

Abstract
We exploited the differential activation of hypoxia-inducible factor (HIF)-dependent gene expression in tumors versus normal tissue for the design of a targeted oncolytic Herpes simplex virus type-1 (HSV-1). A gene that is essential for viral replication, ICP4, was placed under the regulation of a HIF-responsive promoter and then introduced into the thymidine kinase locus (UL23) of HSV d120 which contains partial deletions in the two endogenous ICP4 genes. Recombinant HIF-HSV were isolated and their derivation from d120 was verified by expression of a truncated, nonfunctional form of ICP4 protein. Disruption of the UL23 locus was confirmed by loss of thymidine kinase expression and resistance to acyclovir. Unexpectedly, HIF-HSV expressed ICP4 and induced tumor cell lysis at similar levels under normoxia and hypoxia. The lack of HIF-dependent ICP4 transgene expression by HIF-HSV was due to two factors that have not previously been reported- reversion of the ICP4 gene region to its wild-type configuration and increased HIF-transcriptional activity under normoxia when cells were infected with any strain of HSV-1. The findings that an oncolytic HSV-1 is genetically unstable and can activate a tumor-related promoter in a non-specific manner have important implications for any proposed use of this virus in cancer therapy.
Keywords: hypoxia, hypoxia-inducible factor (HIF), herpes simplex virus, HSV, oncolytic, ICP4
Introduction
The hypoxia-inducible factor (HIF) pathway is activated in many human primary tumors, metastases, and cancer stem cells 1-4. This is associated with increased patient mortality and resistance to chemo- and radio-therapies 5,6. In contrast, the HIF pathway is inhibited in normal, healthy tissues. This provides an opportunity to develop novel anti-cancer therapeutics. HIF is a heterodimeric transcription factor composed of an alpha- and beta-subunit 4. Normal tissues have undetectable levels of the alpha subunit due to its rapid ubiquitination and proteasomal degradation while the beta subunit is constitutively present 7. In tumors, the alpha subunit is stabilized and overexpressed in response to (i) intratumoral hypoxia and (ii) tumor-specific dysregulation of oncogene and tumor suppressor gene pathways. HIF regulates target gene expression by binding to a HIF-responsive DNA element (HRE) 4. Many HIF-target genes have key functions in tumor biology including tumor cell proliferation, tumor cell survival, angiogenesis, invasion/metastasis, and glycolytic metabolism. The ability of HIF to activate a protumorigenic phenotype has led to intense efforts to identify agents that will inhibit its activity. One drawback to the anti-HIF agents discovered to date is that they function indirectly by targeting cellular pathways that regulate HIF 6. Thus, these agents have multiple cellular targets which could lead to unwanted toxicity in normal tissues. This necessitates the continued development of HIF inhibitors with greater specificity.
Oncolytic Herpes Simplex Virus type-1 (HSV-1) is a promising cancer therapy approach which utilizes the cytolytic replication cycle of the virus to specifically kill tumor cells (oncolysis) 8,9. A wild-type HSV-1 cannot be used for cancer therapy because it lacks tumor-specificity and is toxic to normal healthy tissues. To overcome this, the virus has been genetically redesigned such that its cytolytic replication cycle is specifically targeted to tumor cells. One strategy involves transcriptional regulation of infected cell polypeptide 4 (ICP4) 10. ICP4 is essential for the initiation of HSV gene expression and null mutants do not undergo viral replication 11. Two strategies have been employed to transcriptionally regulate ICP4, which is present in two copies per HSV genome. One strategy involves replacing endogenous ICP4 transcriptional regulatory elements with other regulatory elements. This approach has been largely unsuccessful [reviewed in 12]. A second strategy involves inserting into an ICP4 null HSV-1 an exogenous ICP4 under the regulation of a promoter that is preferentially active in tumor cells compared to normal tissues. This second strategy has been more successful. In the G92A 12,13, d12.CALP 14, bM24-TE 15, and CEAICP4 16 oncolytic HSVs, transcriptional regulation of an exogenous ICP4 gene was accomplished using albumin, calponin, Wnt/T-cell factor (Tcf), and carcinoembryonic antigen (CEA) promoters, respectively. This led to selective virus replication and associated cytolysis against hepatocellular (G92A), soft tissue & bone tumors (d12.CALP), and colorectal and hepatoblastomas (bM24-TE). While these three viruses demonstrated anti-tumor activity against their respective tumor types, they are limited in that (i) these tissue-specific promoters are active in some normal cells and (ii) viral replication is primarily targeted to only a specific tumor type and this is further restricted to only a subgroup. CEAICP4 can be potentially used against a broader range of tumor types since CEA is overexpressed in a wide variety of epithelial cancers. However, the replication of CEAICP4 is greatly attenuated even in several CEA-positive cell lines 16. bM24-TE 15 and G92A 12 also exhibited attenuated replication in permissive cells. An alternative approach is to use a viral, rather than cellular, promoter to regulate ICP4 expression. HSPV-1 contains the human papillomavirus type-16 upstream regulatory region (URR16) promoter for the targeted expression of ICP4 in oral cancer 17. Unfortunately, viral replication did not correlate with the tumor origin or the presence of HPV sequences in the cell. This was attributed to altered transcriptional regulation of the URR16 promoter following HSV-1 infection. Therefore, there is a continued need to evaluate other promoters for their ability to support efficient and tumor-specific replication of an oncolytic HSV-1.
We previously developed HIF-responsive promoters which specifically and strongly induce target gene expression in HIF-active tumor cells 18. In this study, we evaluated whether these promoters could be utilized to create an oncolytic HSV-1 which is conditionally activated by HIF. For this, we introduced an ICP4 gene under the regulation of a HIF-responsive promoter into the HSV mutant d120, which contains partial deletions in both copies of the ICP4 gene.
Materials & Methods
Cells
Human LN229 glioma 19, human U251MG-T2 glioma 20, human LN319 glioma 19, monkey kidney Vero [American Type Culture Collection (ATCC) catalog #CCL-81, Manassas, VA], E5 (generously provided by Neil DeLuca, Univ. of Pittsburgh, PA), and N33 (a gift of Dr. PA Schaffer, Harvard Medical School, Boston, MA) cells were cultured in DMEM containing 10% FCS under normoxia. E5 cells stably express HSV-1 ICP4 and are derived from Vero cells as described in 11. N33 cells stably express HSV-2 ICP4 and are derived from Vero cells as described in 21. Normoxic (21% O2) and hypoxic (1% O2) cell culture conditions were generated as described in 20. LN229, U251MG-T2, and LN319 cells were selected because they have a hypoxia-activated HIF-pathway (22 and not shown, generously provided by Erwin Van Meir, Emory Univ., Atlanta, GA).
Viruses and generation of HIF-HSV
Wild-type HSV-1 (KOS strain, ATCC) was grown in Vero cells. The d120 HSV-1 mutant (KOS strain) contains a deletion in both copies of the ICP4 gene (kindly provided by Neil DeLuca, Univ. of Pittsburgh, PA) and was grown in E5 cells. Prior to use, all virus stocks were titered by a plaque assay using Vero (HSV-1) or E5 (d120, HIF-V6R-HSV, HIF-E6L-HSV) cells.
