

Abstract
We have previously shown that immunodeficient mice exhibit significant facial motoneuron (FMN) loss compared to wild-type (WT) mice after a facial nerve axotomy. Interleukin-10 (IL-10) is known as a regulatory cytokine that plays an important role in maintaining the anti-inflammatory environment within the central nervous system (CNS). IL-10 is produced by a number of different cells, including Th2 cells, and may exert an anti-apoptotic action on neurons directly. In the present study, the role of IL-10 in mediating neuroprotection following a facial nerve axotomy model in Rag2- and IL-10-deficient mice was investigated. Results indicate that IL-10 is neuroprotective, but only in the presence of CD4+ T cells that are not the requisite source of IL-10. In addition, using real-time PCR analysis of laser microdissected brainstem sections, results show that IL-10 mRNA is constitutively expressed in the facial nucleus and that a transient, significant reduction of IL-10 mRNA occurs following axotomy under immunodeficient conditions. Dual labeling immunofluorescence data show, unexpectedly, that the IL-10 receptor (IL-10R) is constitutively expressed by facial motoneurons, but is selectively induced in astrocytes within the facial nucleus after axotomy. Thus, a non-CD4+ T cell source of IL-10 is necessary for modulating both glial and neuronal events that mediate neuroprotection of injured motoneurons, but only with the cooperation of CD4+ T cells, providing an avenue of novel investigation into therapeutic approaches to prevent or reverse motoneuron diseases, such as amyotrophic lateral sclerosis (ALS).
Keywords: Interleukin-10, CD4+ T cell, facial motoneuron, survival, axotomy, neuroprotection
Introduction
Neurons are differentiated cells that lack the capacity to divide. Therefore, the loss of a neuron due to nerve injury prevents nerve regeneration and functional recovery from taking place, primarily because essential proteins needed for nerve repair are no longer produced in the absence of the cell body. Therefore, supporting neuronal survival during a neuroinflammatory disease process or after a nerve injury is vitally important to prevent loss of nerve function. One mechanism that appears to determine if neurodestruction or neuroprotection occurs within the CNS involves the nature and extent of the immune response that develops. It is generally thought that pro-inflammatory responses are detrimental, while anti-inflammatory responses are beneficial (Steinman, 2004; Griffiths et al., 2007; Stoll et al., 2002). Several immunotherapies have been investigated and proven to be effective for the treatment of neurological disorders, e.g., glatiramer acetate (GA) for multiple sclerosis (MS) and beta-amyloid vaccine for Alzheimer’s disease (Ruggieri et al., 2007, Hawkes et al., 2007). As a result, preventing or ameliorating neurological disease through immunotherapy has become an important research topic for drug discovery and clinical treatment of neurological disorders (Hawkes et al., 2007; De Jager et al., 2007), although the mechanisms by which current immunotherapies exert their effects are not well understood.
Previous studies utilizing the facial nerve axotomy model have revealed that a competent immune system, particularly with respect to the CD4+ T cell, is necessary for promoting facial motoneuron (FMN) survival after facial nerve injury (Serpe et al., 1999, 2000, 2003). Recently, we reported that multiple subsets of CD4+ cells, including Th1, Th2, Th17, type 1 regulatory (Tr1), and forkhead box P3 (Foxp3) positive regulatory T cells (Foxp3+ Treg) develop and expand following facial nerve axotomy (Xin et al.,2008). Th1 cells have also been shown to be neuroprotective following trauma (Kipnis et al., 2002), yet, when using Stat6- and Stat4-deficient mice, we found that Th2, but not Th1, cells, respectively, are critical for promoting FMN survival after facial nerve axotomy (Deboy et al., 2006a), and that both activation in the periphery and re-activation within the CNS are necessary to exert the neuroprotective effect (Byram et al., 2004). In a parallel study, CD4+CD25+ T regulatory cells, i.e., Foxp3+ Treg cells, were found to be unnecessary for FMN survival after axotomy (Deboy et al., 2006b), which is consistent with the findings of Kipnis et al (2004) that Foxp3+ Treg cells have a dual effect in neurodegeneration. Based on these data, we proposed that the anti-inflammatory effects of Th2 cells, which need to be re-activated within the CNS following facial nerve axotomy, promote FMN survival.
IL-10 is the phenotype-defining cytokine of Tr1 cells and is also produced by Th2 cells. IL-10 is a potent inhibitor of inflammation due to its ability to dampen Th1 cell activity and subsequent inflammatory cytokine production. In the past ten years, the beneficial effects of IL-10 have been observed in several neuroinflammatory disease models, such as experimental autoimmune encephalomyelitis (EAE), traumatic or excitotoxic spinal cord injury, stroke and Parkinson’s disease (Cua et al., 2001, Brewer et al., 1999; Bethea et al., 1999; Frenkel et al., 2005; Qian et al., 2006). Collectively, these findings indicate that IL-10 plays a role in regulating CNS inflammation, and perhaps the normal maintenance of neuron viability (Strle et al., 2001). Based on the potent ability of IL-10 to inhibit the severity of neuroinflammatory diseases, we hypothesize that IL-10 is a critical factor involved in CD4+ T cell-mediated neuroprotection, and that the CD4+ T cell is the likely source of this IL-10. In the present study, we demonstrate a requirement for both CD4+ T cells and IL-10 in promoting FMN survival after axotomy. We discovered that resident CNS cells, not CD4+ T cells, are the requisite source of the neuroprotective IL-10. However, CD4+ T cells are essential for the regulation and maintenance of IL-10 expression in the facial nucleus after facial nerve axotomy. Furthermore, while IL-10R was found to be constitutively expressed in FMN, it was induced in astrocytes activated by peripheral axotomy. We discuss the hypothesis that IL-10 and CD4+ T cells work cooperatively within the CNS through modulation of anti-inflammatory events in the environment surrounding the injured neurons and, perhaps, through direct action on anti-apoptotic pathways within the neuron itself.
Materials and Methods
Animals and surgical procedures
All mice used in the present study were on a C57BL/6 background. Seven-week-old female wild-type, Rag-2 knockout (Rag-2-/-) mice were obtained from Taconic Laboratories (Germantown, NY) and IL-10 knockout (IL-10-/-) mice from the Jackson laboratory (Bar Harbor, ME). Mice were housed under a 12-h light/dark cycle in microisolater cages contained within a laminar flow system to maintain a pathogen-free environment. All surgical procedures were completed in accordance with National Institutes of Health guidelines on the care and use of laboratory animals for research purposes. Mice were anesthetized with 3% halothane for all surgical procedures. Using aseptic techniques, the right facial nerve of each animal was exposed at its exit from the stylomastoid foramen and completely transected (Serpe et al., 1999; Jones and LaVelle, 1985). The distal nerve stump was resected and pushed away from the proximal nerve stump, thereby, preventing reconnection of the facial nerve.
