

Abstract
Cationic, amphiphilic polymers are currently being used as antimicrobial agents which disrupt biomembranes, although their mechanism(s) remain poorly understood. Herein, membrane association and disruption by amphiphilic polymers bearing primary, tertiary, or quaternary ammonium salt groups reveals the role of cationic group structure in the polymer-membrane interaction. The dissociation constants of polymers to liposomes of POPC were obtained by a fluorometric assay, exploiting the environmental sensitivity of dansyl moieties in the polymer end groups. Dye leakage from liposomes and solid-state NMR provided further insights into the polymer-induced membrane disruption. Interestingly, the polymers with primary amine groups induced reorganization of the bilayer structure to align lipid headgroups perpendicular to the membrane. The results showed that polymers bearing primary amines exceed the tertiary and quaternary ammonium counterparts in membrane binding and disrupting abilities. This is likely due to enhanced complexation of primary amines to the phosphate groups in the lipids, through a combination of hydrogen bonding and electrostatic interactions.
Keywords: antimicrobial, hemolytic, polymer, host defense peptide, quaternary ammonium salt
Introduction
The need for antimicrobial compounds which exert their effects by novel mechanisms is urgent, due to the rapidly increasing number of bacteria resistant to conventional drugs.1 In nature, many host defense peptides putatively combat infection by disruption of bacterial cell membranes.2–6 Because the peptides lack sequence homology and because the all-D- and all-L-amino acid forms are similarly active, it is believed that the primary mechanism of antimicrobial action does not rely on specific receptor-mediated interactions.7 Therefore, the function of these peptides has been generalized to other macromolecular platforms, such as designed synthetic peptides,8 oligomers,9 dendrimers,10 and polymers.11 While a variety of designed antimicrobial peptides have been shown to be active against a broad-spectrum of bacteria, pharmaceutical applications of these peptides are limited due to cost- and labor-intensive synthesis as well as poorly understood pharmacokinetics.12 Therefore, it is important to pursue alternate routes toward the design of antimicrobial compounds which can mimic host defense peptides. Recent studies demonstrated the capabilities of amphiphilic polymers to kill a broad-spectrum of bacteria by directly interacting with the cell membrane lipids.13–15 While these polymers are far less expensive compared to peptides and therefore may be reasonable candidates for biomedical applications, their toxicity remains a major concern. In order to enhance their antibacterial activities and reduce toxicity, it is important to understand the details of their interaction with biomembranes and to identify structural parameters which modulate their selectivity for bacterial versus human cell membranes. In this study, we present a thorough investigation of the role of cationic group structure on the functional properties of these amphiphilic polymers using a variety of biophysical and biochemical approaches.
Polymeric surfactants have long been known to kill bacteria by a putative mechanism of membrane disruption,16 rendering these polymers attractive candidates for disinfecting applications. In general, these materials are high molecular weight (MW typically 10–100 kDa) polymers bearing quaternary ammonium salt (QAS) groups attached to hydrophobic long alkyl chains (6–12 carbons).17, 18 They are frequently immobilized on surfaces in order to kill bacteria on contact.19, 20 Recently, a novel class of cationic and amphiphilic macromolecules, which are intended to mimic host defense peptides found in nature, have been developed.14, 15 The host defense peptides are particularly interesting prototypes due to their ability to selectively kill bacteria while minimizing disruption of human cells; an ability ascribed to their fine balance of cationic and hydrophobic residues combined with their low MW.21 The peptide-mimetic polymers are distinguishable from conventional polysurfactants by their characteristically low MWs (typically < 10 kDa), short alkyl groups (1–4 carbons), and protonated ammonium groups (e.g. primary amines), which are the specific structural features designed to mimic the salient physiochemical properties of host defense peptides. Peptide-mimetic polymers of methacrylates,11, 22 methacrylamides,23 norbornenes,24, 25 and β-lactams26 have been reported to kill bacteria by a putative mechanism of membrane disruption. By extensive optimization, a select few examples of such polymers displayed the ability to kill bacteria while causing minimal harm to human red blood cells. The favorable activity profiles of these prototypes are rather evocative of the host defense peptides, despite the fact that polymers lack precisely defined sequence and secondary structures.
An interesting feature of host defense peptides and their synthetic polymer mimics is the source of their cationic charge: they contain primary amine groups which are protonated at physiological pH, while polysurfactants typically contain permanently charged quaternary ammonium salt (QAS) groups. We have recently reported a direct comparison of polymethacrylate derivatives containing either protonated amines or quaternary ammonium units, which revealed that the primary amines are most effective at conferring favorable antimicrobial properties.22 Additionally, polystyrenes27 and polydiallyamines28 have shown greater antimicrobial activity when functionalized with tertiary ammonium, rather than QAS groups. Based on the structure-activity data, we hypothesized that the polymer interaction consists of a binding equilibrium and subsequent membrane permeabilization, which is profoundly impacted by the chemical structure of the cationic ammonium groups. This led us to inquire: what is the role of the cationic groups in the binding and membrane disruption processes?
The central hypothesis of this field is that cell membrane-disruption induced by amphiphilic polymers leads to the observed biocidal activities. Although a great deal of effort has focused on the interaction of high molecular weight polymers with model membranes,29 relatively little is known about the membrane disruption induced by this emerging class of low molecular weight amphiphilic polymers, which mimic host defense peptides. Accordingly, recent investigations on the propensity of such antimicrobial polymers to permeabilize model membranes have been carried out. Recently, the effect of lipid composition on the ability of antimicrobial oligomers to induce dye leakage from lipid bilayers by forming transmembrane pores was reported.30 Epand et al demonstrated that the lipid composition is a main determinant of dye leakage induced by peptide-mimetic antimicrobial nylon-3 polymers.13 In addition, our laboratory recently showed that amphiphilic copolymers based on methacrylamides induced leakage from liposomes depending on the lipid composition and hydrophobicity of the polymers, which correlated well with their antimicrobial and hemolytic activies.23 However, a complete understanding of the structural features of the polymers, rather than the lipids, which control the membrane activity has not been achieved to date. Particularly, it would interesting to assess the role of cationic group structure in the membrane disruption process, as a representative structural difference between peptide-mimetic and conventional polysurfactant antimicrobials.
To that end, we quantify the partitioning to and disruption of model lipid membranes by representative dye-labeled copolymers, which contained different ammonium structures (primary, tertiary, or quaternary ammonium). We examined the molecular interactions between the ammonium groups and the phosphate groups of lipids by solid-state NMR and water-octanol partitioning in the presence of a phosphate surfactant. We chose to investigate the interaction of these polymers with membranes composed of POPC, a zwitterionic lipid which is a major phospholipid component in the outer leaflet of mammalian cell membranes. While several reports have shown the interaction of polymers with vesicles composed of bacterial lipids, their interaction with mammalian-type lipids remains largely ambiguous. Understanding the role of cationic groups in the disruption of these model membranes will provide a biophysical basis for understanding their observed hemolytic activities. This knowledge will aid in the future design of antimicrobial polymers with a minimal propensity to disrupt mammalian cells. Additionally, understanding the role of cationic group structure in the polymer-membrane interaction will augment the design principles being actively developed in the fields of gene- and drug delivery, which also require materials with controlled membrane activities.