The E6L and V6R HIF-responsive promoters were derived from the pBI-E6L and pBI-V6R plasmids18. These plasmids contain a HIF-responsive promoter upstream of a multiple cloning site and can be used for targeted gene expression in HIF-active cells. The E6L and V6R promoters contain 6 tandem copies of the HIF-response elements from the erythropoietin or vascular endothelial growth factor genes, respectively. The cloning process for the generation of HIF-V6R-HSV and HIF-E6L-HSV required the creation of two SpeI restriction enzyme recognition sites in the pBI-V6R and pBI-E6L plasmids. A SpeI recognition sequence was created in pBI-V6R and pBI-E6L at the 3′ end of the SV40 polyadenylation sequence using the QuikChange Site-directed mutagenesis kit (Stratagene, La Jolla, CA). Primers for mutagenesis were Spemutf (5′-CCCTTTCGTCTTCACTAGTCGCTGGTCGAGCTG-3′) and Spemutr (5′-CAGCTCGACCAGCGACTAGTGAAGACGAAAGGG-3′). PCR conditions were one cycle at 95°C for 30 sec; sixteen cycles at 95°C for 30 sec, 55°C for 1 min, 68°C for 9 min; and then held at 4°C. Generation of the SpeI site was confirmed by restriction enzyme analysis. pBI-V6R and pBI-E6L were further modified to contain HindIII and SpeI recognition sequences at the 5′ end of the β-globin polyadenylation sequence. The single stranded oligonucleotides EHSlinkR (5′-ACTAGTAAGCTTGAT-3′) and EHSlinkf (5′-ATCAAGCTTACTAGT-3′) were mixed, heated to 95°C for 5 min, allowed to cool to room temperature to generate double stranded oligonucleotides, and then ligated into EcoRV-digested pBI-V6R and pBI-E6L plasmids. Insertion of the oligonucleotide and recreation of the EcoRV recognition site were confirmed by restriction enzyme analysis. All plasmids had more than one oligonucleotide insert based on DNA sequencing using the bglobinseq primer (5′-AACAACACCCTGAAAACTTT-3′). Extra inserts were removed by EcoRV restriction enzyme digestion, gel purification of the vector, and ligation. The presence of one oligonucleotide insert was confirmed by restriction enzyme analysis and DNA sequencing using the bglobinseq primer. The modified plasmids were called pBI-V6R-(double SpeI) and pBI-E6L-(double SpeI). The complete coding sequence for ICP4 was amplified by PCR with Pfu Turbo DNA polymerase (Stratagene) and DMSO (10% final concentration in PCR reaction) using wild-type HSV-1 template DNA and primers ICP4F (5′-CCGATCGTCCACACG-3′) and ICP4R (5′-TACTGCAAAACTTAATCAGG-3′). PCR conditions were one cycle at 95°C for 1 min.; thirty cycles at 95°C for 30 sec, 57°C for 30 sec, and 72°C for 5 min; one cycle at 72°C for 10 min; and then held at 4°C. The resulting 4.3 kb PCR product was gel purified and then cloned into the pCR-Blunt II-TOPO (Invitrogen) to generate pCR-Blunt II-TOPO-ICP4. The presence and orientation of ICP4 coding sequences in pCR-Blunt II-TOPO were confirmed by restriction endonuclease digestion and DNA sequencing using M13 forward and reverse primers. A 4.4 kb HindIII/XbaI fragment containing the ICP4 coding region from pCR-Blunt II-TOPO-ICP4 was ligated into pBI-V6R-(double SpeI) and pBI-E6L-(double SpeI) digested with HindIII/NheI to generate pBI-V6R-ICP4 and pBI-E6L-ICP4. A 5.4 kb SpeI fragment from pBI-V6R-ICP4 and pBI-E6L-ICP4 was ligated into XbaI digested pTKΔL [12, generously provided by Samuel Rabkin, Harvard Medical School, Boston, MA] to generate pTKΔL-V6R-ICP4 and pTKΔL-E6L-ICP4. To create recombinant HIF-V6R-HSV, E5 cells were transfected using Effectene (Qiagen, Valencia, CA) with HindIII-linearized pTKΔL-V6R-ICP4. Five hours later, cells were infected with d120 at multiplicity of infection (MOI) 1. Eighteen hours later, 75 μM acyclovir (ACV) was added to the media and the cells were incubated until plaques were observed. Virus was recovered from cells by three freeze-thaw cycles and grown on Vero cells under hypoxia with 75 μM ACV. Virus was amplified in E5 cells and then grown in (i) Vero cells under hypoxia with 75 μM ACV or (ii) E5 cells. Ten HIF-V6R-HSV plaques (clone numbers 6-15) were selected from the Vero plates and seven HIF-V6R-HSV plaques (clone numbers 16-22) were selected from the E5 plates. To create recombinant HIF-E6L-HSV, E5 cells were transfected using Effectene with HindIII-linearized pTKΔL-E6L-ICP4. Twenty-four hours later, cells were infected with d120 at MOI 1. Five hours later, 75μM ACV was added to the media and the cells were incubated until plaques were observed. Virus was recovered from cells by three freeze-thaw cycles and then grown in Vero cells under hypoxia with 75 μM ACV. Five HIF-E6L-HSV plaques were selected (clone numbers 1-5). All selected virus clones were amplified and titered.
ICP4 and thymidine kinase (TK) expression
LN229 and E5 cells were mock- or virus-infected at the MOI indicated in the figure legends. The cells were placed under normoxia or hypoxia. One day later, cells were collected in PBS using a cell scraper and then pelleted by microcentrifugation for 5 minutes at 2300 × g. Collected cells were lysed using lysing buffer [20 mM Sodium Phosphate, 5mM EDTA, 150mM NaCl, 50mM NaF, 1% Triton X-100, 1× protease inhibitor cocktail and 1× phosphatase inhibitor cocktail (Sigma, St. Louis, MO)]. Protein concentrations were determined using Protein Assay Dye (BioRad, Hercules, CA). Proteins were separated by electrophoresis using 10% Criterion Precast gels (BioRad) and blotted to PVDF membrane (BioRad). Molecular weight standards were Precision Plus Dual Color (BioRad) and/or Page Ruler Prestained ladder (Fermentas, Glen Burnie, MD). The primary antibodies were anti-HSV-1 ICP4 (Virusys, Taneytown, MD), anti-HSV-1 thymidine kinase (Santa Cruz Biotechnology, Santa Cruz, CA), anti-GAPDH (Calbiochem, La Jolla, CA) or anti-β-actin (Sigma, St. Loius, MO). The secondary antibodies were anti-mouse IgG + IgM alkaline phosphate linked whole antibody from goat (Amersham Biosciences, Piscataway, NJ) or AP-rabbit anti-goat IgG (H+L) conjugate (Zymed Laboratories, Carlsbad, CA). Signal was detected using ECF substrate (Amersham Biosciences). Membranes were stripped of signal using 1× Stripping buffer (Boston BioProducts, Worcester, MA) and then reprobed.
ACV-sensitivity and cytopathic effect (CPE) assays
For ACV-sensitivity assays, LN229 cells were mock- or virus- infected at MOI 0.05. Following infection, cells were grown in the absence or presence of 110 μM ACV under normoxia or hypoxia. For CPE assays, LN229, LN319, and U251MG-T2 cells were mock or virus infected at MOI 0.25 and then grown under normoxia or hypoxia. Following infection, cells were visually monitored for CPE (cell lysis/detachment) and then analyzed when extensive CPE was seen. Cells were fixed with ice cold 100% methanol, stained with crystal violet solution (1% crystal violet, 10% formaldehyde, 20% ethanol, in water) for 20 min, destained in water, and allowed to dry at room temperature. Photographs were taken at 100× magnification. The percent cytopathic effect was quantified by a 3-(4,5 dimethlyliazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) assay from three replicates/experimental condition. The values obtained with virus infection were normalized to mock infected cells. Percent CPE=[1-(average optical density of mock infected cells/average optical density of virus infected cells)] ×100.