Isolation of CD4+ T cells and Adoptive transfer
Spleen and lymph nodes (LNs) were collected from donor mice (n = 3-4). Single cell suspensions were prepared and red blood cells were removed by treatment with ammonium chloride for four minutes at 37°C. A cell separation media, Lympholyte-M (Cedarlane Lab, Ontario, Canada) was used to remove dead cells and enrich for viable lymphocytes. Cells were washed with phosphate buffered saline (PBS), pH 7.2, and resuspended in 90 μl of PBS + 5% bovine serum albumin (BSA) per 107 total cells. To isolate CD4+ T cells, 10 μl of magnetic cell sorting (MACS) CD4 (L3T4) microbeads (Miltenyi Biotec, California) per 107 total cells were added, mixed, and incubated for 15 minutes at 6-12 °C. Cells were washed with PBS + 5% BSA, centrifuged at 300×g for 10 minutes, supernatant removed, and cell pellet resuspended in 500 μl PBS + 5% BSA per 108 total cells. Cells were magnetically sorted using an automated cell sorter, autoMACS (Miltenyi Biotec, Bergisch-Gladbach, Germany). Purified CD4+ T cells in PBS were intravenously transferred to recipient mice (5×106 in 500 μl per animal) through the tail vein one week prior to axotomy. The details of the procedure and the repopulation of transferred cells in the host lymphoid tissues have been described in our previous work (Serpe et al., 1999, 2000). Adoptively transferred mouse groups included CD4+ T cells derived from WT mice injected into Rag-2-/- or IL-10-/- mice and CD4+ T cells isolated from WT and IL-10-/- mice injected into Rag-2-/- mice (all recipient mice n=6/group).
Tissue sectioning and cell counts
All FMN survival assays were performed four weeks postoperative (wpo), the time point when significant neuronal loss is observed in the facial nucleus, as previously published (Serpe et al., 2000). Brains were collected and sectioned at a thickness of 25 μm. These sections were stained with thionin and counted under blind conditions as previously described (Serpe et al., 2000). For the determination of basal numbers of FMN in the uninjured facial nucleus between WT and IL-10-/- mice, we calculated the total number of the FMN in each facial nucleus, and compared left and right absolute values through one-way ANOVA. For the comparison of the FMN survival levels after axotomy, FMN counts from aligned sections were added together to determine the total number on each side, and then the percentage of FMN survival for each animal was calculated by dividing FMN counts on the right (injured) side by counts on left (uninjured) side. The counting correction factor and section alignment procedures have been described in our previous publications (Serpe et al., 1999, 2000).
Il-10 administration via gel foam at injury site
Due to fast clearance by the kidneys, we did not intravenously inject IL-10 to mice (Radwanski et al., 1998). To slow the turnover and prolong the half-life of IL-10, gel foam impregnated with recombinant murine IL-10 (20μl, 50ng/ml, PeproTech, Rocky Hill, NJ) was placed at the nerve stump immediately following axotomy and the wound was closed as usual (Serpe et al., 2005). To maintain the continuous release of IL-10 from gel foam, another 3 injections of IL-10 (30μl per injection) were given at 1, 2 and 3 wpo. This approach has been successfully used with neurotrophic factors, and rescue with these factors has been achieved when applied peripherally at the nerve stump (Serpe et al., 2005).
RT-PCR of IL-10, IL-10Rα and IL-10Rβ
Laser microdissection
Brains were collected from WT and Rag-2-/- (n = 3-4) mice at 7 dpo following axotomy. Twenty-five μm coronal cryostat sections were collected throughout the caudal-rostral extent of the facial motor nucleus. The tissue sections were thaw-mounted onto glass PEN foil slides (Leica, Germany). The sections were fixed in 100% ethanol and stained with thionin. The whole facial motor nuclei were laser microdissected using a Leica AS laser microdissector (Nuhsbaum, McHenry, IL) controlled manually. The control and axotomized facial nuclei were collected separately for each animal. The laser microdissected samples were collected in 50 μL of PicoPure RNA Isolation Kit extraction buffer (Arcturus, Sunnyvale, CA) within the caps of PCR epitubes.
RNA isolation and Reverse-Transcription (RT)-PCR
From laser microdissected facial motor nucleus collected samples, total cellular RNA was isolated with the PicoPure RNA Isolation Kit (Arcturus, Sunnyvale, CA), which contained a DNase treatment step. The RNA was then reverse transcribed using Superscript First Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA) with oligo d(T) primers according to manufacturer’s instructions. Complementary DNA was treated with RNase-H and used in real-time PCR reactions. Amplification was detected with SYBR green fluorescence using the iCycler iQ detection system (Applied Biosystems, Foster City, CA). Twenty-five μL PCR reactions contained 1X SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), 1 μL of the aforementioned cDNA dilution, and 200 nM forward and reverse primers. PCR primers (mouse IL-10, IL-10Rαand IL-10Rβ) were purchased from SABiosciences Corp (Frederick, MD). RT-PCR was conducted with a 95°C initial denaturing step for 10 minutes, followed by 45 repeated cycles for 30 seconds denaturing at 95°C, 30 seconds annealing at the optimal annealing temperature (TA), and 30 seconds extension at 65°C. The TA for each gene of interest was determined using either whole mouse brain cDNA or mouse spleen cDNA as the template using a proper temperature gradient according to the designed primer’s estimated annealing temperature. Fluorescence was monitored during the annealing steps. To determine the specificity of the amplified product, a melt curve analysis was performed immediately after the amplification protocol and compared to the predicted melt temperatures for each gene of interest investigated. Relative mRNA expression levels acquired during the PCR amplification protocol were analyzed using the ΔΔCT method against the internal standard, GAPDH.
Electrophoresis
PCR amplified products were run on Criterion precast 10% nondenaturing polyacrylamide TBE gels (BioRad, Hercules, CA) in a Criterion cell vertical electrophoresis system (BioRad, Hercules, CA) for 90 minutes at 100V. PCR amplified products were extracted from 96 well PCR plates and diluted 4:1 with nucleic acid sample loading buffer (BioRad, Hercules, CA). TBE gels were stained with SYBR Green I nucleic acid gel stain (Invitrogen, Carlsbad, CA), with 100μL added to the 1X TBE nucleic acid electrophoresis running buffer (BioRad, Hercules, CA). Gels were imaged on a STORM 860 Phosphoimager using Storm Scanner and ImageQuant programs.