Experimental Methods
Materials
2,2'-azobisisobutyronitrile (AIBN), N,N-dimethylaminoethyl methacrylate (DMAEMA), and methyliodide were purchased from Sigma-Aldrich. Dansyl chloride, methylmethacrylate (MMA), ethanolamine, di-tert-butyldicarbonate, and methacryloyl chloride were purchased from Acros. Reagent grade solvents were purchased from Fisher and used without further purification. Standardized solutions of 0.100N sodium hydroxide and hydrogen chloride were purchased from Ricca Chemical Company. All reagents were used without further purification. The lipid 1-palmitoyl-2oleoyl-sn-glycero-3-phosphocholine (POPC) was purchased as a lyophilized powder from Avanti Polar Lipids (Alabaster, AL). Lipid stock solutions (10 mM) in chloroform were stored at −80 °C, under a nitrogen blanket, until use. The mono-n-dodecyl phosphate was purchased from the Wake Pure Chemicals (Japan).
Synthesis of the Dye-Labeled Copolymers
A dye-conjugated chain transfer agent was synthesized by a previously described procedure.31 Briefly, cystamine hydrochloride and dansyl chloride were stirred in DMF in the presence of triethylamine at room temperature overnight. Then, the purified product was mixed with Tris-(2-carboxyethyl)phosphine in order to reduce the disulfide bond. The random copolymers were prepared by previously described procedures.31 Briefly, methacrylate derivatives were dissolved in acetonitirle in various ratios. AIBN and the chain transfer agent were added and the solution was purged with nitrogen. The reaction mixture was heated at 70 °C overnight. Subsequently, the obtained polymers were purified by size exclusion chromatography (Sephadex LH20, GE Healthcare) to remove unreacted monomer and chain transfer agent. Polymers bearing primary amines in the side chains were obtained by cleavage of Boc protecting groups by HCl in methanol while polymers containing quaternary ammonium salt units were obtained by reaction of the DMAEMA units with methyliodide. Finally, the cationic polymers were purified by precipitation from methanol into diethylether and collected by centrifugation. Subsequently, they were dissolved in water and lyophilized, affording yellow/green powders. 1H NMR confirmed that the desired polymers were obtained. Comparison of integrated peak areas arising from the polymer side chains and the dansyl end group enabled calculation of the number average degree of polymerization and fraction of hydrophobic repeat units.
Potentiometric Titration
Polymers were titrated according to the procedure of Thomas and Tirrell.32 Briefly, polymers (~ 5 mg) were dissolved in aqueous saline (10 mL, [NaCl] = 150 mM) in a glass scintillation vial and purged with N2. A nitrogen blanket was maintained over the sample throughout the titration and the temperature was held at 24 ± 0.2 °C using a water bath. Aliquots of standardized 0.100 N sodium hydroxide or 0.100 N hydrochloric acid (5 or 10uL) were injected using a Hamilton syringe and the solution was stirred for 5 min to allow thermal and chemical equilibrium. The pH was recorded after each injection using an Accumet Basic AB15 pH meter. The degree of ionization of the polymer was calculated from the charge neutrality condition as,
| (1) |
where α is the fraction of amine groups in solution that are positively charged and [amines]total is the total concentration of all the amine groups in the solution. The raw data, pH as a function of α, were then fit to the generalized Henderson-Hasselbalch equation,33 below, using KaleidaGraph software in order to estimate the apparent average pKa of the amine groups in the polymer side chains.
| (2) |
Polymer 1 could not be titrated to the equivalence point due to the isomerization reactions which occur in basic conditions (pH > 9),34 and polymer 2 could not be titrated to the equivalence point because it precipitates from solution (turbidity was observed) above pH 8.
Partition Coefficient
Polymer solutions (500 uL, 100 uM) were prepared in aqueous buffer (10 mM HEPES or MES, 150 mM NaCl, pH ranging from 6 to 8) in an Eppindorf centrifuge tube and octanol (500 uL) was added. The tube was vortexed for 5 minutes and then allowed to sit overnight in the dark. After 5 minutes of centrifugation at 3000 rpm, an aliquot of each phase was diluted 100-fold into methanol and the fluorescence spectra were recorded. The concentration of polymer in each phase was determined from comparison to calibration curves obtained by measuring the fluorescence spectra of the polymers at various concentrations in methanol (supporting information). The partition coefficient was defined as,
| (3) |
where [P]oct and [P]aq are the concentration of the polymer in the octanol and aqueous phases, respectively. Measurements were performed three times from each phase.
Measurement of logP in the presence of mono-n-dodecylphosphate(DDP) were performed by the same procedure as above, except that the aqueous buffer (10 mM MES, 150 mM NaCl, pH 6) contained 0, 0.1, or 1 mM DDP surfactant. The concentrations of DDP were well below the literature values of the critical micelle concentration for this surfactant, which vary from 3 to 9 mM depending on the temperature, salt concentration and pH.
Polymer-Liposome Binding
A solution of lipid (100 μL, 10 mM) in chloroform was slowly evaporated under a gentle N2 stream and subsequently dried under vacuum for 12 hours. The lipid film was resuspended in an aqueous buffer (10mM HEPES or MES, pH 6–8, osmolality = 290±5 mmol/kg), agitated on a vortex mixer for 5 minutes, and subjected to ten freeze/thaw cycles between acetone containing dry ice and a 60 °C water bath. The turbid suspension was then extruded twenty-one times through two stacked polycarbonate membrane with an average pore size of 100 nm, yielding a clear suspension of large unilaminar vesicles (LUVs). An aliquot of the liposome stock solution (2.5 or 5 μL) was injected into a cuvette containing polymer solution (2 mL, 1 μM) and the fluorescence intensity was recorded after 5 minutes of mixing. An excitation wavelength of 330 nm and an emission wavelength of 510 nm were used. The fluorescence data were corrected for dilution as well as the inner-filter effect. The corrected fluorescence intensities were plotted versus the total concentration of lipid and the dissociation constant was determined by curve-fitting to the following expression for single-site binding isotherm using KaleidaGraph software, according to a previously established method.31
| (4) |
where F0 is the fluorescence intensity before the addition of liposomes, ΔF is the change in fluorescence intensity from F0 to the fluorescence when the lipid concentration approaches infinity, [P]0 is the total polymer concentration, [L]0 is the total lipid concentration, n is the number of lipids per binding site, and KD is the dissociation constant. The number of lipids per binding site was fixed in every case to n=6 to enable accurate curve fitting, although similarly good fits were obtained with n = 4–9, as discussed in a previous report.31
Liposome Dye Leakage
The liposome dye leakage assay was performed according to a literature procedure.23 Briefly, a solution of lipid (100 μL, 10 mM) in chloroform was slowly evaporated under a gentle N2 stream and subsequently dried under vacuum for 12 hours. A set of aqueous buffers (10mM HEPES or MES, 50mM sulforhodamine B (SRB), pH 6–8) were adjusted to an osmolality of 290±5 mmol/kg by addition of saturated NaCl and measured using a vapor-pressure osmometer. The dry lipid film was resuspended in this buffer and vigorously vortexed for 5 min. Then, the suspension was subjected to ten freeze/thaw cycles between dry ice in acetone and a 60 °C water bath. Then it was passed twenty-one times through a mini-extruder equipped with two stacked polycarbonate membranes of 400 nm average pore size. Unincorporated dye was removed by size exclusion chromatography over Sepharose Cl-4B gel from Amersham Biosciences (Uppsala, Sweden). The concentration of lipid in the obtained suspension was determined by an established colorimetric phosphorous assay. This solution was diluted using a buffer containing no dye (10 mM HEPES or MES, 150 mM NaCl, pH 6–8, 290±5 mmol/kg) to a lipid concentration of 11.11μM. This suspension (90 μL) was mixed with polymer stock solutions (10 μL) on a 96-well black microplate to give a final lipid concentration of 10 μM in each well. The assay buffer (10 μL) and Triton X (0.1% v/v, 10 μL) were employed as the negative and positive controls. After 1 hour, the fluorescence intensity in each well was recorded using a microplate reader (Thermo scientific Varioskan Flash) with excitation and emission wavelengths of 565 and 586 nm, respectively, with a bandpass of 5 nm each. The fraction of leaked SRB in each well was calculated according to the expression: L = (F−F0)/(FTX−F0) where F is the fluorescence intensity recorded in the well, F0 is the intensity in the negative control well, and FTX is the intensity in the positive control well. For the measurement of dye leakage kinetics, the liposome stock was diluted to 10μM in a 2mL quartz cuvette and fluorescence signal was monitored. Then, an aliquot of polymer solution was injected into the cuvette via a syringe port in the spectroflourometer lid and fluorescence intensity was recorded for 10 minutes. Triton X-100 solution (4% v/v, 5 uL) was injected as the positive lysis control for 100% leakage.