PCR for endogenous ICP4 locus
N33 cells were mock- or virus- infected at MOI 0.5. DNA was extracted from cells with the Gentra Puregene genomic DNA isolation kit (Qiagen). The presence of amplifiable viral DNA was analyzed using RIGR-5′ (5′-CCTAGCACATGTGTGGGGTTAGGGCCGG-3′) and RIGR-3′ (5′-CGACCCACATGTTTACTTAAAAGGCGTG-3′) primers which amplify a 1053 bp region from the right intergenic region of HSV-1. The presence of a wild-type ICP4 gene locus was analyzed using ICP4 P-5′ (5′-GACTATATGAGCCCGAGGACG-3′) and ICP4-3′ (5′-CTCTGGTTGTCAAACAGCAGG-3′) primers. These primers amplify a 2418 bp region spanning from the ICP4 promoter to the ICP4 3′-coding region, a region that is deleted in d120. Amplification of DNA sequences were performed using the GC Rich PCR System (Roche, Mannheim, Germany). PCR conditions were one cycle at 95°C for 5 min.; forty cycles at 95°C for 30 sec, 55°C for 30 sec, and 70°C for 5 min; one cycle at 70°C for 20 min; and then held at 4°C. PCR reactions were run with 1kb and 100 bp molecular weight standards (BioRad) on 1% agarose gels containing ethidium bromide and were visualized by UV light.
HIF transcriptional activity
LN229 cells (2.25 × 105) were transfected as described in 18 using GenePORTER transfection reagent (Genlantis, San Diego, CA) with 1μg pBI-GL-V6R or pBI-GL-E6L which contain the luciferase reporter gene under the control of the V6R or E6L HIF-responsive promoters, respectively. The next day, cells were mock- or virus- (HSV-1, d120, HIF-V6R-HSV, or HIF-E6L-HSV) infected in triplicate at MOI 0.05, 0.25, or 1. Cells were placed under normoxia or hypoxia. One day later, luciferase activity was measured using a Luc-screen kit (Applied Biosystems, Foster City, CA) and normalized to total cellular protein (light units/μg protein). Data was statistically analyzed using a Students t test. A p<0.05 was considered to be significantly different.
Results
Construction of a HIF-activated HSV-1
We utilized the pTKΔL recombination plasmid and d120 virus to construct a recombinant HSV-1 that selectively replicates in HIF-active tumor cells (Figure 1). pTKΔL allows for the introduction of exogenous DNA sequences into the thymidine kinase locus (UL23, TK) of HSV-1 12. This results in the disruption of the TK gene and absence of TK protein expression by recombinant viruses. d120 is a replication-deficient HSV-1 that contains deletions in both copies of the endogenous ICP4 gene. Propagation of d120 is achieved by growth in ICP4-complementing cell lines, such as E5 11. To generate a HIF-activated HSV-1 (HIF-HSV), we introduced the ICP4 gene under the regulation of a HIF-responsive promoter into the TK locus of d120. Two independent HIF-responsive promoters, V6R and E6L, were chosen because of their ability to tightly regulate transgene expression under HIF-active conditions relative to normoxia 18. Multiple recombinant HIF-E6L-HSV (clone numbers 1-5) and HIF-V6R-HSV (clone numbers 6-22) were prepared from independent plaques and characterized further.
Figure 1. Schematic of HIF-HSV.

The HSV-1 ICP4 gene, which is essential for virus replication, was placed under the regulation of the V6R or E6L HIF-responsive promoters 18 and then cloned into pTKΔL 12 to generate pTKΔL-V6R-ICP4 and pTKΔL-E6L-ICP4 recombination plasmids. The ICP4 gene expression cassette was then inserted into the thymidine kinase (TK) gene region of the HSV-1 d210 genome (ICP4 gene deleted, white triangles) by homologous recombination in E5 cells to generate HIF-V6R-HSV and HIF-E6L-HSV.
d120 expresses a unique truncated (34kDa) nonfunctional form of ICP4 protein 11. Therefore, the origin of HIF-V6R-HSV and HIF-E6L-HSV from d120 was examined by western blotting for the 34kDa ICP4 protein using infected E5 cells (Figure 2). d120-infected cells served as a positive control and exhibited expression of the 34 kDa ICP4 protein. HIF-E6L-HSV (Figure 2a) and HIF-V6R-HSV (Figure 2b) also expressed the 34 kDa ICP4. In contrast, HSV-1- and mock- infected cells served as a negative control and displayed no expression of the truncated ICP4 protein. Similar results were obtained using infected LN229 cells (Supplemental Figure 1 and not shown). In summary, these results demonstrate that HIF-V6R-HSV and HIF-E6L-HSV are derived from d120.
Figure 2. ICP4 (34kDa) and thymidine kinase (TK) expression by HIF-E6L-HSV and HIF-V6R-HSV.
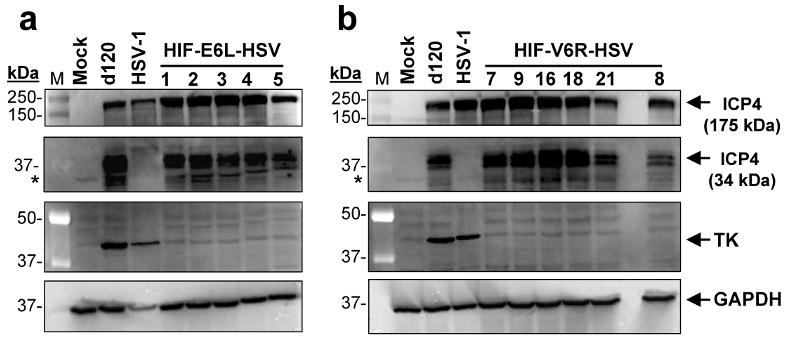
E5 cells were infected at MOI 0.25 with the indicated (a) HIF-E6L-HSV (clones 1-5) or (b) HIF-V6R-HSV (clones 7, 9, 16, 18, 21, 8) and then grown under normoxia. As controls, cells were mock- or virus-infected with HSV-1 strain KOS or d120. One day later, 30μg of total cell lysate was examined by immunoblotting for the presence of full length ICP4 (175 kDa), truncated ICP4 (34kDa), TK (40 kDa), and GAPDH (loading control, 36 kDa) protein expression. The ICP4 bands are shown at two different exposures to better visualize the 175 kDa and 34 kDa ICP4 proteins. Asterisks indicate a non-specific band that is cross-reactive with the anti-ICP4 antibody.
Of note, the expression of full-length ICP4 protein was visible in E5 cells infected with HSV-1, HIF-HSV, and d120, but not mock-infected cells. The expression of full-length ICP4 protein by d120-infected and not mock-infected E5 cells is consistent with other reports 23. E5 cells contain a stably transfected ICP4 gene under the control of its endogenous promoter 11. One explanation for these results is that the ICP4 promoter present in E5 cells is activated by the trans-activator virion-associated VP16 protein in d120 virions, thereby allowing the detection of full-length ICP4 protein 23. To confirm that d120 does not express full-length ICP4 protein, we infected LN229 (ICP4 null) with d120 or HSV-1 at increasing MOIs and then analyzed expression of the 175 kDa and 34 KDa ICP4 protein by western blot analysis (Supplemental Figure 1). Mock- and virus-infected E5 cells were included for comparison. Mock infected cells did not express ICP4 protein. HSV-1 infected E5 and LN229 cells expressed only the 175 kDa ICP4. d120 infected LN229 cells exclusively expressed the 34 kDa ICP4 protein. In contrast, d120 infected E5 cells expressed both the 175 kDa and 34 kDa ICP4 proteins. These results demonstrate that d120 is capable of expressing only the truncated 34 kDa ICP4 variant.