Immunohistochemistry staining
WT mouse brains (n=3) were cryostat sectioned at a thickness of 8 μm and thaw-mounted (37°C for 5–10 min) onto pre-cleaned SuperFrost slides (Fisher Scientific). Sections were fixed in 4% paraformaldehyde in PBS (pH 7.2) for 15 min then subjected to immunochemistry or double immunofluorescence staining following the below protocols. For regular immunohistochemistry staining, the sections were blocked for endogenous biotin using 1% H2O2 in PBS for 5 min, and for non-specific staining using 10% BSA in PBS for 1 hr. Sections were incubated overnight with rabbit anti-IL10Rα (1:200, Rabbit IgG; Santa Cruz) in PBS at 4°C. Sections were washed extensively in PBS and incubated with biotinylated donkey anti-rabbit (1:500, Santa Cruz;) at room temperature for 1 hour, followed by an avidin-biotin-HRP complex (Standard Ultra-Sensitive ABC Staining Kit, Pierce, Rockford, IL), and chromagenic reaction with 3, 3′-diaminobenzidine (DAB, Peroxidase Substrate Kit; Vector Laboratories, Burlingame, CA). Sections were covered with Permount (Fisher-Scientific), coverslipped and viewed using an Olympus BX50 microscope (Tokyo, Japan).
For double immunofluorescence staining, sections were blocked for non-specific staining using 10% BSA in PBS for 1 hr. Sections were incubated overnight with rabbit anti-IL10Rα (1:200, Rabbit IgG; Santa Cruz) in combination of cell marker antibodies, activated astrocytes (GFAP), activated microglia (CD68), and FMN (NeuN in PBS at 4°C. Sections were washed extensively in PBS and incubated with the secondary antibody at room temperature for 1 hour. The following primary and secondary antibodies were used: Alexa Fluor® 488 anti-GFAP (1:800; Invitrogen), Alexa Fluor® 488 CD68 (1:500, Invitrogen), biotinylated NeuN (1:400, Chemicon International), Alexa Fluor® 488 donkey anti-rabbit (1:500, Invitrogen), Alexa Fluor® 555 strepavidin (1:400, Invitrogen). The resultant slides were viewed using an Olympus BX50 microscope (Olympus America Inc, PA) equipped with a digital camera system (Qimaging Retiga 2000R).
IL-10 ELISA
WT, Rag-2-/-, and Rag-2-/- mice received WT or IL-10-/- CD4+ T cells (n=6 per group), then were axotomized. At 7 day postoperative (dpo) following axotomy, brains of these mice were set into a mouse brain matrix (Redding, CA) to provide stability and support during sectioning. The matrix was iced at all times. After the mouse brain had cooled in the matrix for 2 minutes (or until firm), 2 razor blades, spaced 2 mm apart (according to matrix spacing) were placed using a gentle sawing motion and light pressure beginning at the boundary of the pontine band and moving more caudal. Blades were allowed to sit for 15 seconds followed by lifting both blades simultaneously resulting in a 2 mm thick coronal section that included both facial nuclei. A 1 mm concentric tissue punch was then used to punch out the facial nucleus. The punched tissue was blown into a 0.65 mL microcentrifuge tube containing 40 μL lysis buffer with protease inhibitors (Millipore, Billerica, MA) and homogenizing beads (Q-Biogene, Morgan Irvine, CA). Cervical lymph nodes, cerebella, and cervical spinal cord were also collected with the punch to take the same amount of tissues. Punches were immediately homogenized and centrifuged, then stored at -80°C until all samples were extracted and used for ELISA. IL-10 concentrations were determined with a mouse IL-10 ELISA (BioSource, Camarillo, CA) according to the manufacture’s protocol.
Statistical analysis
One-way ANOVA was performed to determine significant difference between groups. P < 0.05 was considered statistically significant.
Results
IL-10 is essential for facial motoneuron (FMN) survival after facial nerve injury
IL-10 is reported to be beneficial in reducing the severity of several pathogenic conditions within the CNS (Cua et al., 2001, Brewer et al., 1999; Bethea et al., 1999; Frenkel et al., 2005; Qian et al., 2006). The goal of our experimental design was to determine if IL-10 played a role in regulating motoneuron survival after direct axonal trauma. WT and IL-10-/- mice received a facial nerve axotomy, brains were collected, sectioned, and stained at 4 wpo, and surviving FMN was counted microscopically. No significant differences in the total number of viable FMN on the uninjured side of IL-10-/- and WT mice were detected [2185±95 vs. 2065±148 (Fig. 1A, left upper panel)], suggesting that a deficiency of IL-10 had no effect on the basal number of FMN in the facial nucleus prior to facial nerve axotomy. In contrast, at 4 weeks after facial nerve axotomy, a deficiency of IL-10 significantly decreased FMN survival (61.05% ± 6.55) as compared to WT mice (85.28% ± 1.17; Fig. 1A middle panel and Fig. 1B). Thus, axotomy in IL-10-deficient mice is associated with a significant decrease in FMN survival at 4 wpo, suggesting that IL-10 is necessary for maintaining FMN survival following a facial nerve injury, even though it was unnecessary for the maintenance of baseline survival in the absence of injury.
Fig. 1. IL-10-deficiency potentiates axotomy-induced FMN death.

A, Micrographs of thionin-stained FMN in the facial nucleus on uninjured (left) and injured (right) side in WT (upper panel), IL-10-/- (middle panel), and IL-10-/- mice + WT CD4+ T cells (lower panel;×40 magnification; scale bar=100μm). The FMNs in the inset at higher magnification (×200; scale bar=20μm) are shown in the left upper corner of WT uninjured facial nucleus. B, Bar graph of the percentage of FMN survival on injured relative to uninjured sides at 4 weeks after axotomy. No statistical difference was detected between the two groups of IL-10-/- mice with and without WT CD4+ T cells. Data are presented as Mean ± SEM (* P <0.05, compared to WT).
A non-CD4+ T cell source of IL-10 is critical for FMN survival
We have previously reported that CD4+ T cells are able to protect FMN from axotomy-induced cell death in immunodeficient mice (Serpe et al., 2003), yet in Fig. 1 we show that IL-10-/- mice, which possess “functional” CD4+ T cells (Kuhn et al., 1993), exhibited a significant FMN loss after facial nerve injury compared to WT. This finding suggested that a deficiency of IL-10 in CD4+ T cells might be responsible for the loss of neuroprotection after nerve axotomy, resulting in the subsequent increase in axotomy-induced FMN death. To test this possibility, we adoptively transferred WT CD4+ T cells into IL-10-/- mice and examined FMN survival at 4 wpo (Fig. 1A, bottom panel). The percentage of FMN survival in WT CD4+ T cell-adoptively transferred IL-10-/- mice was significantly decreased (48.58 % ± 14.58; Fig. 1B) relative to WT levels, and there was no statistical difference between the two groups of IL-10-/- mice with or without WT CD4+ T cells (P> 0.05). These results indicated that even with WT CD4+ T cells capable of producing IL-10, rescue of FMN to WT levels could not be achieved in the absence of IL-10 from other cellular sources. The failure of WT CD4+ T cells to rescue FMN in IL-10-/- mice may result from two possible reasons: 1) the WT CD4+ T cells could not differentiate into neuroprotective subsets in the absence of IL-10 in the host, and 2) the neuroprotective CD4+ T subset did develop but they cannot counteract the lack of IL-10 in the CNS. Furthermore, we speculate that these adoptively transferred WT CD4+ T cells may develop into detrimental CD4+ effector subsets in some IL-10-/- hosts, which in turn exacerbate axotomy-induced FMN death (an average of 48.85% FMN survival in those mice). However, these detrimental responses of WT CD4+ T cells might not occur in all of IL-10-/- host mice, and the differential response of CD4+ T cells in IL-10-/- host (SD=14.58%) might prevent us from detecting a significant difference in comparison to IL-10-/- mice without WT CD4+ T cells.
