

Abstract
Activation of the Escherichia coli malEp promoter relies on the formation of a higher order structure involving cooperative binding of MalT to promoter-proximal and promoter-distal sites as well as CRP binding to three sites located in between. MalT is the primary activator and one function of CRP is to facilitate cooperative binding of MalT to its cognate sites by bending the intervening DNA. It is shown here that CRP also participates directly in malEp activation. This function is carried out by the molecule of CRP bound to the CRP site centered at –139.5 (CRP site 3). This molecule of CRP recruits RNA polymerase by promoting the binding of the RNA polymerase α subunit C-terminal domain (αCTD) to DNA immediately downstream from CRP site 3, via a contact between αCTD and activating region I of CRP. The action of MalT and CRP at malEp hence provides the example of a novel and complex mechanism for transcriptional synergy in prokaryotes whereby one activator both helps the primary activator to form a productive complex with promoter DNA and interacts directly with RNA polymerase holoenzyme.
Keywords: CRP/Escherichia coli/MalT/RNA polymerase α subunit/transcription synergy
Introduction
Prokaryotic transcription activators act either alone or synergistically, depending on promoter architecture. Activation involving only one activator is beginning to be well understood at the mechanistic level for the σ70-dependent promoters (Ishihama, 1992; Lonetto et al., 1998; Rhodius and Busby, 1998; Busby and Ebright, 1999). Most often, transcription activation involves a contact between the regulatory protein and the RNA polymerase holoenzyme (RNAP; subunit composition α2ββ′σ70). Schematically, at class I promoters, the activator facilitates RNAP recruitment by interacting with the C-terminal domain of one α subunit (αCTD, 81 amino acids) and by promoting its binding to adjacent DNA. αCTD is linked to the N-terminal domain (αNTD), which is responsible for the main interactions with the other RNAP subunits, via a flexible linker of at least 13 amino acids. The activator binding site can be located at various distances from the –35/–10 RNAP site elements, provided that the activator site is correctly phased with respect to the RNAP site. At class II promoters, the activator binds a DNA motif overlapping the –35 region and interacts with σ70, αNTD and/or αCTD, the latter interaction promoting αCTD binding to promoter DNA immediately upstream of the activator binding site. While the interaction with αCTD facilitates RNAP recruitment, as observed at the class I promoters, the interaction with σ70 or αNTD facilitates isomerization of closed complexes to open complexes.
In contrast, much less is known about the mechanisms underlying synergistic activation. Dissection of a few natural or synthetic co-activation systems has so far revealed two scenarios. According to the first one, synergy results from the independent action of two activators interacting directly with RNAP, as observed at promoters at which they act alone, such as two class I activators, each of them interacting with one αCTD, or one class I activator and one class II activator (Joung et al., 1993, 1994; Busby et al., 1994; Scott et al., 1995; Belyaeva et al., 1998). According to the second scenario, only one activator, the primary activator, interacts directly with RNAP while the second aids the primary activator to form a productive complex with promoter DNA (Richet et al., 1991; Belyaeva et al., 2000).
This paper deals with the mechanism whereby the Escherichia coli malEp promoter is synergistically activated by MalT, the activator of the maltose regulon, and by the cAMP receptor protein (CRP; also referred to as CAP), a global regulator controlling the utilization of carbon sources (Richet, 1996; Boos and Shuman, 1998). malEp is divergent from the malKp promoter whose activation also depends on the concerted action of MalT and CRP, both of the promoters directing the synthesis of components of the maltodextrin transport system. While malEp and malKp display some residual activity in the presence of MalT alone, they are inert in the presence of CRP alone (Schwartz, 1987; Richet and Søgaard-Andersen, 1994; Richet, 1996). MalT is a monomeric protein (103 kDa) that self-associates in the presence of both of its positive effectors, maltotriose and ATP (Schreiber and Richet, 1999), and recognizes a 10 bp asymmetric DNA motif (Vidal-Ingigliardi et al., 1991). CRP is a dimeric protein (2 × 23 600 kDa) that, in the presence of cAMP, specific ally binds a 22 bp symmetric DNA site and bends DNA to an angle of ∼90°C in the complex formed with DNA (Schultz et al., 1991). The activation of malEp and malKp is a coupled process involving a unique 210 bp regulatory region comprising, from malEp towards malKp: two MalT sites, three CRP sites and two overlapping sets of three MalT sites staggered by 3 bp (Figure 1). The mechanism whereby MalT and CRP co-activate malEp and malKp involves a CRP-induced switch between a repressor state and an active state responsible for the repression and activation of both promoters, respectively (Richet et al., 1991; Richet, 1996). Schematically, MalT can form either a repression complex by binding cooperatively the high-affinity sites 3/4/5 or an activation complex by binding cooperatively the two low-affinity sets of sites, 1/2 and 3′/4′/5′, from which it activates malEp and malKp, respectively. MalT is thought to contact RNAP via the MalT protomer bound to the MalT site overlapping the –35 element (Danot et al., 1996). The repression state predominates in the absence of CRP while assembly of the activation complex is favored in the presence of CRP. Negative supercoiling also plays a critical role in the assembly of the activation complex (Richet and Raibaud, 1991). As inferred from bend-swap experiments showing that the three CRP-induced bends can be replaced by a static bend or an IHF (integration host factor)-induced bend without altering the mechanism of malKp activation, CRP promotes the cooperative binding of MalT onto sites 1/2 and 3′/4′/5′ simply by bending the intervening DNA (Richet and Søgaard-Andersen, 1994). This is the primary role played by CRP in malKp activation. Co-activation of malKp by MalT and CRP therefore follows the second of the scenarios described above.

Fig. 1. Structure of the regulatory region of malEp and malKp. The malEp–malKp region is drawn to scale with the promoter –10 and –35 elements (hatched boxes), the MalT sites (arrowed boxes) and the CRP sites (open boxes) indicated. The transcription start sites of malEp and malKp are 271 bp apart. The nucleotide sequence (Raibaud et al., 1989) is numbered with respect to the malEp transcription start site. MalT sites 3/4/5 (gray arrowed boxes) are involved in promoter repression while MalT sites 1/2 and 3′/4′/5′ (black arrowed boxes) are involved in promoter activation. The 3/4/5 series of sites is staggered by 3 bp from the 3′/4′/5′ series of sites. For the exact location of all of the MalT sites, see Richet et al. (1991) and Vidal-Ingigliardi et al. (1991). The location of the malEpKpI3 insertion and the end points of deletions malKpΔ240 and malEpΔ261 are indicated.
In this paper, I investigate the role played by CRP in malEp activation and show that malEp co-activation by MalT and CRP obeys a new scenario in which CRP, the second activator, both assists MalT, the primary activator, and interacts directly with RNAP.