Disruption of the TK gene locus in HIF-V6R-HSV and HIF-E6L-HSV was evaluated by western blotting for TK expression using infected E5 cells (Figure 2). HSV-1 and d120, which contain an intact TK gene, expressed detectable levels of TK protein. In contrast, HIF-E6L-HSV (Figure 2a) and HIF-V6R-HSV (Figure 2b) did not express TK. Similar results were obtained using infected LN229 cells (Supplemental Figure 2). Of note, a TK band was not evident in d120-infected LN229 cells which is consistent with the replication-deficient phenotype of this virus in non-permissive (ICP4 null) cell lines. We also evaluated disruption of the TK locus in the HIF-HSVs by an acyclovir (ACV) sensitivity assay using infected LN229 cells (Figure 3 and Supplemental Figure 3). ACV is a prodrug which is converted by viral TK into an active inhibitor of viral and cellular DNA synthesis. HSVs with an intact TK gene are sensitive to the anti-viral effects of ACV and do not replicate whereas TK null-HSVs are resistant to the drug and undergo viral replication. ACV has no effect on the growth of mock infected LN229 cells (Figure 3a). d120 did not induce CPE of LN229 cells in the absence or presence of ACV, consistent with its replication-deficient phenotype (Figure 3b). As expected, HSV-1 induced extensive CPE of infected cells in the absence of ACV while no evidence of CPE was evident in HSV-1 infected cells grown in the presence of ACV (Figure 3a and 3b). This demonstrates that ACV is working to inhibit HSV-1. In contrast, HIF-E6L-HSV and HIF-V6R-HSV induced CPE of infected cells in the presence and absence of ACV (Figure 3a and 3b, Supplemental Figure 3). These results are consistent with the disruption of the TK gene locus in HIF-V6R-HSV and HIF-E6L-HSV. Unexpectedly, these studies also revealed that the HIF-HSVs induce CPE of infected cells maintained under normoxia and hypoxia. This was the first indication that the HIF-HSVs are not regulated in a HIF-dependent manner. This is further explored below.
Figure 3. HIF-E6L-HSV and HIF-V6R-HSV are resistant to acyclovir (ACV).
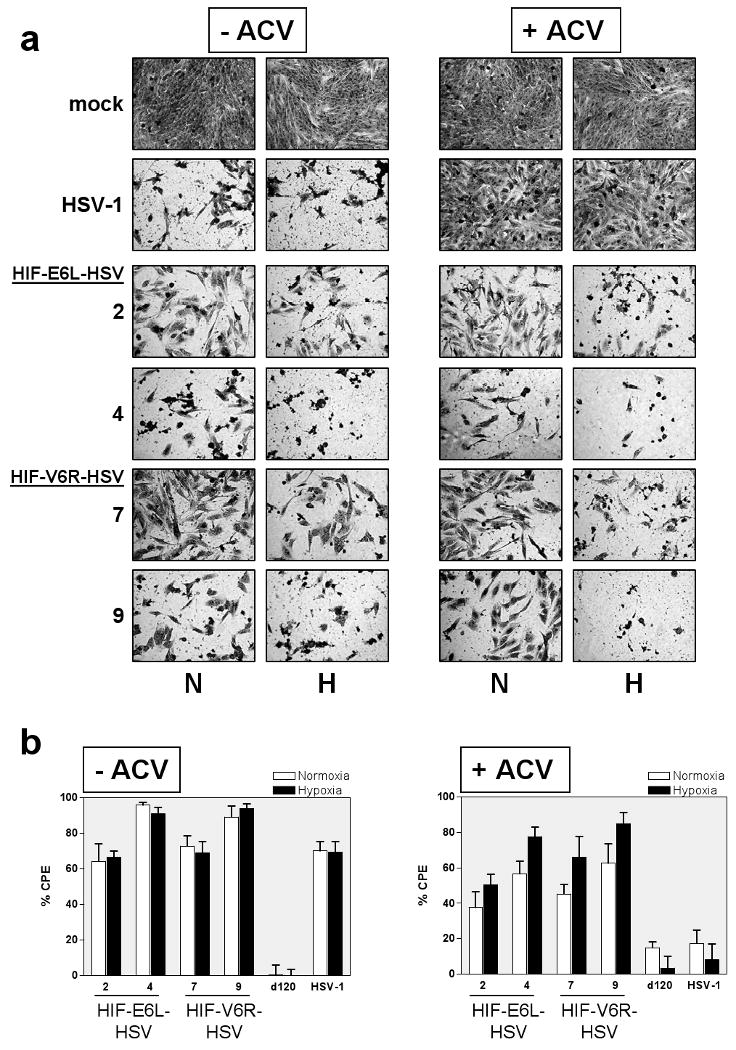
LN229 cells were infected with the indicated HIF-E6L-HSV (clones 2 and 4) or HIF-V6R-HSV (clones 7 and 9). As controls, cells were mock- or virus-infected with HSV-1 strain KOS or d120. Cells were grown in the absence or presence of ACV under normoxia (N) or hypoxia (H). (a) Five days later, cells were fixed and stained with crystal violet to visualize viral mediated CPE. Representative images were photographed at 100× magnification. Data for additional HIF-HSV clones are shown in Supplemental Figure 2. (b) CPE (cytolysis/detachment) was measured by an MTT assay at 5 days post infection from three replicates/experimental condition. The CPE obtained with each virus was normalized to mock infected cells. Percent CPE=[1-(average optical density of mock infected cells/average optical density of virus infected cells)] ×100. The data represents the mean ± SD.
Full length ICP4 (175 kDa) expression by HIF-E6L-HSV and HIF-V6R-HSV is not HIF-dependent
The ability of HIF-E6L-HSV and HIF-V6R-HSV to express full length ICP4 (175 kDa) under normoxia versus hypoxia was evaluated by western blotting using infected LN229 cells (Figure 4). Mock, d120, and HSV-1 infected cells were used as controls. Mock- and d120- infected cells did not express detectable levels of ICP4 protein whereas cells infected with HSV-1 expressed full-length ICP4. Unexpectedly, all of the HIF-E6L-HSV and HIF-V6R-HSV expressed ICP4 at similar levels under normoxia and hypoxia. These results demonstrate a lack of HIF-dependent ICP4 transgene expression by HIF-E6L-HSV and HIF-V6R-HSV.
Figure 4. Strains HIF-E6L-HSV and HIF-V6R-HSV express full length ICP4 (175 kDa) and expression is not HIF-dependent.
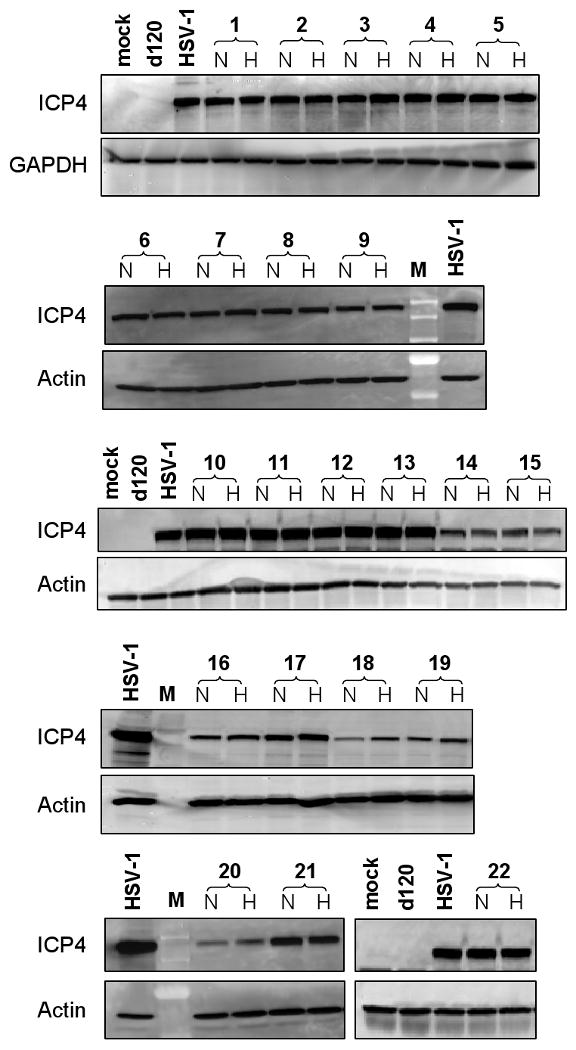
LN229 cells were infected at MOI 0.05 with the indicated HIF-E6L-HSV (clones 1-5) or HIF-V6R-HSV (clones 6-22) clone and then grown under normoxia (N) or hypoxia (H). As controls, cells were mock- or virus-infected with HSV-1 strain KOS or d120. One day later, 20μg of total cell lysate was western blotted for full length ICP4 (175 kDa) protein expression. GAPDH (36 kDa) or actin (42 kDa) protein expression was used as a loading control. M=molecular weight standard lane.