Solid-state NMR
Uniaxially aligned POPC bilayer samples for NMR experiments were prepared using the naphthalene procedure as explained elsewhere.35 In brief, four milligrams of lipids and an appropriate amount of antimicrobial polymer were dissolved in CHCl3/CH3OH (2:1) mixture containing equimolar amounts of naphthalene. The solution was spread on thin glass plates (7 mm × 11 mm × 50 μm, Paul Marienfeld, Bad Mergentheim, Germany) to have approximately 2 mg of lipid per plate and then dried under N2 gas at room temperature. The lipid films were then dried in the vacuum oven at 35 °C for ~10 hrs to remove naphthalene and any residual organic solvents. The glass plates were placed in a hydration chamber (saturated K2SO4 solution) that was maintained at 96% relative humidity for 2–3 days at 37 °C. Then, approximately 3 μL of deionized water was added onto the surface of the lipid-polymer film on glass plates. The glass plates were stacked, wrapped with parafilm, and sealed in plastic bags (Plastic Bagmart, Marietta, GA) to prevent dehydration of the sample during NMR data acquisition. The sealed samples were incubated at 37 °C for 6–24 h.
All 31P experiments were performed on a Varian solid-state NMR spectrometer (Varian inc, Palo Alto, CA) operating with resonance frequencies of 400.138 MHz and 161.979 MHz for 1H and 31P nuclei, respectively. A Varian temperature-controller unit was used to maintain the sample temperature and the sample was equilibrated for at least 30 min before acquiring FID. All experiments on uniaxially aligned samples were performed with the orientation of the lipid bilayer normal parallel to the external magnetic field using a home-built 1H/31P double-resonance probe, which has a four-turn square coil (12 mm × 12 mm × 4 mm) constructed using a 2-mm-wide flat-wire with a spacing of 1 mm between turns. 31P chemical shift spectra were obtained using a spin-echo sequence (90°-t-180°-t-acquire) under a 30 kHz proton-decoupling radio frequency field with a 50 kHz spectral width. A typical 31P 90° pulse length of 3.1 μs was used. All spectra were acquired using 512 transients and a 5 s recycle delay. 31P chemical shift values were referenced relative to 85% H3PO4 on thin glass plates (0 ppm).
Results and Discussion
Polymer Synthesis and Characterization
We prepared three model polymethacrylate derivatives, each containing a different type of ammonium group as the source of cationic charge. The polymer side chains consist of hydrophobic groups and primary, tertiary or quaternary ammonium salt groups (Figure 1). The polymer end groups are conjugated to dansyl, a fluorophore which can be exploited as a probe of the microenvironment polarity.36 The mole fraction of methyl groups in the side chains, fmethyl, and the degree of polymerization (DP) values are similar in the three polymers, such that the effect of the cationic group structure can be investigated directly.
Figure 1.
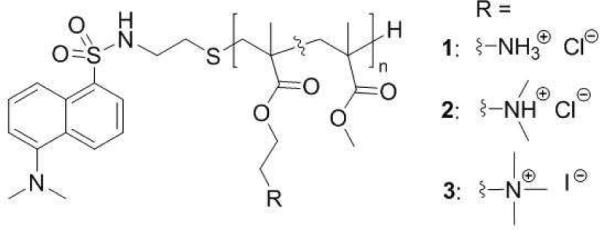
Structure of random copolymers containing methyl groups as hydrophobic moieties and different cationic groups in the side chains as well as a dansyl moiety at the polymer terminus
We chose to prepare relatively short polymers (DP = 19–22; Mn ~ 2–4 kDa) with an average fraction of methyl methacrylate relative to all monomer units in a polymer chain, fmethyl of 0.56–0.57 for this study (Table 1) because similar parameters gave rise to hemolytic polymers in our previous work.22 The molecular weights were determined by comparing the integrated areas in the 1H NMR spectra. The wavelengths of maximum absorbance and fluorescence emission of the polymers in methanol, in which the polymer chains are expected to be well solvated, were in the ranges of 334–339 and 508–512 nm, respectively (Table 1). The molar extinction coefficients (ε) of the polymers in methanol were in the range of 4400–4700 M−1cm−1. These values are comparable to free dansyl as well as dansyl-conjugated proteins reported in the literature,37, 38 which indicated that the conjugation to these polymers did not diminish the absorbance and emission properties of the dansyl end groups.
Table 1.
Characterization of the Random Copolymers with Dansyl End Groups
| Polymer | fmethyl[a] | DP[a] | Mn[a] (kDa) | ε[b] (M−1cm−1) | λmax[b] (nm) | HC50 | |
|---|---|---|---|---|---|---|---|
| Abs. | Em. | (μM, μg/mL) | |||||
| 1 | 0.56 | 20 | 2.6 | 4700 | 334 | 508 | 36, 94 |
| 2 | 0.57 | 19 | 2.7 | 4400 | 336 | 512 | 230, 620 |
| 3 | 0.56 | 22 | 4.2 | 4600 | 339 | 511 | >480, >2000 |
Mole fraction of methyl repeat units (fmethyl), number average degree of polymerization (DP) and molecular weight (Mn) determined by peak integration analysis of the lH NMR spectra in methanol-d4.