Results
CRP does not play a purely structural role in malEp activation
To test the hypothesis that CRP plays a purely architectural role in malEp activation, as found for malKp, I examined whether an IHF-stabilized activation complex activates malEp. The chimera assayed was the V94 variant in which the three CRP binding sites have been replaced by one IHF binding site (Figure 2) and which exhibits a highly active malKp promoter depending on both MalT and IHF and functioning like wild-type malKp (Richet and Søgaard-Andersen, 1994). The assays were performed in vivo by comparing the amount of β-galactosidase made from various malEp–lac transcriptional fusions carried by a single-copy-number plasmid in a Δlac strain. The bacteria were grown in minimal medium in the presence of glycerol and maltose, i.e. under conditions in which both MalT and CRP are active. As shown in Figure 2, the malEp activity of the V94 chimera represents only 13% of the wild-type level, scarcely higher than observed with a malEp variant in which the three CRP binding sites have been inactivated by point mutations (malEpKp1, -2, -3). Moreover, while the weak malEp activity of the chimera was MalT dependent, as indicated by assays performed in the presence of glycerol only (i.e. under conditions in which the concentration of active MalT is low), it was IHF independent (Figure 2). This contrasts with the high activity of the V94 malKp promoter (nearly 50% of the wild-type level), which is both IHF- and MalT-dependent, and is 24-fold higher than for a malKp variant in which the three CRP binding sites have been disrupted by point mutations (Richet and Søgaard-Andersen, 1994; see Figure 2).

Fig. 2. In vivo activity of the chimeric malEp variant. The amount of β-galactosidase made by isogenic hip+ and hip– strains (pop2226 and pop4103, respectively) harboring pOM82 (or derivatives thereof) and grown in the presence or absence of maltose was determined as described in Materials and methods. The results are expressed relative to the values obtained for the induced wild-type malEp (14 000 and 14 100 U in the hip+ and the hip– strains, respectively). The asterisks symbolize the malEpKp1, -2, -3 and -4 mutations that correspond to 3 bp substitutions inactivating CRP sites 1, 2, 3 and 4, respectively (CRP site 4 is a low affinity CRP binding site that does not play any role in malEp and malKp activation) (Vidal-Ingigliardi and Raibaud, 1991). The values obtained for malKp (in parentheses) are from Richet and Søgaard-Andersen (1994).
Similar results were obtained when the malEp chimeric variant was tested in a purified transcription system in which malEp and malKp are known to behave as in vivo (Richet and Raibaud, 1991). The assay used was a single-round transcription assay that monitors the rate of open complex formation. The template was a supercoiled plasmid containing the malEp–malKp region cloned between two divergent terminators (Figure 3A). As shown in Figure 3B, the V94 chimera displayed little malEp activity (<5% of the wild-type level), in contrast to the high activity level of the divergent malKp promoter (34% of the wild-type level). Altogether, the in vivo and in vitro data demonstrate that an IHF-stabilized activation complex does not efficiently activate malEp. This result strongly suggests that, besides its structural role in the stabilization of the activation complex, CRP plays an additional role in malEp activation that involves critical protein–protein interactions.
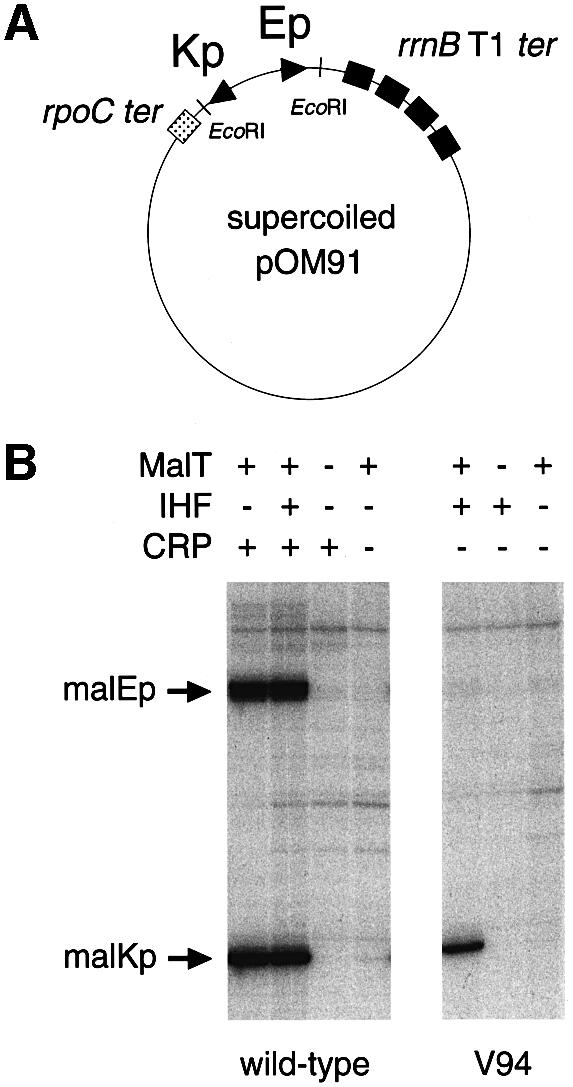
Fig. 3. In vitro activity of the chimeric malEp variant. In vitro transcription assays were carried out in the presence of supercoiled pOM91 variants, 150 nM MalT, 12.5 nM IHF or 150 nM CRP, and 15 nM RNAP. The transcripts made from malEp and malKp are ∼230 and ∼180 nucleotides long, respectively.
The HL159 substitution in CRP specifically interferes with malEp activation
Several studies have shown that a CRP molecule can operate via a class I mechanism (i.e. via a contact with one of the αCTD) from a DNA site located quite far from the –35/–10 promoter elements (Busby and Ebright, 1999). The CRP surface determinant thereby contacted by αCTD is activating region 1 (AR1), an exposed β-turn consisting of residues 156–164 (Busby and Ebright, 1999). The HL159 substitution has been shown to impair strongly the interaction between αCTD and CRP without altering the ability of the protein to bind and bend DNA (Zhou et al., 1993, 1994; Attey et al., 1994). To test the hypothesis that the additional role of CRP at malEp involves a class I contact with RNAP, the effect of the HL159 substitution on malEp activation was analyzed in vitro by using malKp as a control. In order to avoid indirect effects, the templates used were promoter variants carrying an upstream deletion (malEpΔ261 or malKpΔ240) that inactivates the divergent promoter without altering the malEp–malKp regulatory region (see Figure 1) (Raibaud et al., 1989). In vitro transcription assays were performed as described above by using supercoiled templates (Figure 4A).
