esophageal adenocarcinoma. Cancer Res., 56, 4351–4353. [PubMed] [Google Scholar]
- Bloom B.B., Humphries,D.E., Kuang,P.P., Fine,A. and Goldstein,R.H. (1996) Structure and expression of the promoter for the R4/ALK5 human type I transforming growth factor-β receptor: regulation by TGF-β. Biochim. Biophys. Acta, 1312, 243–248. [DOI] [PubMed] [Google Scholar]
- Brummel T., Abdollah,S., Haerry,T.E., Shimell,M.J., Merriam,J., Raftery,L., Wrana,J.L. and O’Connor,M.B. (1999) The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev., 13, 98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Rubock,M.J. and Whitman,M. (1996) A transcriptional partner for MAD proteins in TGF-β signaling. Nature, 383, 691–696. [DOI] [PubMed] [Google Scholar]
- Chen X., Weisberg,E., Fridmacher,V., Watanabe,M., Naco,G., and Whitman,M. (1997) Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature, 389, 85–89. [DOI] [PubMed] [Google Scholar]
- Courey A.J. and Tjian,R. (1988) Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell, 55, 887–898. [DOI] [PubMed] [Google Scholar]
- Das P., Maduzia,L.L., Wang,H., Finelli,A.L., Cho,S.H., Smith,M.M. and Padgett,R.W. (1998) The Drosophila gene Medea demonstrates the requirement for different classes of Smads in dpp signaling. Development, 125, 1519–1528. [DOI] [PubMed] [Google Scholar]
- Das P., Inoue,H., Baker,J.C., Beppu,H., Kawabata,M., Harland,R.M., Miyazono,K. and Padgett,R.W. (1999) Drosophila dSmad2 and Atr-I transmit activin/TGFβ signals. Genes Cells, 4, 123–134. [DOI] [PubMed] [Google Scholar]
- Datto M.B., Li,Y., Panus,J.F., Howe,D.J., Xiong,Y. and Wang,X.-F. (1995) Transforming growth factor β induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl Acad. Sci. USA, 92, 5545–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datto M.B., Hu,P.P., Kowalik,T.F., Yingling,J. and Wang,X.-F. (1997) The viral oncoprotein E1A blocks transforming growth factor β-mediated induction of p21/WAF1/Cip1 and p15/INK4B. Mol. Cell. Biol., 17, 2030–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S., Itoh,S., Vivien,D., ten Dijke,P., Huet,S. and Gauthier,J.M. (1998) Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J., 17, 3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R. and Feng,X.-H. (1997) TGF-β receptor signaling. Biochim. Biophys. Acta, 1333, F105–F150. [DOI] [PubMed] [Google Scholar]
- Derynck R., Zhang,Y. and Feng,X.-H. (1998) Smads: transcriptional activators of TGF-β responses. Cell, 95, 737–740. [DOI] [PubMed] [Google Scholar]
- Eppert K. et al. (1996) MADR2 maps to 18q21 and encodes a TGFβ-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell, 86, 543–552. [DOI] [PubMed] [Google Scholar]
- Feng X.-H. and Derynck,R. (1996) Ligand-independent activation of TGF-β signaling pathways by heteromeric cytoplasmic domains of TGF-β receptors. J. Biol. Chem., 271, 13123–13129. [DOI] [PubMed] [Google Scholar]
- Feng X.-H., Filvaroff,E.H. and Derynck,R. (1995) Transforming growth factor-β (TGF-β)-induced down-regulation of cyclin A expression requires a functional TGF-β receptor complex. Characterization of chimeric and truncated type I and type II receptors. J. Biol. Chem., 270, 24237–24245. [DOI] [PubMed] [Google Scholar]
- Feng X.-H., Zhang,Y., Wu,R.-Y. and Derynck,R. (1998) The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-β-induced transcriptional activation. Genes Dev., 12, 2153–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser A.G., Busam,K.J., Kim,S.-J., Lafyatis,R., O’Reilly,M.A., Webbink,R., Roberts,A.B. and Sporn,M.B. (1993) Regulation of the transforming growth factor-β1 and -β3 promoters by transcription factor Sp1. Gene, 129, 223–228. [DOI] [PubMed] [Google Scholar]