Molar extinction coefficient, ε, at the wavelength of maximum absorbance, Abs. λmas, and wavelength of maximum fluorescence emission intensity, Em. λmax, recorded in methanol
The hemolytic properties of the polymers followed the same trend as the polymers without dansyl labels, which were shown in a previous report.22 Polymer 1 showed the lowest HC50 value of 36 μM while 2 was much less hemolytic with an HC50 value of 230 μM. The QAS-containing polymer 3 did not induce any observable hemolysis up to the highest concentration tested (~ 1% hemolysis at 480 μM). In exactly the same assay conditions, the bee venom toxin melittin showed an HC50 value of 4.4 μM. Based on their characterization and hemolytic activities, the representative copolymers in this study are suitable for investigating the role of cationic group structure in the membrane binding and disruption exerted by amphiphilic polymethacrylates.
Potentiometric Titration
Before studying the interaction of the polymers with lipid vesicles, we performed potentiometric titrations of 1 and 2 in order to quantify their extent of ionization (α) in buffers of various pH. Our previous studies revealed that the hemolytic activities of such polymers are dependent on α.22 The titration of these polymers was limited at high pH due to isomerization34 in the case of 1 or precipitation in the case of 2, as previously described.22 Despite these limitations, 1 and 2 were titrated to pH 8 and 7.5, respectively, in good agreement with the back-titration data, indicating that the polymers are stable in this pH range (Figure 2). The apparent pKa values of 1 and 2 are 8.3 and 7.5, respectively, according to extrapolation of curve fits to the generalized Henderson-Hasselbalch equation (eq. 2 in Experimental). At pH 6, the α values of 1 and 2 are > 0.95 whereas, at pH 7, the α value of 1 was about 0.9 and that of 2 decreased to about 0.7, indicating these polymers display a fraction of neutral amine groups at physiological pH. The presence of neutral (deprotonated) amine groups at physiological pH might play a role in the membrane-binding and disruption exerted by the copolymers.
Figure 2.
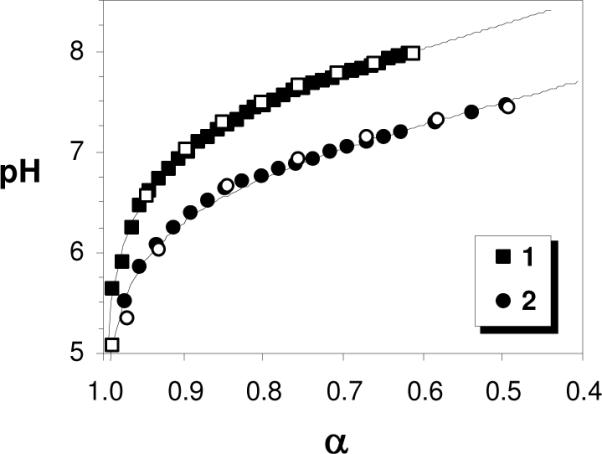
Potentiometric titration curves for polymers 1 and 2. Filled symbols represent the forward titration with NaOH aq. and empty symbols denote the back-titration with HCl aq. in good agreement. The curves are best fits to the generalized Henderson-Hasselbalch equation for polyelectrolytes.
Water-Octanol Partition Coefficients
In the design of membrane-active polymers, the importance of striking a fine-tuned balance between hydrophobic and hydrophilic components, known as “amphiphilic balance”, has been widely appreciated. When the components are present in the appropriate ratio, favorable antimicrobial activity and low toxicity to human cells can be observed.15, 39 When the polymers are too hydrophilic, only weak activity is exerted, whereas toxicity to both bacterial and human cells is observed when the polymers are excessively hydrophobic. The overall hydrophobicity of amphiphilic polymers, however, has been largely limited to qualitative descriptions of chemical structure of polymers, including comonomer ratio11, 26 and the length of alkyl side chains.31
In this study, we determined the water-octanol partition coefficient (logP) by measuring the equilibrium concentration of polymers in water and octanol phases based on the fluorescence intensity of the dansyl-labeled polymers in methanol. The logP is a quantitative measure of the overall hydrophobicity or amphiphilic balance, which is influenced by the cationic ammonium group structure. Traditionally, logP values have been useful to predict the ability of small compounds and drugs to permeate cell membranes in pharmaceutical applications.40
The logP of the polymers containing primary or tertiary amines were found to depend on pH, while those of the polymer containing quaternary ammonium groups were pH-independent (Figure 3). The polymers 1 and 2 displayed the same logP value of about −0.5 at pH 6, a condition in which nearly all of the amine groups are protonated according to the titration data (Figure 1), implying that the copolymers are rather hydrophilic in this condition. The logP for the polymer 1 increased gradually to about 0.0 as the pH was increased to 8. On the other hand, the logP value of 2 increased dramatically around pH 7 and reached a plateau at higher pH, toward a logP value of about +2.0. When the pH axis is re-scaled to the extent of ionization α, the logP values of 1 and 2 increase linearly with decreasing α. This indicates that deprotonation of the ammonium groups in the side chains renders the partitioning of polymer into the octanol phase more favorable, within the physiological pH range. The linear relationship suggests that the contribution of deprotonation of ammonium groups to the partitioning of polymers into octanol is constant. The slope for polymer 2 is roughly three-fold greater in magnitude than that of 1. These results indicate that the deprotonated (neutral) tertiary amines impart significantly greater hydrophobicity to the polymers relative to the primary amines, as would be expected. Hence, the chemical structure of the cationic groups, and not simply their positive charge, profoundly impacts the amphiphilic balance.
Figure 3.

Partition coefficients of each polymer between octanol and aqueous (HEPES or MES 10mM, NaCl 150 mM) phases as functions of (A) buffer pH and (B) extent of ionization α
The logP of 3 is about −1.0 across the entire pH range studied, implying that the polymer containing quaternary ammonium salts is predominantly hydrophilic and pH insensitive in this range. Interestingly, the logP values of 1 and 2 in the pH 6 buffer are roughly −0.5, which suggests slightly lower hydrophilicity relative to 3, even though 1 and 2 are almost completely ionized in that condition. This demonstrates that the overall hydrophilicity is not a simple function of the number of cationic ammonium groups, but also depends on their chemical structure. Specifically, amphiphilic copolymers containing QAS groups are more hydrophilic than exactly analogous polymers containing the same number of cationic primary or tertiary ammonium groups.
Polymer-POPC Dissociation Constants
We exploited the environmentally sensitive dansyl end groups to detect the partitioning of polymers into the hydrophobic region of Zwitterionic lipid bilayers. Using a previously described technique,31 we obtained the binding isotherms of each polymer to LUVs of POPC in buffers of pH ranging from 6 to 8 (Figure 4).
Figure 4.

Binding isotherms of polymers 1 (filled squares), 2 (empty circles), and 3 (empty triangles) to vesicles of POPC in buffer of pH (A) 6, (B) 7, and (C) 8. The initial polymer concentration was 1 μM. Fluorescence intensities are corrected for dilution and the inner filter effect. The curves represent best fits to the single-site binding model (equation 4). The curve fitting was employed with n = 6.