HIF-E6L-HSV and HIF-V6R-HSV lyse normoxic and hypoxic cells
The ability of HIF-E6L-HSV and HIF-V6R-HSV to specifically induce CPE of hypoxic tumor cells was evaluated using LN229 (Figure 5, Figure 3, and Supplemental Figure 3), U251MG-T2 (Figure 5 and Supplemental Figure 4), and LN319 tumor cells (Figure 5). Similar results were obtained for all three cell lines. Mock and replication-deficient d120 infected cells served as negative controls and displayed less than 14% cell death under normoxia and hypoxia. This demonstrates the lack of toxicity due to the infection and/or hypoxic conditions. In contrast, HSV-1 induced extensive cytolysis (>56%) under normoxia and hypoxia. HIF-E6L-HSV and HIF-V6R-HSV infection led to extensive cytolysis (>60%) under normoxia and hypoxia. In general, the degree of cytolysis by HSV-1 and the HIF-HSVs was greater under normoxia than hypoxia. These results demonstrate that the HIF-HSVs do not exhibit HIF-dependent oncolytic activity. This is consistent with the ICP4 (175 kDa) expression data from Figure 4 and shows that the levels of ICP4 produced by the HIF-HSVs is sufficient to initiate the viral replication cycle under normoxia and hypoxia. To better understand the lack of HIF-dependent regulation of the HIF-HSVs, two parallel sets of experiments were performed as described below.
Figure 5. Normoxic and hypoxic cells undergo CPE following infection with HIF-E6L-HSV and HIF-V6R-HSV.

LN229, U251MG-T2, and LN319 cells were infected with HIF-E6L-HSV (clones 1-5) or HIF-V6R-HSV (clones 7, 9, 16, 18, 21) and then grown under normoxia or hypoxia. As controls, cells were mock- or virus-infected with HSV-1 strain KOS or d120. CPE (cytolysis/detachment) was measured by an MTT assay at 5 days post infection from three replicates/experimental condition. The CPE obtained with each virus was normalized to mock infected cells as described in the legend of Figure 3. The data represents the mean ± SD.
Reconstitution of the ICP4 gene region to its wild-type configuration in HIF-E6L-HSV and HIF-V6R-HSV
We evaluated whether the ICP4 gene region reverted to a wild-type configuration in the HIF-HSVs. We performed PCR using viral DNA and primers specific for the HSV-1 ICP4 promoter and its 3′ coding region (Figure 6). For these studies, the N33 cell line was utilized to grow viruses for DNA isolation. This cell line contains HSV-2, rather than HSV-1, ICP4 sequences. Cellular DNA isolated from uninfected N33 cells did not generate the expected 2418 bp ICP4 PCR product, demonstrating the specificity of the primers for HSV-1, and not HSV-2, ICP4 sequences. As expected, the HSV-1 positive control showed a 2418 bp ICP4 PCR product while the d120 negative control did not. All the HIF-E6L-HSV and HIF-V6R-HSV DNA samples showed a 2418 bp ICP4 PCR product. This demonstrates that the HIF-HSVs contain a complete coding sequence for ICP4 downstream of its native promoter. This most likely explains the lack of HIF-dependent regulation of the HIF-HSVs.
Figure 6. Reconstitution of the ICP4 gene region to its wild-type configuration in HIF-V6R-HSV and HIF-E6L-HSV.
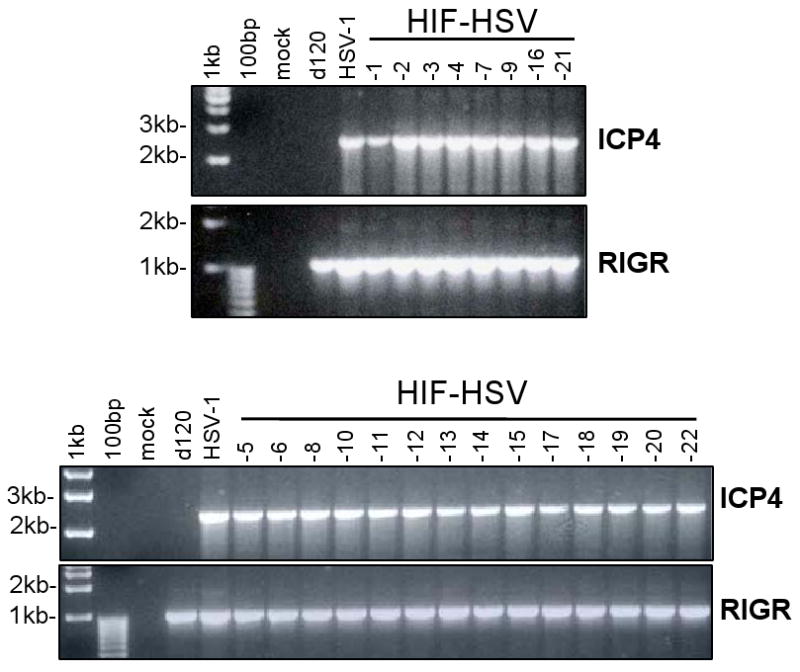
Total DNA isolated from HIF-E6L-HSV (clones 1-5) or HIF-V6R-HSV (clones 6-22) infected N33 cells were subjected to PCR using primers specific for the HSV-1 ICP4 promoter and 3′ coding region. As controls, DNA from uninfected or virus (HSV-1, d120) infected N33 cells were used. The 2418 bp PCR product using the ICP4 primers (specific for the ICP4 promoter and its 3′ coding region) indicate full-length HSV-1 ICP4 coding sequences downstream of its native promoter. The 1053 bp PCR product using the RIGR (right intergenic region) primers demonstrate the presence of amplifiable viral DNA.
Increased HIF-transcriptional activity under normoxia following HSV infection
We evaluated if HIF transcriptional activity was increased under normoxia or hypoxia following infection with d120, HSV-1, or the HIF-HSVs (Figure 7). For this, LN229 cells were transfected with a plasmid containing the luciferase reporter gene under the control of the same E6L or V6R HIF-responsive promoters which are present in HIF-E6L-HSV and HIF-V6R-HSV, respectively. Similar to published studies 18, mock infected cells showed a significant increase in luciferase reporter gene expression under hypoxia compared to normoxia (p<0.013, Supplemental Table 1). This demonstrates that the HIF-activated luciferase reporter plasmid and our hypoxic conditions were working as expected. Infection of cells with d120, HSV-1, HIF-E6L-HSV, or HIF-V6R-HSV led to a significant increase in HIF-transcriptional activity under normoxia by up to 14-fold (p<0.04). In virus infected cells maintained under hypoxia, there was no consistent pattern of HIF-transcriptional activity compared to mock infected cells maintained under hypoxia (not shown). These results demonstrate that HSV infection increases HIF-transcriptional activity under normoxia.
Figure 7. Increased HIF-transcriptional activity under normoxia following HSV infection.