IL-10-deficient CD4+ T cells provide neuroprotection
Although T cells are a major source of IL-10, it has been reported that cells within the CNS, i.e., reactive astrocytes, perivascular macrophages, and parenchymal microglia, are also able to synthesize and release IL-10 (Strle et al., 2001; Hulshof et al., 2002). Since our results suggest that CD4+ T cells do not appear to be the critical source of IL-10 for FMN survival, the next set of experiments was designed to determine if the presence of CD4+ T cells within the CNS triggers IL-10 expression within the CNS following nerve injury. FMN survival was measured in Rag-2-/- mice reconstituted with IL-10-/- CD4+ T cells so that the only cell unable to produce IL-10 would be the CD4+ T cell. As shown in Fig. 2, There was no statistical difference of IL-10 levels between the two groups of Rag-2-/- mice reconstituted with IL-10-/- or WT CD4+ T cells (P>0.05). The presence of IL-10-deficient CD4+ T cells prevented the axotomy-induced decrease in FMN survival measured in the Rag-2-/- mice, just as was seen when using Rag-2-/- mice reconstituted with WT CD4+ T cells. These findings indicate that CD4+ T cells contribute to FMN survival by inducing other cells to produce IL-10.
Fig. 2. IL-10-deficiency does not impair the neuroprotective effect of CD4+ T cells.

A. Micrographs of thionin-stained FMN in the facial nucleus on uninjured (left) and injured (right) sides in Rag-2-/- mice + WT CD4+ T cells (upper panel) or IL-10-deficient CD4+ T cells (lower panel,×40 magnification; scale bar=100μm). B. Bar graph of the percentage of FMN survival on injured relative to uninjured sides at 4 weeks after axotomy. No significant differences were detected between the two group mice that received WT CD4+ T cells or IL-10-deficient CD4+ T cells. In the adoptive transfer, five million CD4+ T cells per mouse were injected through tail vein. Data are presented as Mean ± SEM (* P<0.05, compared to Rag-2-/- mice without treatment).
IL-10 within the CNS microenvironment is critical for FMN survival
Due to a short half-life in vivo, IL-10 is often administered by local injection (Radwanski et al., 1998; Asadullah et al., 2003). Additionally, like many other cytokines, IL-10 cannot cross an intact blood brain barrier (BBB) to enter the CNS parenchyma. Although circulating IL-10 has a wide effect on peripheral immune responses that can affect the nature of the immune response within the CNS, the delivery of IL-10 directly to the CNS appears to be essential for expression of its neuroprotective effects (Cua et al., 2001; Koeberle et al., 2004). However, Taskinen et al. (2000) reported that, after a nerve injury, endoneurial expression of IL-10 mRNA was observed at 1 day postoperative and persisted for up to 4 weeks, suggesting that endoneurial expression of IL-10 might participate in mediating neuronal survival. If such a mechanism were operative after a facial nerve injury, then the injured neuron itself could be the source of IL-10 within the CNS. It might be possible to mimic endoneurial expression of IL-10 via the transport of IL-10 into an injured peripheral nerve. Using such an approach would allow us to determine if the injured neuron itself participated in FMN survival by producing IL-10 or if the cellular source of IL-10 was another cell within the CNS. This approach would also indicate whether or not a peripheral source of IL-10 induced following a facial nerve injury could provide the IL-10 needed for FMN survival to occur within the CNS. To address these possibilities, we applied a gel foam impregnated with IL-10 at the proximal injured facial nerve stump prior to wound closure. This route of application allows retrograde IL-10 transport to the nerve cell body along the proximal nerve (Serpe et al., 2005), and mimics endoneurial production of IL-10. The IL-10 level was maintained by 3 additional injections into the foam after surgery. The percentage of FMN survival in the IL-10-retrograde transfer group was 68.55% ± 11.21, a significant decrease compared to WT mice [Fig. 3A upper panel and 3B; (P<0.05)], and was not significantly different from that measured in the IL-10-/- mice in the absence of IL-10 administration (data not shown). These data indicate that a source of IL-10 from the peripheral injury site does not mediate neuroprotection, and that a neuronal source of IL-10 is an unlikely source of the neuroprotective IL-10, and that IL-10 expression by a non-neuronal, non-T cell source within the CNS is a critical factor for mediating FMN survival.
Fig. 3. Peripheral treatment of IL-10 fails to rescue FMN from death in the presence or absence of CD4+ T cells.
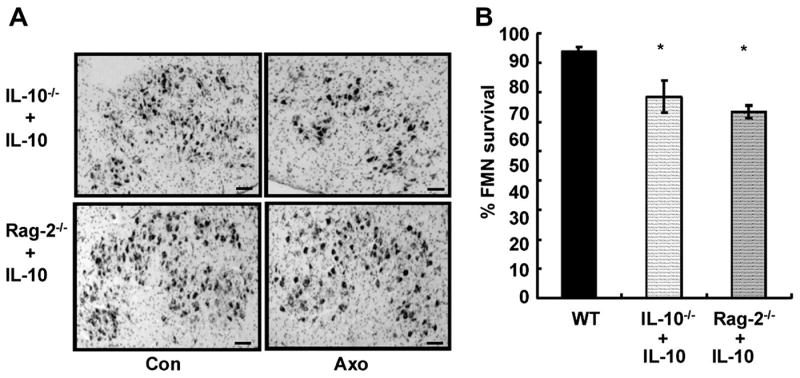
A. Micrographs of thionin-stained FMN in the facial nucleus of uninjured (left) and injured (right) sides in IL-10-/- (upper panel) or Rag-2-/- mice (lower panel) that received IL-10 at injury site (×40 magnification; scale bar=100μm). B. Bar graph of the percentage of FMN survival on injured side is relative to uninjured side at 4 weeks after axotomy. Data are presented as Mean ± SEM. No significant difference was detected between IL-10 treated IL-10-/- and Rag-2-/- mice, but both of them showed a significant decrease in FMN numbers when compared to WT mice (* P<0.05)
Constitutive expression of IL-10 in mouse facial nucleus
Using RT-PCR, we confirmed that IL-10 mRNA is expressed constitutively in the facial nucleus of both WT and Rag-2-/- mice, and its amount is comparable in the two types of mice (Fig. 4A). However, the gel results show a visually detectable reduction of IL-10 expression in the axotomized side of Rag-2-/- mice. To verify this difference following axotomy, we measured the amount of IL-10 protein present in facial nucleus tissue with ELISA. Our results indicate that IL-10 levels were not altered by axotomy in the WT mice, with the levels comparable between the axotomized and control sides (Fig. 4 B). In contrast, IL-10 protein was significantly reduced on the axotomized side of Rag-2-/- mice at 7 dpo, suggesting that the T cell may play an important role in maintaining IL-10 levels in the facial nucleus after facial nerve injury. As a control, we measured IL-10 in draining cervical lymph nodes and other areas within the CNS, i.e., cerebella and cervical spinal cord anterior horn. Compared to WT mice, Rag-2-/- mice had comparable IL-10 levels in all these areas (data for 7 dpo is shown in Fig 4 C), suggesting that the reduction in IL-10 within the CNS of Rag-2-/- mice 7 days after facial nerve axotomy was limited to the facial nucleus.