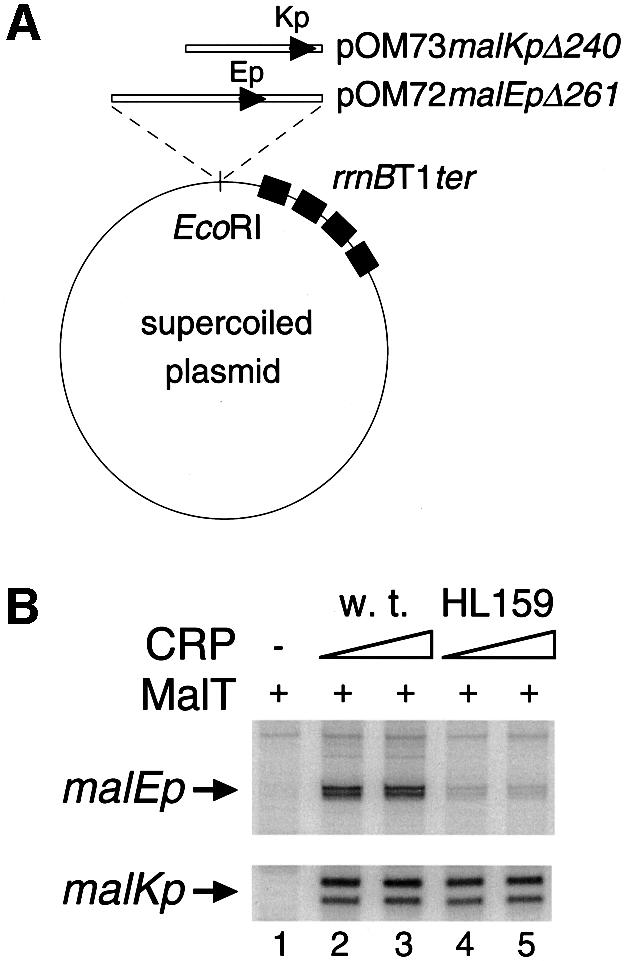
Fig. 4. Effect of the HL159 substitution in CRP on malEp and malKp in vitro. In vitro transcription assays were performed in the presence of supercoiled pOM72malEpΔ261 (or pOM73malKpΔ240), 100 nM MalT, various concentrations of wild-type CRP (or CRP HL159) and 20 nM RNAP. The concentrations of wild-type CRP were 130 nM (lane 2) and 260 nM (lane 3); the concentrations of CRP HL159 were 200 nM (lane 4) and 400 nM (lane 5). The malEp and malKp transcripts are ∼230 and ∼120 nucleotides long, respectively.
Interestingly, the HL159 substitution in CRP caused a severe defect in malEp activation, whereas, as expected, it scarcely affected malKp (Figure 4B): malEp was only 15% active when the mutant CRP protein was substituted for wild-type CRP while malKp retained 83% of the activity level obtained with the wild-type protein. The low malEp activity level observed in the presence of the HL159 CRP protein remained unchanged when the concentration of the mutated protein was increased 2- to 4-fold (Figure 4B and data not shown). Both this fact and the absence of a significant effect on malKp exclude the possibility that the defect caused by the HL159 substitution results from a reduced occupation of the CRP sites due to the slightly reduced affinity of the HL159 CRP protein for its DNA site (Zhou et al., 1993). Thus, these data strongly suggest that at least one of the three molecules of CRP involved in the stabilization of the activation complex also participates in malEp activation via a protein–protein contact involving AR1 and possibly αCTD.
Construction of a model malEp variant
To make the malEp activation process amenable to mechanistic studies, a mutation (malEpKpI3) was introduced that alleviates the requirement for negative DNA supercoiling at the step of activation complex formation. malEpKpI3 corresponds to a 3 bp insertion in the central region, between CRP site 3 and MalT site 3, which brings the high affinity repressor set of MalT sites (3/4/5) in register with MalT sites 1/2 (Figure 5A) (it should be recalled that the two overlapping sets of MalT sites, 3/4/5 and 3′/4′/5′, are staggered by 3 bp). As a result, activation of this malEp variant involves cooperative binding of MalT onto sites 1/2 and 3/4/5 instead of 3′/4′/5′ (see below). Most likely, both the absence of competing repressor sites and the replacement of the original activator sites by higher affinity sites make assembly of the activation complex independent of negative supercoiling. A mutation in the malKp –10 element (malEpKp70) was also introduced to inactivate the malKp promoter.

Fig. 5. Effect of the HL159 substitution in CRP on open complex formation in the presence of various concentrations of RNAP. (A) Structure of the linear malEp substrate. Owing to the 3 bp insertion at –183, formation of the activation complex involves cooperative binding of MalT onto sites 1/2 and 3/4/5 and no longer depends on negative supercoiling. (B) Kinetics of open complex formation at malEp in the presence of 15 nM RNAP. Open complex formation was monitored by the abortive initiation assay described in Materials and methods using the linear malEp model as template. (C) Kinetics of open complex formation at malEp in the presence of 100 nM RNAP.
When present on a linear DNA fragment, the malEpKpI3,70 variant functioned like supercoiled wild-type malEp. Its activation still depended on the presence of both MalT and CRP, and was strongly depressed by the HL159 substitution in CRP (see Figure 5B). DNase I protection experiments confirmed that MalT binds the 3/4/5 set of MalT sites in the presence of CRP, as expected. The top strand protection pattern of the 3/3′ and 4/4′ regions (see Figure 6B, lanes 1 and 5 and data not shown) was indeed characteristic of site 3 and 4 occupancy (Richet et al., 1991). In addition, assembly of the activation complex by MalT was still dependent on the presence of CRP. Comparison of the protection patterns obtained in the presence of MalT alone, CRP alone or MalT + CRP shows that MalT sites 1/2 were unoccupied and sites 3/4/5 only partially occupied in the presence of MalT alone, while they were fully occupied in the presence of both MalT and CRP (see Figure 6A and B, lanes 1–6 and data not shown). Reciprocally, MalT stabilized CRP onto its binding sites, which further confirms that MalT and CRP cooperatively bind their cognate sites. The malEpKpI3,70 variant, hereafter referred to as the malEp model substrate, was used in all of the experiments described below.


Fig. 6. DNase I and KMnO4 footprinting of the nucleoprotein complexes formed on the linear malEp model promoter. DNase I and KMnO4 footprinting experiments were performed with the malEpKpI3 malEpΔ259 variant. The RNAP concentrations used were 100 nM when noted ×1 and 300 nM when noted ×3. (A) Bottom strand DNase I protection patterns obtained using the pOM53 EcoRI–KpnI fragment 3′ end-labeled on the bottom strand (non-template strand). (B) Top strand DNase I protection patterns obtained using the pOM53 PstI–EcoRI fragment 3′ end-labeled on the top strand. (C) KMnO4 attack patterns obtained using the pOM53 EcoRI–KpnI fragment 3′ end-labeled on the bottom strand (non-template strand). (D) Location of the RNAP-dependent protections against DNase I cleavage (arrows) and of the enhanced cutting sites that are characteristic of the activation complex and that are not protected upon RNAP addition (asterisks).
CRP plays a direct role in RNAP recruitment
To test further the possibility that CRP acts at malEp via a class I mechanism, I examined whether the direct role of CRP in malEp activation is related to RNAP recruitment, i.e. whether the defect caused by the HL159 substitution in CRP depends on RNAP concentration. The rate of open complex formation at the malEp model substrate was measured at two RNAP concentrations in the presence of saturating concentrations of MalT and wild-type CRP or CRP HL159 by performing fixed-time abortive initiation assays (Figure 5B and C). In the presence of CRP HL159, the amount of open complexes formed 5 min after addition of 15 nM RNAP represents 12% of the level reached in the presence of wild-type CRP. In contrast, when 100 nM RNAP was added, the open complex level represented 50% of that achieved with wild-type CRP. The effect of the HL159 substitution on malEp activation is thus clearly more pronounced at low RNAP concentration than at high RNAP concentration, thereby indicating that CRP plays a direct role in the formation of closed complexes.