LN229 cells were transfected with plasmids pBI-GL-E6L or pBI-GL-V6R which contain the luciferase reporter gene under the control of the E6L or V6R HIF-responsive promoters, respectively. Cells were then mock- or virus-infected with d120, HSV-1, HIF-E6L-HSV (clone 1), or HIF-V6R-HSV (clone 16) at MOI 0.05, 0.25, or 1 in triplicate. Cells were grown under normoxic conditions. The next day, luciferase activity was measured and normalized to total protein concentration. The data represents the mean ± SD. Asterisks indicate a significant increase in HIF-transcriptional activity (luciferase activity) in virus versus mock infected cells (p<0.04).
Discussion
We evaluated the feasibility of generating a HIF-activated oncolytic HSV (HIF-HSV) for the specific killing of tumor cells that are hypoxic. Since hypoxia is a common feature of tumors, but is not seen in normal tissues, such a virus could have very wide applications in the treatment of cancer. We selected the Herpes Simplex virus type-1 as the backbone for the new vector since this virus is highly cytotoxic, has a broad host range, and has shown promising results in clinical trials of several types of tumor 8,9. An early viral gene, ICP4, which is essential for virus replication, was placed under the regulation of a HIF-responsive promoter and then introduced into the TK locus of the ICP4-null HSV mutant d120. Derivation of recombinant HIF-HSV from d120 was verified by expression of a truncated, nonfunctional form of the ICP4 protein. The insertion of the novel construct into the TK locus was confirmed by the loss of TK protein expression and by the development of resistance to acyclovir. We have previously shown that the V6R and E6L HIF-responsive promoters exhibit extremely tight regulation of transgene expression under hypoxic conditions 18. In addition, we recently generated a new form of oncolytic adenovirus using the V6R promoter and this virus shows very precise hypoxia-regulated viral replication and anti-tumor effects 22,24. The failure of our novel HIF-HSVs to show similar hypoxic specificity was therefore not expected.
Two factors were identified which explained the lack of hypoxia-regulated specificity of the HIF-HSVs. The first was the restoration of the ICP4 gene under the control of its native promoter. This occurred during the generation of both HIF-E6L-HSV and HIF-V6R-HSV and in multiple, independently isolated virus clones. d120 can generate wild-type ICP4 recombinants at a low frequency when grown in ICP4 complementing cell lines 11. This most likely reflects a recombination event between d120 and exogenous ICP4 gene sequences which are stably integrated in the cellular genome. In our studies, we utilized the E5 ICP4 complementing cell line. We then transfected these cells with a HIF promoter:ICP4 expression construct and d120 virus. It is possible that these experimental conditions may favor the generation of wild-type ICP4 recombinants at a higher frequency. One possibility is that during the generation of HIF-HSV, a recombination event occurred between ICP4 sequences in the linearized plasmid construct and ICP4 sequences in the d120 genome. Interestingly, the recombination event was restricted to only one of the ICP4 loci, since all HIF-HSVs retained the truncated (34 KDa) ICP4. The next step is to better characterize the recombination event to distinguish recombination of the d120 genome with ICP4 sequence in the plasmid DNA versus E5 cells. One approach would be to use a plasmid construct that has point mutations in ICP4. Another option is to use N33 cells, which contain ICP4 sequences from HSV-2 rather than HSV-1. Four other transcriptionally regulated ICP4 oncolytic HSVs have been generated using E5 cells, d120, and an individualized promoter:ICP4 expression construct 12-16, but none of these reports commented on the generation of wild-type ICP4 recombinants. Thus, the prevalence of generating wild-type ICP4 recombinants using this experimental approach is unknown. Differences between these four viruses exists in the recombination construct used (pKpX2Δ plasmid, pTKΔL plasmid, Flip-Flop-BAC system) and target site for integration of the promoter:ICP4 sequence (TK vs. ICP6). Further analysis of these oncolytic HSVs may reveal whether these factors play a role in the frequency of generating wild-type ICP4 recombinants. Genetic stability of oncolytic viruses is an important safety issue. With regards to oncolytic HSV, the generation of “wild-type” HSV by recombination or genetic instability has the potential to cause serious toxicities in patients including corneal blindness and encephalitis. Our studies highlight the need to (i) better understand the generation of “wild-type” recombinants during the production of oncolytic HSV-1 and (ii) develop new vector systems for producing transcriptionally regulated ICP4 oncolytic HSVs that minimize the generation of wild-type ICP4 recombinants.
The second factor that prevented the HIF-HSV strains from showing the required specificity was that HIF transcriptional activity is induced following HSV infection. This was evident under normoxia following infection with HSV, d120, HIF-V6R-HSV, and HIF-E6L-HSV. In addition, two independent HIF-responsive promoters, V6R and E6L, were activated in the context of a plasmid following HSV infection. This effect is specific to HSV since activation of these promoters was not seen following infection with an adenovirus 25. Thus, the induction of HIF by HSV-1 argues against the use of a HIF-responsive promoter for the creation of a tumor-specific oncolytic HSV-1. Since all of the HIF-HSVs had reconstitution of the ICP4 locus to its wild-type configuration, we were not able to evaluate the impact of increased HIF-transcriptional activity under normoxia on the replication cycle of the HIF-HSVs. The regulation of HIF is complex and occurs at multiple levels including transcription, translation, post-translational modification, protein:protein interaction, and degradation 7. Many positive and negative regulators of HIF have been identified including hypoxia, growth factors, cytokines, hormones, oncogenes, and tumor suppressor genes. In addition to these factors, HIF expression and activity is also activated following bacterial, viral, and parasitic infections 26. This includes infection by two members of the Herpesviridae family, Epstein-Barr virus 27,28 and Kaposi's sarcoma-associated herpes virus (human herpes virus 8) 29-33. Our studies with HSV-1 identify a third member of the Herpesviridae family that induces HIF under normoxia. It will be interesting to determine if all members of this virus family have this feature in common. Viruses induce HIF under normoxia by multiple mechanisms including (i) increased HIF gene transcription 29,32,34, (ii) increased HIF protein synthesis by activating cellular signaling pathways 27,30,35,36, and/or (iii) stabilization of the HIF alpha subunit 28,31,33,37,38. In addition, closely related viruses within the same family, such as Epstein-Barr virus and Kaposi's sarcoma-associated herpes virus, have distinct mechanisms for regulating HIF 27-33. The ability of the replication-deficient d120 virus to activate HIF suggests that viral replication is not required. It is possible that an immediate early protein expressed by d120 may be responsible for this effect 17. d120 expresses four immediate early proteins, ICP0, ICP6, ICP22, and ICP27, following infection of non-permissive cells 11. ICP0 is a ubiquitin ligase and its expression leads to the promiscuous activation of HSV and heterologous promoters 39. Thus, it is possible that ICP0, or one of the other immediate early proteins, activates the HIF-pathway. A similar mechanism was proposed by Griffith et al. during the generation of an oncolytic HSV-1 containing ICP4 under the control of the human papillomavirus type-16 upstream regulatory region (URR16) promoter 17. In that virus, altered regulation of the URR16 promoter was demonstrated following HSV infection. Understanding the mechanism(s) underlying HSV-1 mediated induction of the HIF pathway will be critical in determining whether modifications can be made to improve the tumor specificity of a HIF-regulated oncolytic HSV-1.
Experimental manipulation of HIF levels in tumor models leads to changes in tumor growth, angiogenesis and metastasis, thereby demonstrating its importance as a protumorigenic factor 40-48. In addition, many of the viruses which activate HIF are also oncogenic such as Hepatitis B 37,38,49, Hepatitis C 35, Kaposi's sarcoma-associated herpes virus 29-33, Epstein-Barr Virus 27,28, and human T-cell leukemia virus 36. While HSV-1 infection is not associated with cancer in humans, treatment of experimental tumor models with oncolytic HSV induces tumor angiogenesis 50. In addition, a subset of the pro-angiogenic factors induced in the HSV-treated tumors were previously identified as HIF target genes 4. These findings coupled with our studies demonstrating induction of HIF transcriptional activity by HSV-1 suggest that the pro-angiogenic phenotype of this virus may be mediated, in part, by HIF. Thus, it is possible that activation of the HIF pathway by oncolytic HSV-1 treatment may promote a more aggressive tumor phenotype (increased tumor growth, angiogenesis, and tumor cell invasion/metastasis). This may be most evident in situations where the oncolytic activity of the virus is weak, due to attenuated viral replication or an inability to counter host anti-viral defenses.