- Gidoni D., Dynan,W.S. and Tjian,R. (1984) Multiple specific contacts between a mammalian transcription factor and its cognate promoters. Nature, 312, 409–413. [DOI] [PubMed] [Google Scholar]
- Graycar J.L., Miller,D.A., Arrick,B.A., Lyons,R.M., Moses,H.L. and Derynck,R. (1989) Human transforming growth factor-β 3: recombinant expression, purification and biological activities in comparison with transforming growth factors-β1 and -β2. Mol. Endocrinol., 3, 1977–1986. [DOI] [PubMed] [Google Scholar]
- Greenwel P., Inagaki,Y., Hu,W., Walsh,M. and Ramirez,F. (1997) Sp1 is required for the early response of α2(I) collagen to transforming growth factor-β1. J. Biol. Chem., 272, 19738–19745. [DOI] [PubMed] [Google Scholar]
- Hahn S.A. et al. (1996a) Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res., 56, 490–494. [PubMed] [Google Scholar]
- Hahn S.A. et al. (1996b) DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science, 271, 350–353. [DOI] [PubMed] [Google Scholar]
- Hannon G.J. and Beach,D. (1994) p15ink4B is a potential effector of TGF-β-induced cell cycle arrest. Nature, 371, 257–260. [DOI] [PubMed] [Google Scholar]
- Hata A., Lo,R.S., Wotton,D., Lagna,G. and Massagué,J. (1997) Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature, 388, 82–87. [DOI] [PubMed] [Google Scholar]
- Heldin C.-H., Miyazono,K. and ten Dijke,P. (1997) TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature, 390, 465–471. [DOI] [PubMed] [Google Scholar]
- Hu P.P., Datto,M.B. and Wang,X.-F. (1998) Molecular mechanisms of transforming growth factor-β signaling. Endocr. Rev., 19, 349–363. [DOI] [PubMed] [Google Scholar]
- Hua X., Liu,X., Ansari,D.O. and Lodish,H.F. (1998) Synergistic cooperation of TFE3 and Smad proteins in TGF-β-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev., 12, 3084–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson J.B., Podos,S.D., Keith,K., Simpson,S.L. and Ferguson,E.L. (1998) The Drosophila Medea gene is required downstream of dpp and encodes a functional homolog of human Smad4. Development, 125, 1407–1420. [DOI] [PubMed] [Google Scholar]
- Humphries D.E., Bloom,B.B., Fine,A. and Goldstein,R.H. (1994) Structure and expression of the promoter for the human type II transforming growth factor-β receptor. Biochem. Biophys. Res. Commun., 203, 1020–1027. [DOI] [PubMed] [Google Scholar]
- Inoue H.T., et al. (1998) Interplay of signal mediators of decapentaplegic (Dpp): molecular characterization of mothers against dpp, Medea and daughters against dpp. Mol. Biol. Cell, 9, 2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R., Wells,N.J. and Hunter,T. (1998) TGF-β-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev., 12, 2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C., Casinghino,S., McCarthy,T.L. and Centrella,M. (1996) Cloning, characterization and expression of the transforming growth factor-β type I receptor promoter in fetal rat bone cells. J. Cell. Biochem., 63, 478–490. [DOI] [PubMed] [Google Scholar]
- Jonk L.J., Itoh,S., Heldin,C.H., ten Dijke,P. and Kruijer,W. (1998) Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-β, activin and bone morphogenetic protein-inducible enhancer. J. Biol. Chem., 273, 21145–21152. [DOI] [PubMed] [Google Scholar]