DNase I footprinting studies of the open complexes formed at malEp: RNAP-induced protection in the –120/–125 and –155/–164 regions
To determine whether CRP promotes αCTD binding to upstream positions upon RNAP binding to malEp, I used the same approach as Kolb et al. (1993) and performed DNase I protection experiments with the malEp model substrate under the conditions of the abortive initiation assays described above. Comparison of the top and bottom strand footprints obtained in the absence of RNAP with MalT + wild-type CRP or CRP HL159 shows that the protection patterns were the same, i.e. that, as expected, the CRP HL159 substitution does not impair assembly of the activation complex (Figure 6A and B, lanes 5 and 6). Note also that DNase I hypersensitivity at –120/–125 on both strands as well as at –155, –185 and –186 on the top strand, which characterizes the activation complex, was not altered upon substitution of CRP HL159 for wild-type CRP (enhanced DNase I cleavage most likely reflects minor groove widening due to local DNA bends or structural changes of the double helix accompanying cooperative binding of MalT onto sites 1/2 and 3/4/5). In the presence of MalT and wild-type CRP, RNAP binding to the core promoter was accompanied by a clear protection of the –120/–125 region against DNase attack on both strands (Figure 6A and B, lanes 5 and 7). Most interestingly, this RNAP-dependent protection was not observed when CRP HL159 was substituted for wild-type CRP, even when a higher concentration of RNAP (× 3) was added to achieve the same degree of protection of the core promoter region as in the presence of MalT and wild-type CRP (Figure 6A and B, lanes 5–9). The simplest interpretation of these data is that RNAP binding to malEp involves binding of αCTD to –120/–125 and that αCTD binding depends on an interaction with the AR1 determinant of one of the adjacent CRP, as observed at class I promoters.
The top strand footprints also revealed RNAP-dependent protection at three positions (–155, –162 and –164) upstream from CRP site 3 (Figure 6B, lanes 5–7). The most striking protection affects the phosphodiester bond between nucleotides –155 and –156. Interestingly, these RNAP-dependent protections were still observed when CRP HL159 was substituted for wild-type CRP (Figure 6B, lanes 5–9). These results are best explained by proposing that the other αCTD binds the –155/–164 region and that this binding does not involve any interaction with the AR1 surface element of CRP bound to site 3.
To verify that the complexes formed at malEp upon RNAP addition are open complexes regardless of the CRP variant present, the nucleoprotein complexes were probed with KMnO4, a reagent that reacts preferentially with unpaired thymine. The assays involving CRP HL159 were performed in the presence of a 3-fold higher concentration of RNAP to ensure equal protection of the core promoter (see Figure 6A, lanes 7 and 9). As shown in Figure 6C, RNAP binding to malEp resulted in the same KMnO4 hypersensitivity of the thymine at position –3 irrespective of whether malEp had been pre-incubated in the presence of MalT + wild-type CRP or MalT + CRP HL159. Thus, these results clearly indicate that the RNAP complexes analyzed were open complexes in both cases and that the open complexes formed in the presence of CRP HL159 have the same characteristic as those formed in the presence of wild-type CRP.
αCTD is involved in the RNAP-dependent protection observed upstream and downstream from CRP 3
To determine whether the protection observed at –120/ –125 and –155/–164 depends on αCTD, DNase I protection experiments were performed with reconstituted RNAPs containing wild-type α or an α variant lacking the C-terminal domain (αΔ235) (Hayward et al., 1991; Blatter et al., 1994). The concentration of αΔ235 RNAP was adjusted so as to give the same protection (∼50%) of the core promoter region and the same amount of open complexes as reconstituted wild-type RNAP (Figure 7A and C). The bottom and top strand patterns obtained when MalT and wild-type CRP were incubated in the absence or presence of the reconstituted holoenzymes showed that, unlike wild-type RNAP, αΔ235 RNAP binding did not lead to protection of the –120/–125 region (Figure 7A and B). Interestingly, protection of the –154/–165 region on the top strand also disappeared when αΔ235 RNAP was substituted for wild-type RNAP (Figure 7B). Altogether, these data thus indicate that the RNAP-dependent protection observed on both sides of CRP 3 involves αCTD.

Fig. 7. DNase I and KMnO4 footprinting of the complexes formed at malEp in the presence of reconstituted wild-type or mutant RNAP. (A) Bottom strand DNase I protection patterns obtained using the pOM53 EcoRI–KpnI fragment 3′ end-labeled on the bottom strand. (B) Top strand DNase I protection patterns obtained using the pOM53 PstI–EcoRI fragment 3′ end-labeled on the top strand. (C) KMnO4 attack patterns obtained using the pOM53 EcoRI–KpnI fragment 3′ end-labeled on the bottom strand.
Purified α and CRP cooperatively bind region –120/–125 and CRP site 3, respectively
As shown by Attey et al. (1994), cooperative binding of CRP and purified α can be detected by DNase I footprinting. To obtain direct evidence that protection of the –120/–125 region is due to αCTD binding to –120/ –125 and depends on an interaction between αCTD and one of the adjacently bound CRP molecules, I examined whether CRP alone, at a saturating concentration, promotes the binding of purified α to region –120/–125. As shown by Figure 8A (lanes 6–9), α-dependent protection of region –120/–125 was observed on the bottom strand when the malEp model substrate was incubated in the presence of wild-type CRP and purified α. The protection pattern was similar to that induced by RNAP in the presence of MalT and wild-type CRP (Figure 8A, compare lanes 1–4 with lanes 6–9). Moreover, protection of region –120/–125 was strongly reduced when purified α was incubated in the presence of CRP HL159 (Figure 8A, lanes 10–12). Similar protection experiments were also performed in the presence of subsaturating concentrations of CRP. Figure 8B shows the top strand patterns obtained. As observed for the bottom strand, purified α protected region –120/–125 in the presence of wild-type CRP but not in the presence of CRP HL159. Most importantly, α binding to –120/–125 was accompanied by a better protection of CRP site 3 and a lower occupation of CRP site 2, an effect that was not observed with CRP HL159. This observation provides clear evidence that α binding to –120/–125 does not involve CRP 2, but involves a direct contact between α and the AR1 region of the CRP 3 downstream subunit. The lower occupancy of CRP site 2 observed in the presence of wild-type CRP and purified α is most likely due to the fact that CRP 1 promotes α binding to DNA upstream of CRP site 1, as suggested by the observation that addition of purified α to wild-type CRP led to clear protection of the –97/–100 region, i.e. the proximal half of CRP site 2 (Figure 8B, lanes 1–4) [note that CRP affinity for CRP site 1 is 6-fold higher than its affinity for CRP site 2 (Vidal-Ingigliardi and Raibaud, 1991)].

Fig. 8. Recruitment of purified α by CRP. (A) Bottom strand DNase I protection patterns obtained using the pOM53 PstI–EcoRI fragment 3′ end-labeled on the bottom strand. Lanes 1–5: the DNase I footprinting assays were performed as described in Materials and methods in the presence of 75 nM wild-type CRP or 300 nM CRP HL159. Lanes 6–12: the assays were performed in a low ionic strength buffer (36 mM HEPES–KOH pH 8.0, 40 mM potassium acetate, 10 mM magnesium acetate, 1 mM CaCl2, 1 mM DTT, 100 µg/ml acetylated BSA, 0.2 mM cAMP) in the presence of 150 nM wild-type CRP or 460 nM CRP HL159. (B) Top strand DNase I protection patterns obtained using the pOM53 PstI–EcoRI fragment 3′ end-labeled on the top strand. The assays were performed as described in Materials and methods in the presence of 75 nM wild-type CRP or 300 nM CRP HL159. In both (A) and (B), the concentrations of purified α used were 1.2 µM when noted ×1 and 3.7 µM when noted ×3.