In the absence of lipids ([POPC] = 0 μM), each of the polymers exhibited fluorescence emission even in the aqueous buffers (F0). Although dansyl is typically non-fluorescent in the aqueous microenvironment, the amphiphilic polymers may adopt conformations or aggregates which partially shield the dansyl end groups from the aqueous surroundings. As the pH was elevated from 6 to 8, the initial fluorescence intensities F0 of 1 and 3 did not change significantly. On the other hand, the polymer 2 showed an F0 value of about 9.4 at pH 6 but increased markedly to about 20.2 in the pH 8 buffer, which suggests that the dansyl end group was transferred to a less polar microenvironment. Such an observation is consistent with the formation of hydrophobic polymer aggregates of 2 in the aqueous environment when the pH is elevated (pH = 8, logP = +2.0).
Upon titration of the polymer solutions with aliquots of POPC stock solution, changes in the fluorescence intensity were monitored in buffers ranging from pH 6 to 8. The fluorescence intensity of polymer 1 increased roughly 1.8 fold when saturated with liposomes ([POPC] = 192 μM) in each of the buffers used. At the same POPC concentration, the fluorescence intensity of polymer 2 increased about 1.6 fold in pH 6 and 1.7 fold in pH 7. However, in pH 8, the emission of 2 was not significantly altered by the addition of liposomes, which implies that the hydrophobic aggregates did not bind to POPC liposomes appreciably. This would imply that excessively hydrophobic (logP ~ +2.0) copolymers are not effective membrane active compounds due to the diminished propensity of their aggregates to insert into lipid bilayers, which has been hypothesized previously for other membrane-disrupting amphiphilic polymers.41, 42 Finally, the QAS-containing polymer 3 showed only slight changes upon liposome titration, regardless of buffer pH, which implies that the polymers did not significantly partition into the hydrophobic region of POPC lipid bilayers. This shows that excessively hydrophilic (logP ~ −1.0) copolymers are favorably solvated in the external aqueous environment and likewise ineffective as membrane active compounds. Between these extremes, copolymers with a logP value near 0.0, such as 1 at pH 7.5, appear to exert superior membrane-binding affinity, which implies that membrane activity can be tuned by quantitatively adjusting the amphiphilic balance. We show for the first time that this adjustment could be made by judiciously choosing the structure of the cationic appendages, in addition to the traditional approach of tuning the hydrophobic groups. This pivotal result may aid in explaining the superior membrane activity of polymers containing protonated, rather than quaternized, ammonium groups in the side chains, such as polymethacrylates,22 polystyrenes,27 and polydiallylamines.28
In order to quantify binding affinity, curve-fitting was performed on the isotherm data using equation 4 for the single-site binding model, as previously described, to obtain dissociation constants (KD) for polymer-liposome binding (Figure 5). It is interesting that the KD of polymer 2 was about 1.7-fold higher than that of 1 in pH 6, although nearly all of the amine groups in both polymers were protonated and their logP values were nearly identical at that pH. This indicates that the absolute number of cationic charges in the polymers and the logP value do not uniquely determine the binding affinity to POPC vesicles. The KD values of polymer 1 and 2 decreased with increasing pH and approached ~12 μM, indicating that the binding of polymers might be predominantly determined by their hydrophobicity at high pH. We further examined the effect of hydrophobicity on the binding of polymers by re-scaling the pH axis to logP (Figure 5, panel B). The KD values decreased with increasing logP values, indicating that an increase of the overall hydrophobicity drives the partitioning of the polymers to lipid bilayers. In the logP range of −0.7 to 0, polymer 1 showed lower KD values than 2. The difference between values of ln(KD) for 1 and 2 was roughly constant at ~ 0.7, corresponding to ΔΔG ~ 0.43 kcal/mol of polymer. This implies that the binding of polymer 1 to POPC vesicles is slightly more favorable than that of 2.
Figure 5.

(A) Dissociation constants for polymer-POPC vesicle binding monitored by dansyl fluorescence as a function of pH and (B) dissociation constant related to water-octanol partition coefficient in the log-log plot. The error bars represent the standard deviation of three KD values calculated from independent binding isotherm measurements.
We hypothesized that the lower KD values of 1 to POPC, relative to those of 2, arise from greater affinity of the primary amine groups for the phosphate lipid headgroups, relative to that of tertiary amines. To test this hypothesis, the water-octanol partition coefficients were measured for each of the polymers in the presence of the surfactant dodecylphosphate (DDP) in the pH 6 buffer (Figure 6). The logP values of each of the copolymers were sensitive to the presence of 1mM dodecylphosphate (DDP), although each polymer showed a distinct magnitude of change in logP upon the addition of the surfactant. It appears that the logP values of 1 displayed the most pronounced sensitivity to the addition of DDP. The magnitude of change in logP for 1 (+0.99) exceeded that of 2 (+0.54) and 3 (+0.46) upon addition of 1mM DDP. Since the pKa values of the phosphate group in DDP are reportedly 2.8 and 7.2, the mono-anion is the predominant species at pH 6.43 Therefore, we speculate that the cationic ammonium groups of polymer and the phosphate groups of DDP form a non-polar amine-phosphate complex. Such complexation would be expected to enhance the partitioning of the polymer to the non-polar octanol phase and, in that context, the primary amine groups appear to have the greatest affinity for phosphate headgroups in the surfactant. On the other hand, the logP of the polymer with quaternary ammonium salt groups, 3, which is expected to have electrostatic interaction with anionic DDP, was less affected by the presence DDP. This result suggests that the interaction between primary ammonium groups and anionic phosphate groups are driven by a combination of electrostatic and hydrogen bonding interactions. The energy is small compared to that of perfect hydrogen bonding (1–7 kcal/mol of amine), so the complexation can be considered weak. Furthermore, this outcome supports the notion that the insertion of dansyl groups on 1 into the hydrophobic core of phospholipid membrane is facilitated by hydrogen bonding to the phosphates in the lipid headgroups, which are reflected by the lower KD values (higher binding affinity) of polymer 1. Because guanidinium groups are known to form more robust complexes with phosphate groups, relative to primary amines, it is reasonable to hypothesize from this result that amphiphilic copolymers displaying guanidine moieties in the side chains would partition into lipid bilayers as well. While a large body of work has focused on the cell-penetrating ability of arginine-rich macromolecules,44, 45 relatively little attention has been paid to amphiphilic guanidine-containing copolymers in the design of membrane-disrupting polymers.
Figure 6.

Partition coefficients between aqueous buffer (10 mM MES, 150 mM NaCl, pH 6) and octanol for each of the copolymers in the presence of 0, 0.1, and 1 mM dodecylphosphate. The total polymer concentration was 0.1 mM.
At pH 7, 1 and 2 showed the same KD, which implies that the total binding energies to POPC vesicles are equivalent in that condition. However, the relative contributions of overall hydrophobicity (logP) and amine-phosphate complexation to the overall binding energy indeed differed. In other words, the amine-phosphate complexation apparently exerted a more significant contribution to the binding for polymer 1 than 2. This may aide in explaining why 1 is more hemolytic than 2 and 3, despite their identical KD values in the pH 7 buffer. That is, the extent of the hemolysis may reflect the action (pore formation or total disruption) exerted by polymers on the membranes subsequent to initial binding, rather than simply the amount of bound polymer.