- Kawabata M., Inoue,H., Hanyu,A., Imanura,T. and Miyazono,K. (1998) Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. EMBO J., 17, 4056–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Johnson,K., Chen,H., Carroll,S. and Laughon,A. (1997) Drosophila Mad binds to DNA and directly mediates activation of vestigial by Dpp. Nature, 388, 304–308. [DOI] [PubMed] [Google Scholar]
- Kim S.-J., Glick,A., Sporn,M.B. and Roberts,A.B. (1989) Characterization of the promoter region of the human transforming growth factor-β1 gene. J. Biol. Chem., 264, 402–408. [PubMed] [Google Scholar]
- Kim Y., Ratziu,V., Choi,S.-G., Lalazar,A., Theiss,G., Dang,Q., Kim,S.-J. and Friedman,S.L. (1998) Transcriptional activation of transforming growth factor β1 and its receptors by the Kruppel-like factor Zf9/core promoter-binding protein and Sp1. J. Biol. Chem., 273, 33750–33758. [DOI] [PubMed] [Google Scholar]
- Labbé E., Silvestri,C., Hoodless,P.A., Wrana,J.L. and Attisano,L. (1998) Smad2 and Smad3 positively and negatively regulate TGFβ-dependent transcription through the forkhead DNA-binding protein FAST2. Mol. Cell, 2, 109–120. [DOI] [PubMed] [Google Scholar]
- Lagna G., Hata,A., Hemmati-Brivanlou,A. and Massagué,J. (1996) Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature, 383, 832–836. [DOI] [PubMed] [Google Scholar]
- Laiho M., Weis,M.B. and Massagué,J. (1990) Concomitant loss of transforming growth factor (TGF)-β receptor types I and II in TGF-β-resistant cell mutants implicates both receptor types in signal transduction. J. Biol. Chem., 265, 18518–18524. [PubMed] [Google Scholar]
- Li C.Y., Suardet,L. and Little,J.B. (1995) Potential role of WAF1/Cip1/p21 as a mediator of TGF-β cytoinhibitory effect. J. Biol. Chem., 270, 4971–4974. [DOI] [PubMed] [Google Scholar]
- Li J.-M., Nichols,M.A., Chandrasekharan,S., Xiong,Y. and Wang,X.-F. (1995) Transforming growth factor β activates the promoter of cyclin-dependent kinase inhibitor p15INK4B through an Sp1 consensus site. J. Biol. Chem., 270, 26750–26753. [DOI] [PubMed] [Google Scholar]
- Liu B., Dou,C.L., Prabhu,L. and Lai,E. (1999) FAST-2 is a mammalian winged-helix protein which mediates TGF-β signals. Mol. Cell. Biol., 19, 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Hata,A., Baker,J.C., Doody,J., Cárcamo,J., Harland,R.M. and Massagué,J. (1996) A human Mad protein acting as a BMP-regulated transcriptional activator. Nature, 381, 620–623. [DOI] [PubMed] [Google Scholar]
- Liu F., Pouponnot,C. and Massagué,J. (1997) Dual role of the Smad4/DPC4 tumor suppressor in TGFβ-inducible transcriptional complexes. Genes Dev., 11, 3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias-Silva M., Abdollah,S., Hoodless,P., Pirone,R., Attisano,L. and Wrana,J.L. (1996) MADR-2 is a substrate of the TGF-β receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell, 87, 1215–1224. [DOI] [PubMed] [Google Scholar]
- Massagué J. (1998) TGF-β signal transduction. Annu. Rev. Biochem., 67, 753–791. [DOI] [PubMed] [Google Scholar]
- Moustakas A. and Kardassis,D. (1998) Regulation of the human p21/WAF1/Cip1 promoter in hepatic cells by functional interactions between Sp1 and Smad family members. Proc. Natl Acad. Sci. USA, 95, 6733–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A. et al. (1997) TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J., 16, 5353–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara A., Hanai,J.I., Okamoto,N., Yanagisawa,J., Kato,S., Miyazono,K. and Kawabata,M. (1998) Role of p300, a transcriptional coactivator, in signalling of TGF-β. Genes Cells, 3, 613–623. [DOI] [PubMed] [Google Scholar]