Discussion
The work presented here provides a clear understanding of the mechanism whereby MalT and CRP co-activate malEp. It establishes that, besides its structural role in the stabilization of the activation complex formed by MalT, CRP participates directly in malEp activation via a class I mechanism and that this function is carried out by the molecule of CRP bound to site 3. Two observations support the conclusion that the role of CRP involves direct protein–protein interactions in addition to its architectural function. First, both in vivo and in vitro, an IHF-stabilized activation complex activates malEp only very weakly while it activates the malKp promoter almost as effectively as a CRP-stabilized activation complex. Secondly, the HL159 substitution in CRP, which is located at the surface of the protein and does not affect its ability to bind and bend its DNA recognition motif (Zhou et al., 1993), strongly impairs malEp activation in vitro while it scarcely has any effect on malKp activation. Several lines of evidence indicate that this additional role of CRP is carried out by the molecule of CRP bound to site 3 and that it consists of the recruitment of αCTD to the –120/–125 site via a direct contact between αCTD and the AR1 region of CRP 3: (i) RNAP binding to the activation complex leads to protection against DNase I attack of the –120/–125 region; (ii) the HL159 substitution in CRP or deletion of the C-terminal domain of the α subunit cause the disappearance of the RNAP-dependent protection of region –120/–125; (iii) wild-type CRP and purified α bind CRP site 3 and region –120/–125 cooperatively in the absence of any other protein; and (iv) the HL159 substitution in CRP suppresses this cooperative effect. Thus, the action of CRP 3 has all of the characteristic features of a class I mechanism (Busby and Ebright, 1999): CRP 3 operates from a distal site; it promotes α binding to a DNA sequence located downstream from the CRP binding site via a contact between the AR1 determinant of CRP and the α subunit; and it facilitates RNAP recruitment, as inferred from the fact that disruption of the AR1 region causes a stronger defect in malEp activation at low RNAP concentration than at high RNAP concentration.
It might seem surprising that the molecule of CRP bound to site 3, which is centered at –139.5, can act as a class I activator although its binding site is not located on the proper face of the DNA helix and is much further away from the RNAP site than classically observed for a class I mechanism. Class I activation by CRP indeed requires that the CRP site be located on the same face of the DNA helix as the core RNAP site, i.e. its binding site has to be centered near positions –61.5, –72.5, –82.5, –92.5. In addition, the most distal position producing some stimulatory effect is –92.5 (Gaston et al., 1990; Ushida and Aiba, 1990). However, the path followed by the DNA double helix within the higher order structure formed by MalT and CRP at malEp most likely allows CRP 3 to interact productively with one of the αCTD. In particular, the two CRP-induced bends interposed between CRP site 3 and the RNAP site probably bring CRP site 3 closer to the RNAP site. As shown by Belyaeva et al. (1998), the presence of a proximal CRP binding site at –41.5 indeed increases the distance from which CRP can act as a class I activator by two helix turns. The CRP-induced DNA unwinding (Douc-Rasy et al., 1989) might also allow for a reorientation of the DNA segment carrying region –120/ –125 and CRP site 3 with respect to the core promoter, and compensate for the improper phasing between CRP site 3 and the core promoter sequence.
This work also revealed RNAP-dependent protections just upstream from CRP site 3, at –155/–164. While this effect clearly depends on the presence of the C-terminal domain of α, it does not depend on the AR1 region of CRP. These protections might result from the binding of the second copy of αCTD to region –155/–164. The sequence of this region does not display any obvious similarity to the consensus of UP element subsites (Estrem et al., 1999), but, as suggested by the DNase I hypersensitivity at –155 that characterizes the activation complex, the DNA double helix might present some local structural features that favor αCTD binding. Binding of the second copy of αCTD to DNA just upstream from the CRP site has also been observed at the lac promoter, a simple class I promoter, by Kolb et al. (1993) and Naryshkin et al. (2000), but in this case it is not known whether αCTD binding to upstream DNA is independent of the AR1 determinant of the upstream CRP subunit.
In conclusion, the mechanism underlying the synergistic action of MalT and CRP at malEp follows a complex and new scenario in which the second activator both assists the primary activator and acts as a class I activator. Such a scheme corresponds to a combination of the two scenarios so far described. A similar strategy might also be at the basis of araBAD promoter co-activation by AraC and CRP: CRP indeed both facilitates the formation of an activation complex by AraC by breaking the repression loop and participates directly in the activation of the araBAD promoter, most likely as a class I activator (Lobell and Schleif, 1991; Zhang and Schleif, 1998). Given the flexibility conferred by the flexible linker tethering αCTD to RNAP, one might predict the existence of even more complex combinations of the two basic synergistic strategies already described, such as one in which two activators acting independently upon RNAP are each assisted by another regulatory protein, helping them to form productive complexes with promoter DNA.
The large size of the higher order structure responsible for malEp and malKp activation, which is made of at least five MalT protomers and three molecules of CRP, i.e. ≥640 kDa of protein, makes malEp and malKp unique among the regulated prokaryotic promoters so far described. Through direct contacts with RNAP mediated by MalT 1 (Danot et al., 1996) and CRP 3 (this work), the activation complex recruits RNAP to malEp and promotes initiation of transcription, turning the inert malEp promoter into a promoter stronger than the synthetic tac promoter (my unpublished data). It is worth noting that malEp activation represents a highly integrated process wherein the functions of the components of the activation complex are multiple and interrelated, e.g. CRP 3 both stabilizes the activation complex and interacts with the RNAP α subunit, while CRP 1 and 2 stabilize the activation complex and most likely also facilitate the interaction between CRP 3 and αCTD. Finally, one should emphazise that, in some respects, the activation complex operating at malEp–malKp resembles the eukaryotic enhanceosomes whose assembly also involves cooperative binding of several transcriptional activators as well as architectural proteins and that recruit general transcription factors and RNA polymerase II holoenzyme to the promoter through specific protein–protein contacts (Giese et al., 1995; Kim and Maniatis, 1997; Kim et al., 1998).
Materials and methods
Strains and plasmids
pop2150 is a derivative of MC4100 (Casadaban, 1976) containing the ΔmalA510 deletion, which removes the beginning of the malT gene (Raibaud et al., 1983). pop2226 is a derivative of MC4100 carrying malPp201, a promoter-down mutation of the malPQ operon (O.Raibaud, unpublished). pop4103 is a Δ[hip]::cat derivative of pop2226 (Richet and Søgaard-Andersen, 1994).