Fig. 4. Constitutive IL-10 expression in the facial nucleus and transient reduction in IL-10 expression in Rag-2-/- mice following facial nerve axotomy.
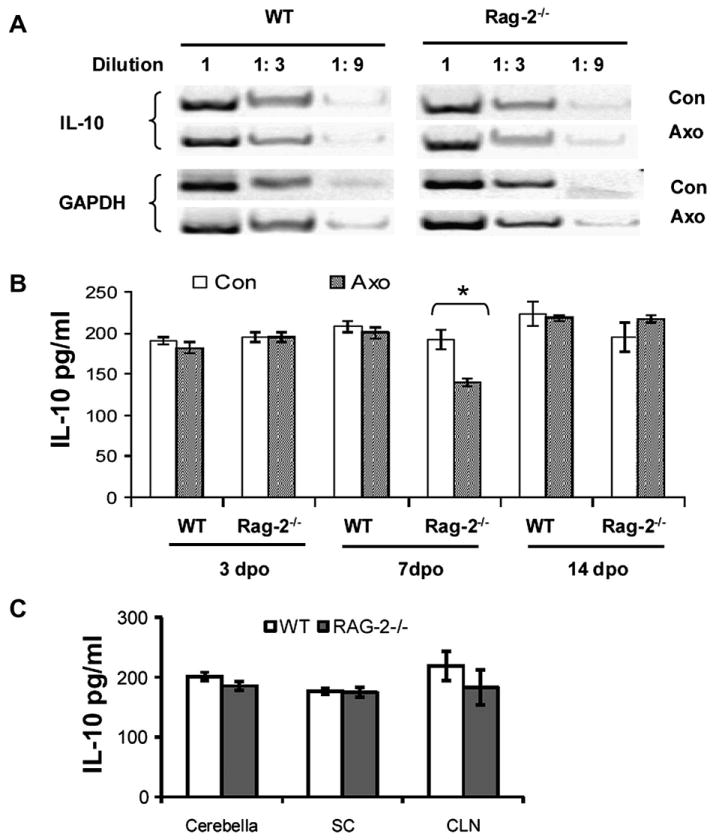
A. Limit dilution gel results of RT-PCR show the presence of IL-10 mRNA in the facial nucleus in both WT and Rag-2-/- mouse prior to and post facial nerve axotomy. B. Bar graph of the IL-10 amount measured with ELISA in the facial nucleus at 3, 7 and 14 days following facial nerve axotomy. Data are presented as Mean ± SEM (* P<0.05, compared to control side). C. Bar graph of the IL-10 amount measured with ELISA in the cerebella, spinal cord (SC) and cervical lymph node (CLN) at 7 days following facial nerve axotomy.
CD4+ T cells restore IL-10 levels in the axotomized facial nucleus of Rag-2-/- mice
To determine if CD4+ T cells were responsible for maintaining IL-10 in the facial nucleus following facial nerve axotomy, we measured IL-10 in the facial nucleus of Rag-2-/- mice to which either IL-10 intact WT or IL-10-deficient CD4+ cells were adoptively transferred one week prior to axotomy. As shown in Fig. 5, at 7 dpo, IL-10 levels in the facial nucleus on the axotomized side of both sets of mice were comparable to that on the control side (approximately 200 pg/ml). In contrast, this level of IL-10 in the facial nucleus was significantly more than the level (approximately 150 pg/ml) in the facial nucleus on the axotomized side of Rag-2-/- mice that did not receive any type of CD4+ T cells (as shown in Fig 4B). Thus, no significant differences in IL-10 were detected between axotomized and control sides in Rag-2-/- receiving an adoptive transfer of either IL-10+/+ or IL-10-/- CD4+ T cells. These results indicate that the reduction of IL-10 expression in Rag-2-/- mice at 7 dpo was prevented by the presence of CD4+ T cells, independent of their ability to produce IL-10, suggesting a potential mechanism by which IL-10-deficient CD4+ T cells possess the comparable neuroprotective effects of WT CD4+ T cells (as shown in Fig. 2).
Fig. 5. Both WT and IL-10-deficiency CD4+ T cells are capable of restoring IL-10 level in Rag-2-/- axotomized facial nucleus.
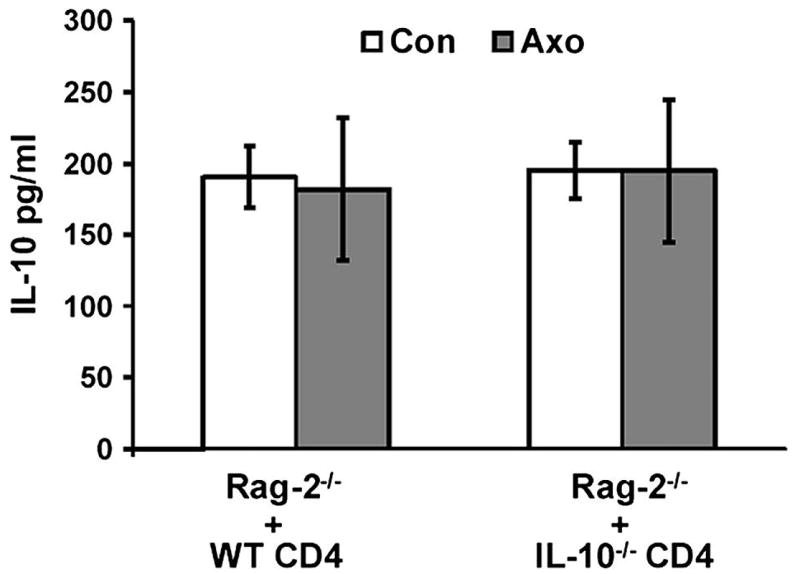
Rag-2-/- mice received WT or IL-10-/- CD4+ T cells was axotomized. Bar graph of IL-10 expression measured with ELISA in the facial nucleus at 7 days following facial nerve axotomy. No significant differences were detected between the two groups of Rag-2-/- mice that received WT CD4+ T cells or IL-10-deficient CD4+ T cells. The baseline IL-10 levels in WT and Rag-2-/- mice at 7 dpo are shown in Fig 4B. In the adoptive transfer, five million of CD4+ T cells per mouse were injected through tail vein.