- Noma T., Glick,A.B., Geiser,A.G., O’Reilly,M.A., Miller,J., Roberts,A.B. and Sporn,M.B. (1991) Molecular cloning and structure of the human transforming growth factor-β2 gene promoter. Growth Factors, 4, 247–255. [DOI] [PubMed] [Google Scholar]
- Pouponnot C., Jayaraman,L. and Massagué,J. (1998) Physical and functional interaction of SMADs and p300/CBP. J. Biol. Chem., 273, 22865–22868. [DOI] [PubMed] [Google Scholar]
- Reynisdóttir I. and Massagué,J. (1997) The subcellular locations of p15(Ink4b) and p27(Kip1) coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev., 11, 492–503. [DOI] [PubMed] [Google Scholar]
- Reynisdóttir I., Polyak,K., Iavarone,A. and Massagué,J. (1995) Kip/Cip and Ink4 cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev., 9, 1831–1845. [DOI] [PubMed] [Google Scholar]
- Rich J.N., Zhang,M., Datto,M.B., Bigner,D.B. and Wang,X.-F. (1999) Transforming growth factor-β-mediated p15Ink4B induction and growth inhibition in astrocytes is SMAD3-dependent and a pathway prominently altered in human glioma cell lines. J. Biol. Chem., 274, 35053–35058. [DOI] [PubMed] [Google Scholar]
- Riggins G.J., Kinzler,K.W., Vogelstein,B. and Thiagalingam,S. (1997) Frequency of Smad gene mutations in human cancers. Cancer Res., 57, 2578–2580. [PubMed] [Google Scholar]
- Roberts A.B. (1998) Molecular and cell biology of TGF-β. Miner. Electrolyte Metab., 24, 111–119. [DOI] [PubMed] [Google Scholar]
- Ryu S. and Tjian,R. (1999) Purification of transcription cofactor complex CRSP. Proc. Natl Acad. Sci. USA, 96, 7137–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu S., Zhou,S., Ladurner,A.G. and Tjian,R. (1999) The transcriptional cofactor complex CRSP is required for activity of the enhancer-binding protein Sp1. Nature, 397, 446–450. [DOI] [PubMed] [Google Scholar]
- Schutte M. et al. (1996) DPC4 gene in various tumor types. Cancer Res., 56, 2527–2530. [PubMed] [Google Scholar]
- Shen X., Hu,P.P., Liberati,N.T., Datto,M.B., Frederick,J.P. and Wang,X.F. (1998) TGF-β-induced phosphorylation of Smad3 regulates its interaction with coactivator p300/CREB-binding protein. Mol. Biol. Cell, 9, 3309–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev., 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Shi Y., Hata,A., Lo,R.S., Massagué,J. and Pavletich,N. (1997) A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature, 388, 87–93. [DOI] [PubMed] [Google Scholar]
- Shi Y., Wang,Y.F., Jayaraman,L., Yang,H., Massagué,J. and Pavletich,N.P. (1998) Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell, 94, 585–594. [DOI] [PubMed] [Google Scholar]
- Song C.Z., Siok,T.E. and Gelehrter,T.D. (1998) Smad4/DPC4 and Smad3 mediate transforming growth factor-β (TGF-β) signaling through direct binding to a novel TGF-β-responsive element in the human plasminogen activator inhibitor-1 promoter. J. Biol. Chem., 273, 29287–29290. [DOI] [PubMed] [Google Scholar]
- Souchelnytskyi S., Tamaki,K., Engström,U., Wernstedt,C., ten Dijke,P. and Heldin,C.-H. (1997) Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-β signaling. J. Biol. Chem., 272, 28107–28115. [DOI] [PubMed] [Google Scholar]
- Stroschein S.L., Wang,W. and Luo,K. (1999) Cooperative binding of Smad proteins to two adjacent DNA elements in the plasminogen activator inhibitor-1 promoter mediates transforming growth factor β-induced Smad-dependent transcriptional activation. J. Biol. Chem., 274, 9431–9441. [DOI] [PubMed] [Google Scholar]