All of the malEp–malKp variants used here are derivatives of the 478 bp EcoRI–EcoRI fragment of pOM18 that carries the wild-type malEp-malKp region (Raibaud et al., 1989). M13malEK1 is an M13mp11 derivative containing the 478 bp EcoRI–EcoRI malEp–malKp fragment from pOM18 (Vidal-Ingigliardi and Raibaud, 1991) inserted into the EcoRI site of M13mp11. pOM53, a pSB118 derivative containing the malEpΔ259 malEpKpI3 variant (from +38 to –259 with respect to malEp), was constructed as follows. A DNA fragment extending from +38 (malEp) to +8 (malKp) was obtained by PCR using the M13mal EK1malEpKpI3 variant as template and primers EK1 (5′-GGTCCT TGTTGGTGAAG-3′) and EK70 (5′-TTCATGATGGGCTGGCTAGC CGTCATCAGGAGAT-3′), which replaces the ‘–10’ sequence of malKp by an NheI site. The PCR DNA fragment was made blunt ended, cut by NheI, and cloned between the HincII and XbaI sites of pSB118, yielding pOM53. The construct was verified by sequencing. pOM72malEpΔ261 and pOM73malKpΔ240 contain the malEpΔ261 variant of malEp and the malKpΔ240 variant of malKp, respectively, inserted upstream of the rrnBT1 terminators of pOM70 (Richet and Raibaud, 1991). pOM82 is a derivative of pJEL126 (Valentin-Hansen et al., 1986) containing the wild-type 478 bp EcoRI–EcoRI malEp–malKp fragment inserted into the EcoRI site of pJEL126, with malEp fused to the promoterless lacZYA operon present on the vector (Richet, 1996). pOM91 is a derivative of pOM90 containing the wild-type EcoRI–EcoRI malEp–malKp fragment inserted into the EcoRI site between the divergent rpoC and the rrnBT1 terminators, with malEp upstream of the rpoC terminator and malKp upstream of rrnBT1 (Richet and Søgaard-Andersen, 1994). Derivatives of pOM82 and pOM91 containing the malEpKp1, -2, -3, -4 mutations or the V94 variant (Vidal-Ingigliardi and Raibaud, 1991; Richet and Søgaard-Andersen, 1994) were obtained by cloning the mutated EcoRI–EcoRI malEp–malKp fragment into pJEL126 and pOM90, respectively.
β-galactosidase assays
Strains pop2226 and pop4103 harboring derivatives of pOM82 were grown in M63 minimal medium (Miller, 1972) supplemented with 1 µg/ml thiamine, 30 µg/ml ampicillin, 0.5% glycerol and, when indicated, 0.4% maltose. The cultures were inoculated to a low cell density (A600 ∼4–9 × 10–4) and grown at 30°C up to an A600 of 0.5–0.9. β-galactosidase was assayed at 28°C as described by Miller (1972), using chloroform and 0.002% SDS to disrupt the cells. Enzyme activities, expressed in Miller units, are the average of assays performed in duplicate on at least two independent cultures and corrected for the background level [324 U, as determined by measuring the amount of β-galactosidase made by strain pop2150 (pOM82)]. The variations observed between two independent experiments were <20%.
Proteins
ATP-free MalT was prepared as described (Schreiber and Richet, 1999). Pure IHF from E.coli was a gift from H.Nash (NIH, Bethesda). Wild-type CRP and CRP HL159 were purified as described (Richet and Raibaud, 1991). Although both preparations of CRP were 30% active in titration assays with a DNA fragment containing CRP site 1, a 3- to 4-fold higher concentration of CRP HL159 had to be used in abortive initiation assays and footprinting assays so as to have the CRP sites occupied to the same extent by both proteins. This difference is most likely due to the fact that the HL159 substitution slightly decreases the affinity of CRP for its DNA site (Zhou et al., 1993).
Native RNA polymerase holoenzyme was purified as described by Hager et al. (1990) except that the cells were broken with a French press and the chromatography on a MonoQ column replaced by chromatography on a Poros HQ/M column (Boehringer Mannheim). Reconstituted RNAP containing His-tagged wild-type α or αΔ235 was prepared as described by Tang et al. (1995) and further purified by chromatography on a MonoQ PC 1.6/5 column (Pharmacia Biotech). As indicated by transcription assays performed with the lacUV5 promoter, the reconstituted αΔ235 RNAP was ∼2.5-fold less active than the reconstituted wild-type enzyme.
Purified His-tagged α subunit was obtained by preparing denatured recombinant protein from strain BL21 (DE3) (pHTT7f1-NHα) as described (Tang et al., 1995). After chromatography on Ni2+-NTA agarose, the α subunit was renatured by dialysis using the same procedure as for RNA polymerase reconstitution (Tang et al., 1995) and further purified by anion-exchange chromatography on a Poros HQ/M column (Boehringer Mannheim) to remove contaminating nucleases. The protein was concentrated by dialysis against a buffer containing 20 mM Tris–HCl pH 8.0, 0.2 M NaCl, 0.1 mM EDTA, 0.1 mM dithiothreitol (DTT) and 50% glycerol, and stored at –20°C.
Single-round transcription assays
Supercoiled plasmid DNA (0.25 nM), purified in a CsCl–ethidium bromide gradient, was pre-incubated at 20°C for 20 min in the presence of MalT and CRP (or IHF) in 18 µl containing 36 mM HEPES–KOH pH 8.0, 200 mM potassium acetate, 10 mM magnesium acetate, 1 mM DTT, 100 µg/ml acetylated bovine serum albumin (BSA), 1 mM maltotriose, 0.1 mM ATP and 0.2 mM cAMP. Two microliters of native RNAP were added and open complex formation was allowed to proceed for 2 min. Two microliters of a solution containing 1 mg/ml heparin (Sigma, H-0880), ATP, GTP and CTP (2 mM each), and 0.1 mM [α-32P]UTP (5 Ci/mmol) were then added to block open complex formation and to allow RNA synthesis. The reaction was quenched 10 min later by adding 80 µl of a solution containing 0.375 M sodium acetate, 12.5 mM EDTA and 12 µg of sonicated plasmid DNA. RNA was deproteinized and ethanol precipitated before electrophoresis through a denaturing polyacrylamide gel. After drying, the gel was autoradiographed using Kodak XAR-5 film without a screen. The relative amounts of transcripts were quantified by scanning the gel on a phosphoimager. Under these conditions, the amounts of open complexes made at malEp and malKp after 2 min incubation in the presence of RNAP represented 50–75% of the maximum.
Abortive initiation assays
ATP-free MalT (175 nM) and CRP (75 nM wild-type protein or 300 nM CRP HL159) were pre-incubated at 30°C for 20 min in 18 µl of 36 mM HEPES–KOH pH 8.0, 100 mM potassium acetate, 10 mM magnesium acetate, 1 mM DTT, 100 µg/ml acetylated BSA, 1 mM maltotriose, 0.1 mM dATP, 0.2 mM cAMP, 1 nM DNA fragment [dATP is as effective as ATP in promoting activation of transcription by MalT in the presence of maltotriose (Richet and Raibaud, 1989)]. Two microliters of a RNAP solution were then added and the mixture incubated for varying times at 30°C. The synthesis of abortive products (GpUpUpU) was initiated by adding 2 µl containing 5 mM GpU, 0.5 mM [α-32P]UTP (0.6 Ci mmol–1) and 500 µg/ml heparin, and allowed to proceed for 30 min. The reaction products were separated from free [α-32P]UTP by chromatography on Whatman 3MM paper as described by McClure (1980). The chromatograms were dried and scanned on a phosphoimager to quantify the amount of GpUpUpU formed. The DNA template used was a 355 bp malEp–malKp DNA fragment containing the malEpKpI3 malEpKp70 mutations and extending from +73 (malEp) to +8 (malKp). It was synthesized by PCR on the M13malEK1malEpKpI3 template using primers EK9 (5′-GGATGCGTGCACCTGTT-3′) and EK70 (see above). The DNA fragment was then purified using the PCR product purification kit from Boehringer Mannheim and its concentration was determined as described (Richet and Raibaud, 1989).