Although HSV-1 shows promise as an anti-tumor virus and has entered several human clinical trials, the present study has revealed possible problems that could arise in its use. Reversion to a wild-type phenotype, loss of specificity due to viral transactivation of the transgenic promoter, and viral activation of tumor angiogenesis must all be taken into account in future research.
Supplementary Material
E5 cells were mock or virus (d120, HSV-1) infected at MOI 0.25. LN229 cells were mock or virus (d120, HSV-1) infected at MOI 0.1, 0.5, 1, 2.5, or 5. The cells were grown under normoxia. One day later, 20μg of total cell lysate was western blotted for full length (175 kDa) or truncated (34 KDa) ICP4 expression. Actin (42 kDa) protein expression was used as a loading control. M=molecular weight standard lane.
LN229 cells were infected at MOI 0.25 with HIF-E6L-HSV (clones 1-5) or HIF-V6R-HSV (clones 7, 9, 16, 18, 21) and then grown under normoxia (N) or hypoxia (H). As controls, cells were mock- or virus-infected with HSV-1 or d120. One day later, 20μg of total cell lysate was western blotted for TK (40 kDa), and GAPDH (loading control, 36 kDa) protein expression.
LN229 cells were infected with HIF-E6L-HSV (clones 1, 3, 5) or HIF-V6R-HSV (clones 16, 18, 21). Infected cells were grown in the absence of presence of ACV under normoxia (N) or hypoxia (H). As controls, cells were mock- or HSV-1-infected. Five days later, cells were fixed and stained with crystal violet to visualize viral mediated CPE. Representative images were photographed at 100× magnification. Data for mock, HSV-1, and additional HIF-HSV clones are shown in Figure 3.
U251MG-T2 cells were infected at MOI 0.25 with (a) HIF-E6L-HSV (clones 1-5) or (b) HIF-V6R-HSV (clones 7, 9, 16, 18, 21) and then grown under normoxia (N) or hypoxia (H). As controls, cells were mock- or virus-infected with d120 or HSV-1. Five (HIF-E6L-HSV study) or three (HIF-V6R-HSV study) days later, cells were fixed and stained with crystal violet to visualize viral mediated CPE. Representative images were photographed at 100× magnification. nd: not determined.
Acknowledgments
Grant support was provided by the NIH to DEP (NS49300) and EJS (DE017611). We thank Neil DeLuca (E5 cells, HSV-1 d120 virus) and Samuel Rabkin (pTKΔL plasmid) for providing critical reagents; Emily Wright, and Amanda Magee for assistance with the western blot and HIF-transcriptional activity assays; and David Padalino and Amanda Magee for critical reading of the manuscript.
Abbreviations
- HSV-1
Herpes Simplex Virus-1
- HIF
hypoxia-inducible factor
- HRE
HIF-responsive promoter
- MOI
multiplicity of infection
- ACV
acyclovir
- TK
thymidine kinase
- ICP4
infected cell polypeptide 4
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830–5835. [PubMed] [Google Scholar]
- 2.Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–421. doi: 10.1016/s0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–685. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JM. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007;435:297–321. doi: 10.1016/S0076-6879(07)35015-5. [DOI] [PubMed] [Google Scholar]
- 6.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12:853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Bardos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta. 2005;1755:107–120. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Grandi P, Peruzzi P, Reinhart B, Cohen JB, Chiocca EA, Glorioso JC. Design and application of oncolytic HSV vectors for glioblastoma therapy. Expert Rev Neurother. 2009;9:505–517. doi: 10.1586/ern.09.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman GK, Pressey JG, Reddy AT, Markert JM, Gillespie GY. Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol Ther. 2009;17:1125–1135. doi: 10.1038/mt.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardcastle J, Kurozumi K, Chiocca EA, Kaur B. Oncolytic viruses driven by tumor-specific promoters. Curr Cancer Drug Targets. 2007;7:181–189. doi: 10.2174/156800907780058880. [DOI] [PubMed] [Google Scholar]
- 11.DeLuca NA, McCarthy AM, Schaffer PA. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J Virol. 1985;56:558–570. doi: 10.1128/jvi.56.2.558-570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyatake S, Iyer A, Martuza RL, Rabkin SD. Transcriptional targeting of herpes simplex virus for cell-specific replication. J Virol. 1997;71:5124–5132. doi: 10.1128/jvi.71.7.5124-5132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyatake SI, Tani S, Feigenbaum F, Sundaresan P, Toda H, Narumi O, et al. Hepatoma-specific antitumor activity of an albumin enhancer/promoter regulated herpes simplex virus in vivo. Gene Ther. 1999;6:564–572. doi: 10.1038/sj.gt.3300861. [DOI] [PubMed] [Google Scholar]
- 14.Yamamura H, Hashio M, Noguchi M, Sugenoya Y, Osakada M, Hirano N, et al. Identification of the transcriptional regulatory sequences of human calponin promoter and their use in targeting a conditionally replicating herpes vector to malignant human soft tissue and bone tumors. Cancer Res. 2001;61:3969–3977. [PubMed] [Google Scholar]
- 15.Kuroda T, Rabkin SD, Martuza RL. Effective treatment of tumors with strong beta-catenin/T-cell factor activity by transcriptionally targeted oncolytic herpes simplex virus vector. Cancer Res. 2006;66:10127–10135. doi: 10.1158/0008-5472.CAN-06-2744. [DOI] [PubMed] [Google Scholar]
- 16.Mullen JT, Kasuya H, Yoon SS, Carroll NM, Pawlik TM, Chandrasekhar S, et al. Regulation of herpes simplex virus 1 replication using tumor-associated promoters. Ann Surg. 2002;236:502–512. doi: 10.1097/00000658-200210000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffith C, Noonan S, Lou E, Shillitoe EJ. An oncolytic mutant of herpes simplex virus type-1 in which replication is governed by a promoter/enhancer of human papillomavirus type-16. Cancer Gene Ther. 2007;14:985–993. doi: 10.1038/sj.cgt.7701089. [DOI] [PubMed] [Google Scholar]
- 18.Post DE, Van Meir EG. Generation of bidirectional hypoxia/HIF-responsive expression vectors to target gene expression to hypoxic cells. Gene Ther. 2001;8:1801–1807. doi: 10.1038/sj.gt.3301605. [DOI] [PubMed] [Google Scholar]
- 19.Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Post DE, Devi NS, Li Z, Brat DJ, Kaur B, Nicholson A, et al. Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin Cancer Res. 2004;10:8603–8612. doi: 10.1158/1078-0432.CCR-04-1432. [DOI] [PubMed] [Google Scholar]
- 21.Smith CA, Schaffer PA. Mutants defective in herpes simplex virus type 2 ICP4: isolation and preliminary characterization. J Virol. 1987;61:1092–1097. doi: 10.1128/jvi.61.4.1092-1097.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Post DE, Van Meir EG. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene. 2003;22:2065–2072. doi: 10.1038/sj.onc.1206464. [DOI] [PubMed] [Google Scholar]