Dye Efflux from Liposomes
In order to assess the ability of the polymers in this study to disrupt the integrity of model membranes, we monitored the leakage of an entrapped fluorophore from within large unilaminar vesicles of POPC upon polymer addition. The polymers 1 and 2 induced dye efflux from the liposomes in a pH-dependent manner, while the polymer 3 was not significantly membrane-lytic regardless of the buffer pH (Figure 7).
Figure 7.

Sulforhodamine B (SRB) leakage from large unilaminar vesicles (LUVs) of POPC induced by each of the copolymers in buffer of pH (A) 6, (B) 7, and (C) 8. The total lipid concentration was fixed at 10 μM. The error bars represent standard deviation of the triplicate measurements.
At pH 6, the polymers have the same number of cationic charges and similar logP values. However, they showed marked differences in their ability to induce dye leakage from vesicles of POPC. This demonstrates that not only the net cationic charge and hydrophobicity, but also the chemical structure of the cationic groups, are important determinants of membrane-lytic potency.
Interestingly, the polymer containing primary amines, 1, retained its membrane-lytic property in buffers ranging from pH 6 to 8. On the other hand, the membrane damage caused by 2 was enhanced by increasing pH in this range (Figure 8). Recalling that the KD value of 2 was enhanced by approximately two-fold upon increasing pH from 6 to 7, it is interesting that the concentration of this polymer which induced ~50% dye leakage seemed to increase by orders of magnitude as the pH was varied in this range. Regardless of pH, the polymer with QAS moieties, 3, did not show the ability to induce dye leakage from POPC vesicles. This is likely due to little or no binding affinity to POPC displayed by this completely hydrophilic polymer (logP ~ −1). Membrane disruption by polymer containing QAS units is indeed possible when longer alkyl chains are attached in order to increase the hydrophobicity; however, as we show here by direct comparison, the QAS units are less effective at conferring membrane-disrupting ability relative to protonated (primary or tertiary) ammonium groups. In addition to the binding curves, this result further corroborates several reports on the enhanced antimicrobial and hemolytic activity of polymers bearing protonated, rather than QAS, ammonium groups.22, 27, 28
Figure 8.
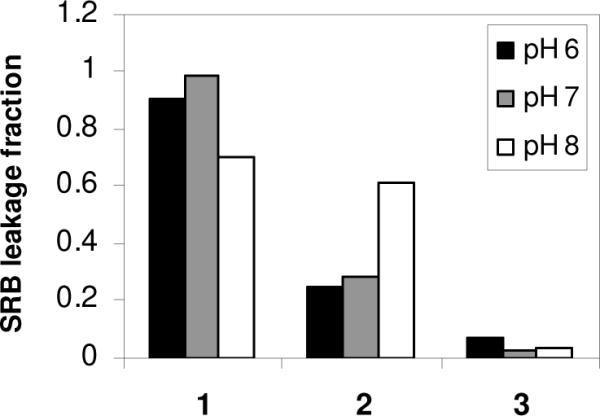
Fraction of SRB leakage induced by the copolymers at a fixed concentration of 62.5 nM in buffers of pH 6, 7, and 8.
At pH 7, polymers 1 and 2 have the same KD value (Fig. 5), which implies that the same concentration of polymers bound to the POPC membranes. However, in this condition, 1 was much more lytic than 2 (Fig. 7, Panel B). This implies that the membrane-permeabilization ability of the bound polymers depends on the structure of their cationic groups, rather than the mass action (total bound amount) of polymers on the membrane. We speculate that this is likely due to the differences in amine-phosphate interactions: the formation of complex between primary ammonium groups and phosphate lipid head groups might facilitate the insertion of polymers into the lipid membrane such that the membrane permeabilization is enhanced.
Solid-state NMR
In order to further probe the role of cationic group structure in the interaction of polymers with model membranes at high-resolution, 31P NMR spectra of mechanically aligned POPC bilayers were obtained. The 31P spectrum of mechanically aligned lipid bilayers is sensitive to the orientation of the lipid molecules, the phase of lipids, and the interaction between lipid molecules and additives such as peptides.46, 47 The pure POPC bilayers (0 mole % polymer) showed a single sharp peak at 31.7 ppm, which is indicative of the well-aligned sample with the orientation of the lipid bilayer normal parallel to the external magnetic field. Increasing the mole percentage of bound polymer relative to lipid in each case caused spectral changes indicative of electrostatic interactions between the phosphate group of POPC and the amine group of polymers leading to disruption in the lipid bilayers (Figure 9).
Figure 9.

Amphiphilic polymer 1, 2, or 3 induced changes in lipid bilayers revealed by 31P NMR spectra. Experimental 31P chemical shift spectra of mechanically-aligned bilayer samples of POPC with mole percentages of polymer relative to lipid of (i) 0%, (ii) 1 mol %, (iii) 2 mol %, and (iv) 3 mol %. All NMR experiments were performed with the bilayer normal was set parallel to the external magnetic field of the spectrometer. Other experimental details are given in the main text.
The presence of the intense peak at 31.7 ppm for lipid bilayers with the polymers 1 or 3 incorporated suggest that a majority of the lipid populations are in lamellar phase and not perturbed by the polymer interaction. On the other hand, POPC bilayers incorporated with the polymer 2 at the concentrations 2 mol% and 3 mol% showed an apparent shift of main peak to 29.2 ppm and 27.8 ppm, respectively. Since the perpendicular-edge of the spectrum arising from unaligned lipids is unchanged due to polymer 2 interaction, the decreasing frequency of the intense peak (~31.7 ppm) suggest conformational changes of POPC head group induced by the interaction of polymer 2. The spectra of POPC bilayers incorporated with the polymer 1 and 2, additional peaks at 25 ppm for all of the polymer 1 and 22 ppm and 21 ppm for the 2 mol% and 3 mol% of the polymer 2, respectively, are clearly seen in the spectra. It is also seen a broad intensity over the range of the 31P chemical shift anisotropy arising from unaligned lipids for most bilayers samples incorporated with polymers. A small isotropic peak at 2 ppm is also seen from the POPC bilayers incorporated with 3 mol% polymer 2. The polymer 3 sample did not give extra peaks, but only a broad powder pattern. These results suggest that polymers 1 and 2 alter the head group conformation of POPC leading to polymer-rich and polymer-poor lipid domains. For example, a peak at 25, 22, or 21 ppm may be attributed to the POPC bilayers containing polymer-rich domains.
Polymer 1, which contains primary ammonium appendages, caused a broadening of the peak near 31 ppm and also gave rise to two distinct peaks near 25 and −13 ppm when the mole percent of bound polymer was 2 or 3%. These changes demonstrate that the polymer alters the conformation of the POPC lipid headgroups. The development of a spectral feature near −13 ppm is indicative of lipid headgroups aligned perpendicular to the membrane, which may result from the formation of pores. The amine-phosphate interactions may enhance the reorganization of lipids upon the insertion of polymers to lipid bilayer.
Mixing of polymer 2 into the POPC bilayers also caused distinct changes in the 31P spectra. As with 1, features also appeared near 25 and −13 ppm, which suggests that mechanism of membrane disruption employed by 1 and 2 are similar to some extent. However, at 3 mole % of 2, the POPC bilayers displayed a very broad spectral feature spanning from 31 to −13 ppm due to polymer-induced disordering of lipids as seen in the case of polymer 3. It is likely that such gross disorganization of the lipid headgroups arises from irregular aggregate of lipids with the hydrophobic polymer2.