IL-10R is expressed constitutively in the facial nucleus and upon axotomy in activated astrocytes
To understand the mechanism by which IL-10 mediates its neuroprotective effect independently of a direct effect mediated by CD4+ T cells, it is necessary to determine which cells express IL-10R. Using RT-PCR, we found that both IL-10Rα and IL-10Rβ mRNA were expressed constitutively in the facial nucleus of WT and Rag-2-/- mice (Fig. 6A), and expression was upregulated after axotomy in both sets of mice over 600% for IL-10Rα, and 100% for IL-10Rβ , respectively (Fig. 6B). Furthermore, IL-10Rα protein expression, as examined with immunohistochemical staining, was also detectable in the uninjured facial nucleus of WT and Rag-2-/- mice. Because noticeable immunoreactivity was observed mainly in cells morphologically similar to neurons (Fig. 6C), we confirmed this co-localization using double immunofluorescence labeling for IL-10Rα and the neuronal marker, NeuN. Our results indicate that some IL-10Rα colocalized with NeuN, while some FMN displayed single staining for IL-10Rα or NeuN and others showed IL-10Rα and NeuN localized in different compartments of the same cell, suggesting heterogeneous expression of IL-10Rα and NeuN in uninjured FMN. Isotype and 2nd antibody only controls staining showed negative results and are not shown.
Fig. 6. Constitutive expression of IL-10 in the facial nucleus and upregulation by facial nerve axotomy.

A. Limit dilution gel results of RT-PCR show the presence of IL-10R mRNA in the facial nucleus in both WT and Rag-2-/- mouse prior to and post facial nerve axotomy. B. Bar graph shows the percentage of axotomy-induced upregulation of IL-10R in two types of mice. Data are presented as Mean ± SEM. C. Double immunofluorescence staining results for IL-10Rα in facial nucleus on uninjured side at 7 days following facial nerve axotomy.
IL-10Rα immunoreactivity was enhanced significantly in the axotomized facial nucleus (Fig 7) in cells with stellate, glial-like appearances. To confirm the type of these IL-10Rα –positive cells, co-localization immunohistochemical staining for IL-10Rα and either GFAP (marker for activated astrocytes) or CD68 (marker for activated microglia) was performed. As illustrated in Fig. 7, compared to the negative staining of GFAP and CD68 in control facial nucleus, axotomized facial nuclei displayed strong immunoreactivity of GFAP and CD68, confirming that astrocytes and microglia are activated following facial nerve axotomy. IL-10Rα was also upregulated in the axotomized facial nucleus, co-localizing primarily with GFAP, but not CD68, indicating that upregulation of IL-10Rα occurred specifically in astrocytes but not microglia. Unfortunately, because axotomy abolishes NeuN expression in FMN to undetectable levels (McPhail et al., 2004), we could not do co-localization with a neuronal marker on the injured side.
Fig. 7. Axotomy-induced upregulation of IL-10Rα was primarily observed in activated astrocytes.
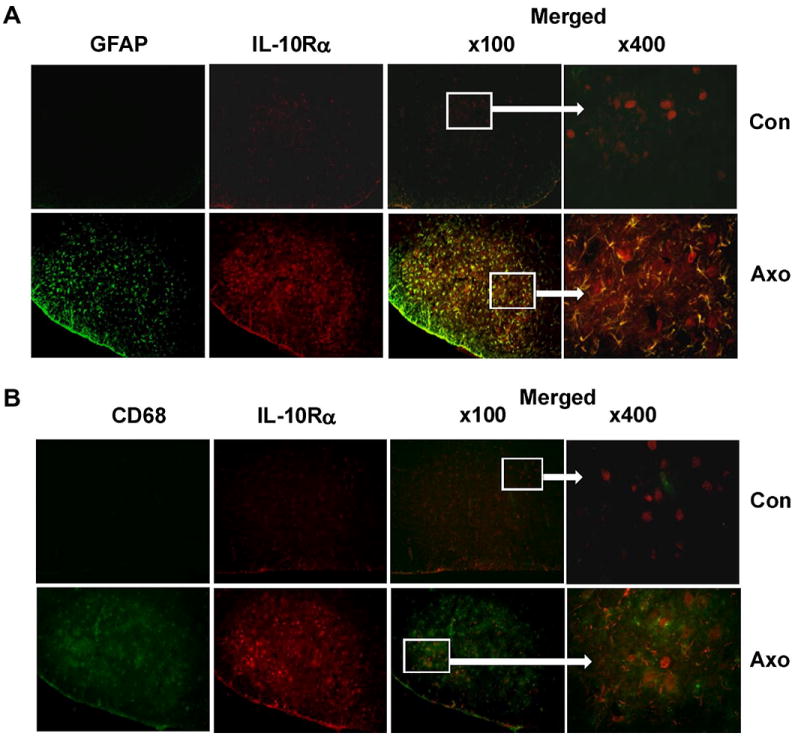
A. Micrographs of double immunofluorescence staining results for IL-10Rα and GFAP in the facial nucleus on uninjured (upper panel) and injured (lower panel) sides in WT mice at 7 days after facial nerve axotomy. B. Micrographs of double immunofluorescence staining results for IL-10Rα and CD68 in the facial nucleus on uninjured (upper panel) and injured (lower panel) sides in WT mice at 7 days after facial nerve axotomy.
Discussion
Our previous findings have established that CD4+ Th2-like cells are neuroprotective after a facial nerve axotomy in the mouse (Deboy et al., 2006a). Our findings also indicated that both peripheral CD4+ T cell initial activation in the draining cervical lymph node and subsequent re-activation within the brainstem facial nucleus are necessary for neuroprotection to occur after axotomy (Byram et al., 2004). The evidence presented in this study indicates that IL-10 and CD4+ T cells work cooperatively, within the CNS, to protect FMN from cell death following injury. The mechanism of neuroprotection appears to involve a role for the T cell in maintaining sufficient, glial-derived IL-10 levels near the injured cell bodies in the ipsilateral facial nucleus and a role for the dual actions of IL-10 to mediate FMN protection both indirectly and directly.