DNase I footprinting
Footprinting experiments were performed with the EcoRI–KpnI or PstI–EcoRI fragment from pOM53, which contains the malEpKpI3 malEpΔ259 derivative of malEp. The DNA fragments were purified by electrophoresis on a polyacrylamide gel and specifically end-labeled at their EcoRI end by filling in with the Klenow fragment of DNA polymerase I in the presence of dTTP and [α-32P]dATP, followed by ethanol precipitation.
In experiments involving native RNAP or purified α, the labeled DNA fragment (<1 nM) was pre-incubated for 15 min at 30°C in the presence of 175 nM MalT and 75 nM wild-type CRP (or 300 nM CRP HL159) in 18 µl of the reaction mixture used for the abortive initiation assays and supplemented with 1 mM CaCl2. Two microliters of native RNAP or purified α were then added and the mixture incubated for 15 min. DNase I attack was performed by adding 2 µl of a DNase I solution and further incubating the mixture for 1 min. Digestion was stopped and the samples processed as described for in vitro transcription assays. After electrophoresis, the gel was dried and autoradiographed on Kodak XAR-5 film without an intensifying screen except for the gel shown in Figure 7A.
The assays involving reconstituted RNAP were performed with a low ionic strength buffer (7 mM Tris–HCl pH 7.7, 18 mM HEPES–KOH pH 8.0, 5 mM potassium acetate, 40 mM NaCl, 3 mM magnesium acetate, 1 mM CaCl2, 1 mM DTT, 300 µg/ml acetylated BSA, 1 mM maltotriose, 0.1 mM dATP, 0.2 mM cAMP) supplemented with 7.5% polyethylene glycol 6000. Labeled DNA was pre-incubated for 10 min at 30°C in the presence of 20 nM CRP and 30 nM MalT; reconstituted RNAP (75 nM wild-type form or 600 nM αΔ235 form) was added and the mixture further incubated for 5 min before adding DNase I.
KMnO4 attack
The assays were performed under the same conditions as for DNase I footprinting, except that after complex formation with RNAP, 2 µl of 66 mM KMnO4 were added and the mixture further incubated for 30 s at 30°C. The reaction was quenched by adding 2 µl of 2.3 M β-mercapto ethanol and 80 µl of the stop solution used for in vitro transcription assays. After deproteinization and ethanol precipitation, DNA was resuspended in 100 µl of 10% piperidine, heated for 30 min at 90°C and vacuum dried. DNA was then resuspended in 50 µl of water, vacuum dried, resuspended in 50 µl of water and vacuum dried before electrophoresis through a denaturing polyacrylamide gel.
Acknowledgments
Acknowledgements
I am grateful to O.Danot and A.Kolb for stimulating discussions and their critical reading of the manuscript. I am obliged to A.Kolb for purified protein CRP HL159 and to H.Nash for purified IHF. I also thank T.Pugsley for his comments on the manuscript.
References
- Attey A., Belyaeva,T., Savery,N., Hoggett,J., Fujita,N., Ishihama,A. and Busby,S. (1994) Interactions between the cyclic AMP receptor protein and the α subunit of RNA polymerase at the Escherichia coli galactose operon P1 promoter. Nucleic Acids Res., 22, 4375–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaeva T.A., Rhodius,V.A., Webster,C.L. and Busby,S.J. (1998) Transcription activation at promoters carrying tandem DNA sites for the Escherichia coli cyclic AMP receptor protein: organisation of the RNA polymerase α subunits. J. Mol. Biol., 277, 789–804. [DOI] [PubMed] [Google Scholar]
- Belyaeva T.A., Wade,J.T., Webster,C.L., Howard,V.J., Thomas,M.S., Hyde,E.I. and Busby,S.J. (2000) Transcription activation at the Escherichia coli melAB promoter: the role of MelR and the cyclic AMP receptor protein. Mol. Microbiol., 36, 211–222. [DOI] [PubMed] [Google Scholar]
- Blatter E.E., Ross,W., Tang,H., Gourse,R.L. and Ebright,R.H. (1994) Domain organization of RNA polymerase α subunit: C-terminal 85 amino acids constitute a domain capable of dimerization and DNA binding. Cell, 78, 889–896. [DOI] [PubMed] [Google Scholar]
- Boos W. and Shuman,H. (1998) Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol. Mol. Biol. Rev., 62, 204–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby S. and Ebright,R.H. (1999) Transcription activation by catabolite activator protein (CAP). J. Mol. Biol., 293, 199–213. [DOI] [PubMed] [Google Scholar]
- Busby S., West,D., Lawes,M., Webster,C., Ishihama,A. and Kolb,A. (1994) Transcription activation by the Escherichia coli cyclic AMP receptor protein—receptors bound in tandem at promoters can interact synergistically. J. Mol. Biol., 241, 341–352. [DOI] [PubMed] [Google Scholar]
- Casadaban M.J. (1976) Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage Lambda and Mu. J. Mol. Biol., 104, 541–555. [DOI] [PubMed] [Google Scholar]
- Danot O., Vidal-Ingigliardi,D. and Raibaud,O. (1996) Two amino acid residues from the DNA-binding domain of MalT play a crucial role in transcriptional activation. J. Mol. Biol., 262, 1–11. [DOI] [PubMed] [Google Scholar]
- Douc-Rasy S., Kolb,A. and Prunell,A. (1989) Protein-induced unwinding of DNA: measurement by gel electrophoresis of complexes with DNA minicircles. Application to restriction endonuclease EcoRI, catabolite gene activator protein and lac repressor. Nucleic Acids Res., 17, 5173–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrem S.T., Ross,W., Gaal,T., Chen,Z.W., Niu,W., Ebright,R.H. and Gourse,R.L. (1999) Bacterial promoter architecture: subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase α subunit. Genes Dev., 13, 2134–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston K., Bell,A., Kolb,A., Buc,H. and Busby,S. (1990) Stringent spacing requirements for transcription activation by CRP. Cell, 62, 733–743. [DOI] [PubMed] [Google Scholar]
- Giese K., Kingsley,C., Kirshner,J.R. and Grosschedl,R. (1995) Assembly and function of a TCR α enhancer complex is dependent on LEF-1-induced DNA bending and multiple protein–protein interactions. Genes Dev., 9, 995–1008. [DOI] [PubMed] [Google Scholar]
- Hager D.A., Jin,D.J. and Burgess,R.R. (1990) Use of Mono Q high-resolution ion-exchange chromatography to obtain highly pure and active Escherichia coli RNA polymerase. Biochemistry, 29, 7890–7894. [DOI] [PubMed] [Google Scholar]
- Hayward R.S., Igarashi,K. and Ishihama,A. (1991) Functional specialization within the α-subunit of Escherichia coli RNA polymerase. J. Mol. Biol., 221, 23–29. [DOI] [PubMed] [Google Scholar]
- Ishihama A. (1992) Role of the RNA polymerase α subunit in transcription activation. Mol. Microbiol., 6, 3283–3288. [DOI] [PubMed] [Google Scholar]