- Topper J.N., DiChiara,M.R., Brown,J.D., Williams,A.J., Falb,D., Collins,T. and Gimbrone,M.A.,Jr (1998) CREB binding protein is a required coactivator for Smad-dependent, transforming growth factor β transcriptional responses in endothelial cells. Proc. Natl Acad. Sci. USA, 95, 9506–9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Obberghen-Schilling E., Roche,N.S., Flanders,K.C., Sporn,M.B. and Roberts,A.B. (1988) Transforming growth factor β1 positively regulates its own expression in normal and transformed cells. J. Biol. Chem., 263, 7741–7746. [PubMed] [Google Scholar]
- Vindevoghel L., Lechleider,R.J., Kon,A., de Caestecker,M.P., Uitto,J., Roberts,A.B. and Mauviel,A. (1998) SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor-β. Proc. Natl Acad. Sci. USA, 95, 14769–14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield L.M., Letterio,J.J., Geiser,A.G., Flanders,K.C., O’Shaughnessy,J., Roberts,A.B. and Sporn,M.B. (1995) Transforming growth factor-βs in mammary tumorigenesis: promoters or antipromoters? Prog. Clin. Biol. Res., 391, 133–148. [PubMed] [Google Scholar]
- Warner B.J., Blain,S.W., Seoane,J. and Massagué,J. (1999) Myc downregulation by transforming growth factor β required for activation of the p15(Ink4b) G(1) arrest pathway. Mol. Cell. Biol., 19, 5913–5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.L. (1998) Tumor suppressing pathways. Cell, 92, 591–592. [DOI] [PubMed] [Google Scholar]
- Whitman M. (1998) Smads and early developmental signaling by the TGFβ superfamily. Genes Dev., 12, 2445–2462. [DOI] [PubMed] [Google Scholar]
- Wisotzkey R.G., Mehra,A., Sutherland,D.J., Dobens,L.L., Liu,X., Dohrmann,C., Attisano,L. and Raftery,L.A. (1998) Medea is a Drosophila Smad4 homolog that is differentially required to potentiate DPP responses. Development, 125, 1433–1445. [DOI] [PubMed] [Google Scholar]
- Wrana J.L., Attisano,L., Wieser,R., Ventura,F. and Massagué,J. (1994) Mechanism of activation of the TGF-β receptor. Nature, 370, 341–347. [DOI] [PubMed] [Google Scholar]
- Yanagisawa J. et al. (1999) Convergence of transforming growth factor-β and vitamin D signaling pathways on SMAD transcriptional coactivators. Science, 283, 1317–1321. [DOI] [PubMed] [Google Scholar]
- Zawel L., Dai,J.L., Buckhaults,P., Zhou,S., Kinzler,K.W., Vogelstein,B. and Kern,S.E. (1998) Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell, 1, 611–617. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Feng,X.-H., Wu,R.-Y. and Derynck,R. (1996) Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature, 383, 168–172. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Musci,T. and Derynck,R. (1997) The tumor suppressor Smad 4/DPC 4 as a central mediator of Smad function: cooperativity with other Smads and interference of a Smad 4/DPC 4 mutant with nuclear translocation of Smads. Curr. Biol., 7, 270–276. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Feng,X.-H. and Derynck,R. (1998) Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature, 394, 909–913. [DOI] [PubMed] [Google Scholar]
- Zhou S., Zawel,L., Lengauer,C., Kinzler,K.W. and Vogelstein,B. (1998) Characterization of human FAST-1, a TGFβ and activin signal transducer. Mol. Cell, 2, 121–127. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Richardson,J.A., Parada,L.F. and Graff,J.M. (1998) Smad3 mutant mice develop metastatic colorectal cancer. Cell, 94, 703–714. [DOI] [PubMed] [Google Scholar]