A neuroprotective role for IL-10 has been noted over the past decade for neuroinflammatory conditions, such as multiple sclerosis (Cua et al., 2001). IL-10 functions within the CNS as an anti-inflammatory cytokine that maintains neuronal homeostasis (Strle et al., 2001). Recently, it has been reported that IL-10 might exert a neuroprotective effect by acting directly on neurons that express the receptor for IL-10 (IL-10R), e.g., cerebellar granule cells (Bachis et al., 2001) and retinal ganglion cells (Boyd et al., 2003), protecting them from chemical-induced apoptosis in vitro. Koeberle et al. (2004) also reported that IL-10 increased the survival of retinal ganglion neurons after axotomy. In the present study, we adopted two mouse models to determine if the CD4+ T cell is the critical source of IL-10 that mediates the neuroprotective effect of CD4+ T cells on FMN after axotomy, and if the FMN is the direct target of the released IL-10. Two separate mouse models were exploited in this study to assess the importance of IL-10 expression and CD4+ T cell functionality, specifically, IL-10-/- mice have CD4+ T cells, but no IL-10 expression, while Rag-2-/- mice have IL-10 expression, but no CD4+ T cells. It has been reported that activated T cells are able to cross the BBB and infiltrate the CNS parenchyma under neuroinflammatory conditions (Engelhardt and Ransohoff, 2005), including after a facial nerve axotomy within the facial nucleus area (Olsson et al.,1992; Raivich et al.,1998; Ha et al.,2006). At present, Th2 and T-reg cells are two well defined CD4+ T cell subsets that are able to produce IL-10 (Taylor et al., 2006). Although both T cell subsets are activated in the draining lymph node following facial nerve axotomy (Xin et al., 2008), only the Th2-like cell has been shown to be involved with FMN survival (Deboy et al., 2006a). Thus, it was possible that Th2-like cells serve as the source of neuroprotective IL-10 within the CNS. However, we found unexpectedly that IL-10-deficient CD4+ T cells retained their neuroprotective function when adoptively transferred to a Rag2-/- mouse prior to axotomy, suggesting that FMN survival requires both the presence of CD4+ T cells and, also, IL-10 from a non-CD4+ T cell source within the facial nucleus in the CNS. Chabot et al. (1999) showed that the interaction of T cells with microglia promotes both T cell and microglial cell production of IL-10. Therefore, we propose that CD4+ T cells are activated in the periphery following nerve injury and cross the BBB to regulate resident cells within the CNS (Byram et al 2004), possibly glial cells, to produce IL-10 that protects FMN from axotomy-induced cell death.
Constitutive expression of IL-10 in the periphery and CNS is one important mechanism by which an anti-inflammatory environment is maintained under normal physiological circumstances (Strle et al., 2001; Taylor et al., 2006). Our data indicate that the baseline homeostatic level of IL-10 within the CNS appears to be maintained independently of activated lymphocytes since the IL-10 levels in the control facial nucleus of Rag-2-/- mice are comparable to those in the control facial nucleus of WT mice. Unexpectedly, basal IL-10 levels decreased in the facial nucleus of immunodeficient animals after facial nerve injury; whereas, the adoptive transfer of IL-10-deficient CD4+ T cells into the Rag2-/- prior to axotomy prevented IL-10 decline in the facial nucleus. We speculate that the transferred WT CD4+ T cell in the Rag-2-/- mice may have a similar response to axotomy as observed in WT mice following facial nerve axotomy (Xin et al., 2008), It is presently unclear as to how the IL-10-/-CD4+ T cell responds in the Rag-2-/- host, but we will investigate this point in the future. These data indicate that the source of FMN protective IL-10 is from a non-CD4+ T cell. But, perhaps more importantly, the kinetic pattern of IL-10 expression following axotomy indicates that the decrease in IL-10 expression one week after axotomy is transient, and returns to control levels 2 weeks after axotomy despite a continued absence of T cells. Because FMN survival levels are reduced and irreversible in immunodeficient animals, the transient loss of IL-10 at 7 days indicates that the timing of the cooperative rescue signal from IL-10 and T cells is critical. The production of IL-10 by CNS resident cells in normal conditions seems independent of CD4+ T cells, since Rag-2-/- mice have a similar level of IL-10 compared to WT controls prior to axotomy. We speculate that the intrinsic IL-10 signaling circuit (IL10- IL10R-Stat3-IL10 axis) functions to maintain basal levels of IL-10, which may explain why IL-10 levels return to normal levels 2 weeks later. Concurrently, facial nerve axotomy induces an inflammatory response in the facial nucleus, which may disrupt the normal IL-10 signaling circuits and result in a drop of IL-10 at 7 dpo in the Rag-2-/- facial nucleus. In contrast, in WT mice, the regulatory effects of CD4+ T cells may be sufficient to counteract the inflammatory response and maintain IL-10 levels. As an essential future direction, we intend to determine the cellular source of IL-10 in the CNS using in situ hybridization, and a transgenic mouse model that expresses GFP in cells undergoing IL-10 transcription.
IL-10-mediated neuroprotection of FMN could result from anti-inflammatory effects mediated either indirectly, through disruption of pro-inflammatory cell activity, or directly on the neuron itself. Consistent with previous studies demonstrating IL-10R expression on all CNS resident cells, including astrocytes, oligodendrocytes, microglial cells, and neurons (Strle et al., 2001; Hulshof et al., 2002), we confirmed in the present study that IL-10R is constitutively expressed by FMN. What we found that is novel and unexpected is that axotomy upregulates IL-10R expression specifically in astrocytes within the injured facial motor nucleus. It is possible that the astrocyte is another main target cell of IL-10. In the future, we will test whether IL-10 stimulation can result in the activation of neurotrophic factors involved in neuroprotection. It has previously been reported that IL-10 binding to microglial IL-10R represses the production of pro-inflammatory mediators, including tumor necrosis factor alpha (TNF-α), IL-1β, nitric oxide synthase (NOS), and specific chemokines (Marques et al., 2004; Kremlev et al., 2005; Wilms et al., 2007; Block and Hong, 2005). However, we found that axotomy did not alter IL-10R expression in activated microglia, but rather only in astrocytes. With regard to direct actions of IL-10 on injured FMN, previous studies have shown that IL-10 binding to IL-10R-positive neurons initiates the neuronal STAT-3 pathway, which activates the expression of anti-apoptotic factors (Bachis et al., 2001; Boyd et al., 2003). Disruption of the STAT-3 pathway in FMN has been found to lead to an increase in axotomy-induced FMN death (Schweizer et al., 2002), suggesting a potential direct role for IL-10 in initiating neuronal anti-apoptotic pathways and promoting survival after injury. Thus, we postulate a dual action of IL-10, involving both indirect anti-inflammatory and direct anti-apoptotic mechanisms, in the CNS after a peripheral nerve lesion.