Finally, it is interesting that the polymer 3, which contains quaternary ammonium salt groups, exerted a fundamentally different effect on the POPC bilayers compared to 1 and 2. The peak near −13 ppm appears more prominent at lower polymer content and the feature observed around 25 to 20 ppm is not evident at all. This suggests that structure of the cationic groups in the polymer modulates the polymer interaction with lipid bilayers. Although 3 clearly disrupts POPC bilayers when binding is forced, the polymer binds only weakly to POPC vesicles in solution and hence does not induce appreciable dye leakage. This shows that the QAS-containing polymers could potentially form pores and thereby permeabilize POPC bilayers, if the binding was enhanced by increasing the overall hydrophobicity.
Membrane Disruption Mechanism
The results from the series of experiments on partitioning and membrane binding of polymers indicated that the interaction of polymer 1 with lipid bilayers likely involves complexation between the primary ammonium groups of the polymers and phosphate head groups of lipids. In addition, while polymer 2 showed the same binding affinity for liposomes at pH 7, polymer 1 displayed a much greater ability to permeabilize lipid bilayers. This indicates that the dye leakage occurs as a result of molecular interactions of the polymer within the lipid bilayers after initial binding, which likely involves amine-phosphate complexation. The solid-state NMR study likewise indicated that the ammonium groups interact with lipid head groups to induce a reorganization of the lipid bilayer structure, including perpendicular alignment of the lipid head groups. Further analysis of these spectra suggests that the polymer 1 induced the formation of polymer-rich and poor domains. It has been proposed that the membrane-lytic peptides, which often display primary amino functionality, operate by the formation of “barrel-stave” or “toroidal” pores in cell membranes,48 in which a fraction of the lipid headgroups are oriented perpendicular to membrane surface. Alternatively, peptides may induce total disruption resulting from accumulation of peptides on the membranes via the “carpet” mechanism.49 The synthetic polymers in this report also efficiently induced the permeabilization of model membranes, when the cationic groups were protonated primary amines rather than quaternary ammonium salt groups. However, the exact mechanism(s) of their membrane-disruption action remains unclear at present and is currently being investigated. Considering that the solid state NMR results indicated significant lipid head group disordering and reorientation due to polymer interaction, it is reasonable to hypothesize that torodial-type pore formation or a carpet mechanism may be operative. Probing the details of the membrane disruption mechanism exerted by polymers containing the primary amines would be highly desirable in the future.
In order to design non-hemolytic antimicrobial polymers, it has been established that minimization of hydrophobicity is a requirement.31 This is largely due to the fact that the partitioning of polymers into zwitterionic membranes is mainly driven by hydrophobic effects, which were quantified in this study. Interestingly, we also found that the structure of the cationic groups contributes to the membrane association even when hydrophobicity is fixed. The ability of host defense peptides to selectively kill bacteria without harming human cells has been ascribed, at least in part, to electrostatic attraction of the cationic peptides to the anionic bacterial membranes.21 While the polymers in this study apparently permeabilize mammalian cell membranes depending on their hydrophobicity, it remains unclear whether the same mechanism is responsible for their antimicrobial activity. For this reason, given that cell membranes of bacteria and mammalian cells consist of distinctively different types and composition of lipids, quantitative studies on the role of the lipid structure in the polymer-lipid interaction will also be important in the future to clarify the design principles for obtaining selective antimicrobials. It has been shown that the polymers containing primary amines can be tuned to kill bacteria while causing no hemolysis, by reducing the fraction of hydrophobic side chains. This method is ineffective for QAS-containing polymers, which require longer alkyl side chains as the hydrophobic groups in order to express antimicrobial activity as well as hemolytic activity.22 Clarifying the mechanism of their antimicrobial action will therefore enable a complete understanding of the role played by cationic groups in the membrane activity of these emerging new materials.
Conclusions
It would appear that the polymers containing primary amines are more potent hemolytic agents due to their enhanced ability to bind and disrupt the organization of lipid bilayers, relative to polymers with tertiary or quaternary ammonium groups. The structure of cationic group structure in the design of membrane-disrupting polymers is therefore considered a factor of paramount importance. As previously found, achieving a fine balance of hydrophobic and hydrophilic groups enables pronounced interaction of the polymers with biomembranes. We found that the cationic group structure affects the pKa and, in turn, the overall hydrophobicity. The dissociation constants of polymers to lipid bilayers showed pH dependence which seems to rely on hydrophobicity combined with hydrogen-bonding effects. Furthermore, interaction of the polymer side chains with the lipid headgroups as revealed by 31P NMR experiments appears to modulate the lipid head group conformation that results in membrane-disrupting behavior of polymers.
While disinfecting synthetic polymers have traditionally employed quaternary ammonium salt (QAS) moieties as their cationic units, primary amines endow a wide variety of host defense peptides with a net cationic charge. We found here that the membrane disrupting ability of polymers is indeed enhanced by using primary amines, rather than QAS units, as the cationic groups at physiological pH. This appears to highlight the design rationale of membrane-disrupting macromolecules found in nature. Such information may prove useful in the future development of non-hemolytic antimicrobials, as well as gene- or drug-delivery agents.
Supplementary Material
Acknowledgements
This work was supported by the Department of Biologic and Materials Sciences, University of Michigan School of Dentistry and NSF CAREER Award (DMR-0845592 to K.K.). This work was partly supported by the National Institutes of Health (AI054515 and RR023597 to A.R.) and CRIF-NSF funding. Dong Kuk Lee was supported by a Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Funds, KRF-2006-331-C00153). We also thank Dr. Robert Davenport of the University of Michigan Hospital for providing a unit of human red blood cells for this work.