- Joung J.K., Le,L.U. and Hochschild,A. (1993) Synergistic activation of transcription by Escherichia coli cAMP receptor protein. Proc. Natl Acad. Sci. USA, 90, 3083–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J.K., Koepp,D.M. and Hochschild,A. (1994) Synergistic activation of transcription by bacteriophage λ cI protein and E.coli cAMP receptor protein. Science, 265, 1863–1866. [DOI] [PubMed] [Google Scholar]
- Kim T.K. and Maniatis,T. (1997) The mechanism of transcriptional synergy of an in vitro assembled interferon-β enhanceosome. Mol. Cell, 1, 119–129. [DOI] [PubMed] [Google Scholar]
- Kim T.K., Kim,T.H. and Maniatis,T. (1998) Efficient recruitment of TFIIB and CBP-RNA polymerase II holoenzyme by an interferon-β enhanceosome in vitro. Proc. Natl Acad. Sci. USA, 95, 12191–12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb A., Igarashi,K., Ishihama,A., Lavigne,M., Buckle,M. and Buc,H. (1993) E.coli RNA polymerase, deleted in the C-terminal part of its α-subunit, interacts differently with cAMP–CRP complex at the lacP1 and at the galP1 promoter. Nucleic Acids Res., 21, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobell R.B. and Schleif,R.F. (1991) AraC-DNA looping: orientation and distance-dependent loop breaking by the cyclic AMP receptor protein. J. Mol. Biol., 218, 45–54. [DOI] [PubMed] [Google Scholar]
- Lonetto M.A., Rhodius,V., Lamberg,K., Kiley,P., Busby,S. and Gross,C. (1998) Identification of a contact site for different transcription activators in region 4 of the Escherichia coli RNA polymerase σ70 subunit. J. Mol. Biol., 284, 1353–1365. [DOI] [PubMed] [Google Scholar]
- McClure W.R. (1980) Rate-limiting steps in RNA chain initiation. Proc. Natl Acad. Sci. USA, 77, 5634–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- Naryshkin N., Revyakin,A., Kim,Y., Mekler,V. and Ebright,R. (2000) Structural organization of the RNA polymerase–promoter open complex. Cell, 101, 601–611. [DOI] [PubMed] [Google Scholar]
- Raibaud O., Débarbouillé,M. and Schwartz,M. (1983) Use of deletions created in vitro to map transcriptional regulatory signals in the malA region of Escherichia coli. J. Mol. Biol., 163, 395–408. [DOI] [PubMed] [Google Scholar]
- Raibaud O., Vidal-Ingigliardi,D. and Richet,E. (1989) A complex nucleoprotein structure involved in activation of transcription of two divergent Escherichia coli promoters. J. Mol. Biol., 205, 471–485. [DOI] [PubMed] [Google Scholar]
- Rhodius V.A. and Busby,S.J. (1998) Positive activation of gene expression. Curr. Opin. Microbiol., 1, 152–159. [DOI] [PubMed] [Google Scholar]
- Richet E. (1996) On the role of the multiple regulatory elements involved in the activation of the Escherichia coli malEp promoter. J. Mol. Biol., 264, 852–862. [DOI] [PubMed] [Google Scholar]
- Richet E. and Raibaud,O. (1989) MalT, the regulatory protein of the Escherichia coli maltose system, is an ATP-dependent transcriptional activator. EMBO J., 8, 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richet E. and Raibaud,O. (1991) Supercoiling is essential for the formation and stability of the initiation complex at the divergent malEp and malKp promoters. J. Mol. Biol., 218, 529–542. [DOI] [PubMed] [Google Scholar]
- Richet E. and Søgaard-Andersen,L. (1994) CRP induces the repositioning of MalT at the Escherichia coli malKp promoter primarily through DNA bending. EMBO J., 13, 4558–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richet E., Vidal-Ingigliardi,D. and Raibaud,O. (1991) A new mechanism for coactivation of transcription initiation: repositioning of an activator triggered by the binding of a second activator. Cell, 66, 1185–1195. [DOI] [PubMed] [Google Scholar]
- Schreiber V. and Richet,E. (1999) Self-association of the Escherichia coli transcription activator MalT in the presence of maltotriose and ATP. J. Biol. Chem., 274, 33220–33226. [DOI] [PubMed] [Google Scholar]
- Schultz S.C., Shields,G.C. and Steitz,T.A. (1991) Crystal structure of a CAP–DNA complex: the DNA is bent by 90°. Science, 253, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Schwartz M. (1987) The maltose regulon. In Neidhardt,F.C., Ingraham,J.L., Low,K.B., Magasanik,B., Schaechter,M. and Umbarger,H.E. (eds), Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. American Society for Microbiology, Washington, DC, pp. 1482–1502. [Google Scholar]
- Scott S., Busby,S. and Beacham,I. (1995) Transcriptional co-activation at the ansB promoters: involvement of the activating regions of CRP and FNR when bound in tandem. Mol. Microbiol., 18, 521–531. [DOI] [PubMed] [Google Scholar]
- Tang H., Severinov,K., Goldfarb,A. and Ebright,R.H. (1995) Rapid RNA polymerase genetics: one-day, no-column preparation of reconstituted recombinant Escherichia coli RNA polymerase. Proc. Natl Acad. Sci. USA, 92, 4902–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushida C. and Aiba,H. (1990) Helical phase dependent action of CRP: effect of the distance between the CRP site and the –35 region on promoter activity. Nucleic Acids Res., 18, 6325–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Hansen P., Albrechtsen,B. and Løve-Larsen,J.E. (1986) DNA–protein recognition: demonstration of the three genetically separated operator elements that are required for repression of the Escherichia coli deoCABD promoters by the DeoR repressor. EMBO J., 5, 2015–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Ingigliardi D. and Raibaud,O. (1991) Three adjacent binding sites for cAMP receptor protein are involved in the activation of the divergent malEp-malKp promoters. Proc. Natl Acad. Sci. USA, 88, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Ingigliardi D., Richet,E. and Raibaud,O. (1991) Two MalT binding sites in direct repeat. A structural motif involved in the activation of all the promoters of the maltose regulons in Escherichia coli and Klebsiella pneumoniae. J. Mol. Biol., 218, 323–334. [DOI] [PubMed] [Google Scholar]
- Zhang X. and Schleif,R. (1998) Catabolite gene activator protein mutations affecting activity of the araBAD promoter. J. Bacteriol., 180, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zhang,X. and Ebright,R.H. (1993) Identification of the activating region of catabolite gene activator protein (CAP): Isolation and characterization of mutants of CAP specifically defective in transcription activation. Proc. Natl Acad. Sci. USA, 90, 6081–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Merkel,T.J. and Ebright,R.H. (1994) Characterization of the activating region of Escherichia coli catabolite gene activator protein (CAP). II. Role at class I and class II CAP-dependent promoters. J. Mol. Biol., 243, 603–610. [DOI] [PubMed] [Google Scholar]