- 23.Su YH, Zhang X, Wang X, Fraser NW, Block TM. Evidence that the immediate-early gene product ICP4 is necessary for the genome of the herpes simplex virus type 1 ICP4 deletion mutant strain d120 to circularize in infected cells. J Virol. 2006;80:11589–11597. doi: 10.1128/JVI.01869-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Post DE, Sandberg EM, Kyle MM, Devi NS, Brat DJ, Xu Z, et al. Targeted Cancer Gene Therapy Using a Hypoxia Inducible Factor Dependent Oncolytic Adenovirus Armed with Interleukin-4. Cancer Res. 2007;67:6872–6881. doi: 10.1158/0008-5472.CAN-06-3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cherry T, Longo SL, Tovar-Spinoza Z, Post DE. Second-generation HIF-activated oncolytic adenoviruses with improved repliction, oncolytic, and anti-tumor efficacy. Gene Therapy. 2010 doi: 10.1038/gt.2010.100. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zinkernagel AS, Johnson RS, Nizet V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J Mol Med. 2007;85:1339–1346. doi: 10.1007/s00109-007-0282-2. [DOI] [PubMed] [Google Scholar]
- 27.Wakisaka N, Kondo S, Yoshizaki T, Murono S, Furukawa M, Pagano JS. Epstein-Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2004;24:5223–5234. doi: 10.1128/MCB.24.12.5223-5234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kondo S, Seo SY, Yoshizaki T, Wakisaka N, Furukawa M, Joab I, et al. EBV latent membrane protein 1 up-regulates hypoxia-inducible factor 1alpha through Siah1-mediated down-regulation of prolyl hydroxylases 1 and 3 in nasopharyngeal epithelial cells. Cancer Res. 2006;66:9870–9877. doi: 10.1158/0008-5472.CAN-06-1679. [DOI] [PubMed] [Google Scholar]
- 29.Carroll PA, Kenerson HL, Yeung RS, Lagunoff M. Latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J Virol. 2006;80:10802–10812. doi: 10.1128/JVI.00673-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, et al. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 2000;60:4873–4880. [PubMed] [Google Scholar]
- 31.Cai Q, Murakami M, Si H, Robertson ES. A potential alpha-helix motif in the amino terminus of LANA encoded by Kaposi's sarcoma-associated herpesvirus is critical for nuclear accumulation of HIF-1alpha in normoxia. J Virol. 2007;81:10413–10423. doi: 10.1128/JVI.00611-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai Q, Lan K, Verma SC, Si H, Lin D, Robertson ES. Kaposi's sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: Latency control under low oxygen conditions. J Virol. 2006;80:7965–7975. doi: 10.1128/JVI.00689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin YC, Joo CH, Gack MU, Lee HR, Jung JU. Kaposi's sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 2008;68:1751–1759. doi: 10.1158/0008-5472.CAN-07-2766. [DOI] [PubMed] [Google Scholar]
- 34.Deshmane SL, Mukerjee R, Fan S, Del VL, Michiels C, Sweet T, et al. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J Biol Chem. 2009;284:11364–11373. doi: 10.1074/jbc.M809266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nasimuzzaman M, Waris G, Mikolon D, Stupack DG, Siddiqui A. Hepatitis C virus stabilizes hypoxia-inducible factor 1alpha and stimulates the synthesis of vascular endothelial growth factor. J Virol. 2007;81:10249–10257. doi: 10.1128/JVI.00763-07. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Tomita M, Semenza GL, Michiels C, Matsuda T, Uchihara JN, Okudaira T, et al. Activation of hypoxia-inducible factor 1 in human T-cell leukaemia virus type 1-infected cell lines and primary adult T-cell leukaemia cells. Biochem J. 2007;406:317–323. doi: 10.1042/BJ20070286. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Yoo YG, Oh SH, Park ES, Cho H, Lee N, Park H, et al. Hepatitis B virus X protein enhances transcriptional activity of hypoxia-inducible factor-1alpha through activation of mitogen-activated protein kinase pathway. J Biol Chem. 2003;278:39076–39084. doi: 10.1074/jbc.M305101200. [DOI] [PubMed] [Google Scholar]
- 38.Yoo YG, Cho S, Park S, Lee MO. The carboxy-terminus of the hepatitis B virus X protein is necessary and sufficient for the activation of hypoxia-inducible factor-1alpha. FEBS Lett. 2004;577:121–126. doi: 10.1016/j.febslet.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Hagglund R, Roizman B. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J Virol. 2004;78:2169–2178. doi: 10.1128/JVI.78.5.2169-2178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 41.Stoeltzing O, McCarty MF, Wey JS, Fan F, Liu W, Belcheva A, et al. Role of hypoxia-inducible factor 1alpha in gastric cancer cell growth, angiogenesis, and vessel maturation. J Natl Cancer Inst. 2004;96:946–956. doi: 10.1093/jnci/djh168. [DOI] [PubMed] [Google Scholar]
- 42.Li L, Lin X, Staver M, Shoemaker A, Semizarov D, Fesik SW, et al. Evaluating hypoxia-inducible factor-1alpha as a cancer therapeutic target via inducible RNA interference in vivo. Cancer Res. 2005;65:7249–7258. doi: 10.1158/0008-5472.CAN-04-4426. [DOI] [PubMed] [Google Scholar]
- 43.Hiraga T, Kizaka-Kondoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67:4157–4163. doi: 10.1158/0008-5472.CAN-06-2355. [DOI] [PubMed] [Google Scholar]
- 44.Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67:563–572. doi: 10.1158/0008-5472.CAN-06-2701. [DOI] [PubMed] [Google Scholar]
- 45.Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
- 47.Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP, et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell. 2003;4:133–146. doi: 10.1016/s1535-6108(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 48.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 49.Moon EJ, Jeong CH, Jeong JW, Kim KR, Yu DY, Murakami S, et al. Hepatitis B virus X protein induces angiogenesis by stabilizing hypoxia-inducible factor-1alpha. FASEB J. 2004;18:382–384. doi: 10.1096/fj.03-0153fje. [DOI] [PubMed] [Google Scholar]
- 50.Kurozumi K, Hardcastle J, Thakur R, Shroll J, Nowicki M, Otsuki A, et al. Oncolytic HSV-1 infection of tumors induces angiogenesis and upregulates CYR61. Mol Ther. 2008;16:1382–1391. doi: 10.1038/mt.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
E5 cells were mock or virus (d120, HSV-1) infected at MOI 0.25. LN229 cells were mock or virus (d120, HSV-1) infected at MOI 0.1, 0.5, 1, 2.5, or 5. The cells were grown under normoxia. One day later, 20μg of total cell lysate was western blotted for full length (175 kDa) or truncated (34 KDa) ICP4 expression. Actin (42 kDa) protein expression was used as a loading control. M=molecular weight standard lane.
LN229 cells were infected at MOI 0.25 with HIF-E6L-HSV (clones 1-5) or HIF-V6R-HSV (clones 7, 9, 16, 18, 21) and then grown under normoxia (N) or hypoxia (H). As controls, cells were mock- or virus-infected with HSV-1 or d120. One day later, 20μg of total cell lysate was western blotted for TK (40 kDa), and GAPDH (loading control, 36 kDa) protein expression.
LN229 cells were infected with HIF-E6L-HSV (clones 1, 3, 5) or HIF-V6R-HSV (clones 16, 18, 21). Infected cells were grown in the absence of presence of ACV under normoxia (N) or hypoxia (H). As controls, cells were mock- or HSV-1-infected. Five days later, cells were fixed and stained with crystal violet to visualize viral mediated CPE. Representative images were photographed at 100× magnification. Data for mock, HSV-1, and additional HIF-HSV clones are shown in Figure 3.
U251MG-T2 cells were infected at MOI 0.25 with (a) HIF-E6L-HSV (clones 1-5) or (b) HIF-V6R-HSV (clones 7, 9, 16, 18, 21) and then grown under normoxia (N) or hypoxia (H). As controls, cells were mock- or virus-infected with d120 or HSV-1. Five (HIF-E6L-HSV study) or three (HIF-V6R-HSV study) days later, cells were fixed and stained with crystal violet to visualize viral mediated CPE. Representative images were photographed at 100× magnification. nd: not determined.