Footnotes
Supporting Information Synthetic schemes for the synthesis of the dansyl-functionalized chain transfer agent and the random copolymers, emission spectra of the polymers, hemolysis raw data and curve fitting, and binding curves at various pH can be found in the supporting information. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J. Infect Dis Soc, A., Clinical Infectious Diseases. 2008;46(2):155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 2.Zasloff M. Nature. 2002;415(6870):389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 3.Shai Y. Biopolymers. 2002;66(4):236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 4.Matsuzaki K. Biochim. Biophys. Acta-Biomembr. 1999;1462(1–2):1–10. doi: 10.1016/s0005-2736(99)00197-2. [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharjya S, Ramamoorthy A. FEBS J. 2009;276(22):6465–6473. doi: 10.1111/j.1742-4658.2009.07357.x. [DOI] [PubMed] [Google Scholar]
- 6.Marsh ENG, Buer BC, Ramamoorthy A. Mol. Biosyst. 2009;5(10):1143–1147. doi: 10.1039/b909864j. [DOI] [PubMed] [Google Scholar]
- 7.Wade D, Boman A, Wahlin B, Drain CM, Andreu D, Boman HG, Merrifield RB. Proc. Nat. Acad. Sci. U.S.A. 1990;87(12):4761–4765. doi: 10.1073/pnas.87.12.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ge YG, MacDonald DL, Holroyd KJ, Thornsberry C, Wexler H, Zasloff M. Antimicrob. Agents Chemother. 1999;43(4):782–788. doi: 10.1128/aac.43.4.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tew GN, Scott RW, Klein ML, Degrado WF. Acc. Chem. Res. 2010;43(1):30–39. doi: 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CZS, Cooper SL. Adv. Mater. 2000;12(11):843–846. [Google Scholar]
- 11.Kuroda K, DeGrado WF. J. Am. Chem. Soc. 2005;127(12):4128–4129. doi: 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]
- 12.Marr AK, Gooderham WJ, Hancock REW. Curr. Opin. Pharmacol. 2006;6(5):468–472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Epand RF, Mowery BP, Lee SE, Stahl SS, Lehrer RI, Gellman SH, Epand RM. J. Mol. Biol. 2008;379(1):38–50. doi: 10.1016/j.jmb.2008.03.047. [DOI] [PubMed] [Google Scholar]
- 14.Gabriel GJ, Som A, Madkour AE, Eren T, Tew GN. Mater. Sci. Eng. R-Reports. 2007;57(1–6):28–64. doi: 10.1016/j.mser.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palermo EF, Kuroda K. Appl. Microbiol. Biotechnol. 2010;87(5):1605–1615. doi: 10.1007/s00253-010-2687-z. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda T, Tazuke S, Suzuki Y. Macromol. Chem. Phys. 1984;185(5):869–876. [Google Scholar]
- 17.Kenawy ER, Worley SD, Broughton R. Biomacromolecules. 2007;8(5):1359–1384. doi: 10.1021/bm061150q. [DOI] [PubMed] [Google Scholar]
- 18.Tashiro T. Macromol. Mater. Eng. 2001;286(2):63–87. [Google Scholar]
- 19.Tiller JC, Liao CJ, Lewis K, Klibanov AM. Proc. Nat. Acad. Sci. U.S.A. 2001;98(11):5981–5985. doi: 10.1073/pnas.111143098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madkour AE, Dabkowski JA, Nusslein K, Tew GN. Langmuir. 2009;25(2):1060–1067. doi: 10.1021/la802953v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuzaki K, Sugishita K, Fujii N, Miyajima K. Biophys. J. 1994;66(2):A223–A223. [Google Scholar]
- 22.Palermo EF, Kuroda K. Biomacromolecules. 2009;10(6):1416–1428. doi: 10.1021/bm900044x. [DOI] [PubMed] [Google Scholar]
- 23.Palermo EF, Sovadinova I, Kuroda K. Biomacromolecules. 2009;10(11):3098–3107. doi: 10.1021/bm900784x. [DOI] [PubMed] [Google Scholar]
- 24.Ilker MF, Nusslein K, Tew GN, Coughlin EB. J. Am. Chem. Soc. 2004;126(48):15870–15875. doi: 10.1021/ja045664d. [DOI] [PubMed] [Google Scholar]
- 25.Lienkamp K, Madkour AE, Musante A, Nelson CF, Nusslein K, Tew GN. J. Am. Chem. Soc. 2008;130(30):9836–9843. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mowery BP, Lee SE, Kissounko DA, Epand RF, Epand RM, Weisblum B, Stahl SS, Gellman SH. J. Am. Chem. Soc. 2007;129(50):15474–+. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- 27.Gelman MA, Weisblum B, Lynn DM, Gellman SH. Org. Lett. 2004;6(4):557–560. doi: 10.1021/ol036341+. [DOI] [PubMed] [Google Scholar]
- 28.Timofeeva LM, Kleshcheva NA, Moroz AF, Didenko LV. Biomacromolecules. 2009;10(11):2976–2986. doi: 10.1021/bm900435v. [DOI] [PubMed] [Google Scholar]
- 29.Binder WH. Angew. Chem. Int. Ed. 2008;47(17):3092–3095. doi: 10.1002/anie.200800269. [DOI] [PubMed] [Google Scholar]
- 30.Yang LH, Gordon VD, Trinkle DR, Schmidt NW, Davis MA, DeVries C, Som A, Cronan JE, Tew GN, Wong GCL. Proc. Nat. Acad. Sci. U.S.A. 2008;105(52):20595–20600. doi: 10.1073/pnas.0806456105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuroda K, Caputo GA, DeGrado WF. Chem. Eur. J. 2009;15(5):1123–33. doi: 10.1002/chem.200801523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas JL, You H, Tirrell DA. J. Am. Chem. Soc. 1995;117(10):2949–2950. [Google Scholar]
- 33.Wen S, Yin XN, Stevenson WTK. J. Appl. Polym. Sci. 1991;43(1):205–212. [Google Scholar]
- 34.He LH, Read ES, Armes SP, Adams DJ. Macromolecules. 2007;40(13):4429–4438. [Google Scholar]
- 35.Hallock KJ, Wildman KH, Lee DK, Ramamoorthy A. Biophys. J. 2002;82(5):2499–2503. doi: 10.1016/S0006-3495(02)75592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen RF. Arch. Biochem. Biophys. 1967;120(3):609. [Google Scholar]
- 37.Chen RF. Anal. Biochem. 1968;25(1–3):412. doi: 10.1016/0003-2697(68)90116-4. [DOI] [PubMed] [Google Scholar]
- 38.Chen RF. Arch. Biochem. Biophys. 1968;128(1):163. doi: 10.1016/0003-9861(68)90019-2. [DOI] [PubMed] [Google Scholar]
- 39.Mowery BP, Lindner AH, Weisblum B, Stahl SS, Gellman SH. J. Am. Chem. Soc. 2009;131(28):9735–9745. doi: 10.1021/ja901613g. [DOI] [PubMed] [Google Scholar]
- 40.Leo AJ, Hansch C. Perspect. Drug Discovery. Des. 1999;17(1):1–25. [Google Scholar]
- 41.Murthy N, Campbell J, Fausto N, Hoffman AS, Stayton PS. Bioconj. Chem. 2003;14(2):412–419. doi: 10.1021/bc020056d. [DOI] [PubMed] [Google Scholar]
- 42.Thomas JL, Barton SW, Tirrell DA. Biophys. J. 1994;67(3):1101–1106. doi: 10.1016/S0006-3495(94)80575-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakayama K, Tari I, Sakai M, Murata Y, Sugihara G. J. Oleo Sci. 2004;53(5) [Google Scholar]
- 44.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. Adv. Drug Delivery Rev. 2008;60(4–5):452–472. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc. Nat. Acad. Sci. U.S.A. 2000;97(24):13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramamoorthy A. Solid State Nucl. Magn. Reson. 2009;35(4):201–207. doi: 10.1016/j.ssnmr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hallock KJ, Lee DK, Ramamoorthy A. Biophys. J. 2003;84(5):3052–3060. doi: 10.1016/S0006-3495(03)70031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsuzaki K. Biochim. Biophys. Acta-Rev. Biomembr. 1998;1376(3):391–400. doi: 10.1016/s0304-4157(98)00014-8. [DOI] [PubMed] [Google Scholar]
- 49.Shai Y. Biophys. J. 2001;80(1):2A–3A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.