Finally, a clinically relevant, therapeutically effective delivery method for neurotrophic or neuroprotective reagents was used for administering IL-10 in the present study. Enhanced neuronal survival has been reported for brain-derived neurotrophic factor (BDNF) and other neurotrophic factors (Serpe et al., 2005; von Bartheld et al., 2001) administered at the nerve lesion site in the periphery. However, the results of our studies indicate that peripherally-administered IL-10 fails to rescue FMN from axotomy-induced cell death, regardless of the presence of CD4+ T cells. This suggests that IL-10 produced in the periphery has a minimal effect on FMN survival, and that endoneurial expression of IL-10 in injured nerves, reported by Taskinen et al. (2000), is unlikely to contribute directly to neuronal survival. Thus far, we cannot exclude the possibility that IL-10, when administered at the site of injury, is metabolized quickly and may require higher doses to confer a neuroprotective effect. Additionally, neurons are capable of retrogradely transporting neurotrophic factors from the periphery to their cell bodies where upon secretion, the neurotrophic factors can mediate neuroprotection through autocrine signaling. (Serpe et al., 2005; von Bartheld et al., 2001). We did not determine whether IL-10 was successfully transported into the neuronal soma and subsequently secreted into the CNS parenchyma. Thus, we cannot rule out the possibility that IL-10 administered at the site of injury may not be efficiently transported retrogradely or released into the CNS parenchyma to bind to IL-10R on CNS resident cells. These issues remain as future directions for improving IL-10 effectiveness after local application.
In summary, these results substantiate our findings of a potential dual action for IL-10 that is both indirect, acting through CNS glial cells to modify the pro-inflammatory environment, and direct, by activating neuronal anti-apoptotic pathways. The co-modulation of both glial and neuronal events by IL-10, in cooperation with T cell activation, in neuroprotection of injured motoneurons provides an avenue of investigation into motoneuron diseases and therapeutic approaches to the prevention of cell loss. A major future direction will be to use both in vitro and in vivo approaches to determine the cellular source(s) of IL-10 with the CNS and the mechanism underlying neuroimmune interaction in CNS-derived IL-10 production.
Acknowledgments
We thank Lisa Tanzer, Tom Alexander and Linda Poggensee for their assistance. We also thank Dr. Cynthia Von Zee and Dr. Stubbs for their assistance.
Abbreviations
- FMN
facial motoneuron
- BBB
blood brain barrier
- WT
wild type
- wpo
weeks postoperative
- dpo
day postoperative
- FoxP3
forkhead box P3
- Treg
T regulatory
- Tr1
type-1 regulatory
- IL-10R
IL-10 receptor
Footnotes
Disclosure We declare no conflict of interest or financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asadullah K, Sterry W, Volk HD. Interleukin-10 therapy--review of a new approach. Pharmacol Rev. 2003;55:241–269. doi: 10.1124/pr.55.2.4. [DOI] [PubMed] [Google Scholar]
- Bachis A, Colangelo AM, Vicini S, Doe PP, De Bernardi MA, Brooker G, Mocchetti I. Interleukin-10 prevents glutamate-mediated cerebellar granule cell death by blocking caspase-3-like activity. J Neurosci. 2001;21:3104–3112. doi: 10.1523/JNEUROSCI.21-09-03104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethea JR, Nagashima H, Acosta MC, Briceno C, Gomez F, Marcillo AE, Loor K, Green J, Dietrich WD. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma. 1999;16:851–863. doi: 10.1089/neu.1999.16.851. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Boyd ZS, Kriatchko A, Yang J, Agarwal N, Wax MB, Patil RV. Interleukin-10 receptor signaling through STAT-3 regulates the apoptosis of retinal ganglion cells in response to stress. Invest Ophthalmol Vis Sci. 2003;44:5206–5211. doi: 10.1167/iovs.03-0534. [DOI] [PubMed] [Google Scholar]
- Brewer KL, Bethea JR, Yezierski RP. Neuroprotective effects of interleukin-10 following excitotoxic spinal cord injury. Exp Neurol. 1999;159:484–493. doi: 10.1006/exnr.1999.7173. [DOI] [PubMed] [Google Scholar]
- Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot S, Williams G, Hamilton M, Sutherland G, Yong VW. Mechanisms of IL-10 production in human microglia-T cell interaction. J Immunol. 1999;162:6819–6828. [PubMed] [Google Scholar]
- Cua DJ, Hutchins B, LaFace DM, Stohlman SA, Coffman RL. Central nervous system expression of IL-10 inhibits autoimmune encephalomyelitis. J Immunol. 2001;166:602–608. doi: 10.4049/jimmunol.166.1.602. [DOI] [PubMed] [Google Scholar]
- De Jager PL, Hafler DA. New therapeutic approaches for multiple sclerosis. Annu Rev Med. 2007;58:417–432. doi: 10.1146/annurev.med.58.071105.111552. [DOI] [PubMed] [Google Scholar]
- DeBoy CA, Byram SC, Serpe CJ, Wisuri D, Sanders VM, Jones KJ. CD4+CD25+ regulatory T cells and CD1-restricted NKT cells do not mediate facial motoneuron survival after axotomy. J Neuroimmunol. 2006a;176:34–38. doi: 10.1016/j.jneuroim.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Deboy CA, Xin J, Byram SC, Serpe CJ, Sanders VM, Jones KJ. Immune-mediated neuroprotection of axotomized mouse facial motoneurons is dependent on the IL-4/STAT6 signaling pathway in CD4(+) T cells. Exp Neurol. 2006b;201:212–224. doi: 10.1016/j.expneurol.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. J Neurol Sci. 2005;233:125–132. doi: 10.1016/j.jns.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Griffiths M, Neal JW, Gasque P. Innate immunity and protective neuroinflammation: new emphasis on the role of neuroimmune regulatory proteins. Int Rev Neurobiol. 2007;82:29–55. doi: 10.1016/S0074-7742(07)82002-2. [DOI] [PubMed] [Google Scholar]
- Ha GK, Huang Z, Streit WJ, Petitto JM. Endogenous T lymphocytes and microglial reactivity in the axotomized facial motor nucleus of mice: effect of genetic background and the RAG2 gene. J Neuroimmunol. 2006;172:1–8. doi: 10.1016/j.jneuroim.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J. Immunotherapy as treatment for Alzheimer’s disease. Expert Rev Neurother. 2007;7:1535–1548. doi: 10.1586/14737175.7.11.1535. [DOI] [PubMed] [Google Scholar]
- Hulshof S, Montagne L, De Groot CJ, Van Der Valk P. Cellular localization and expression patterns of interleukin-10, interleukin-4, and their receptors in multiple sclerosis lesions. Glia. 2002;38:24–35. doi: 10.1002/glia.10050. [DOI] [PubMed] [Google Scholar]
- Jones KJ, LaVelle A. Changes in nuclear envelope invaginations in axotomized immature and mature hamster facial motoneurons. Brain Res. 1985;353:241–249. doi: 10.1016/0165-3806(85)90212-3. [DOI] [PubMed] [Google Scholar]
- Kipnis J, Avidan H, Caspi RR, Schwartz M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14663–14669. doi: 10.1073/pnas.0404842101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipnis J, Mizrahi T, Yoles E, Ben-Nun A, Schwartz M. Myelin specific Th1 cells are necessary for post-traumatic protective autoimmunity. J Neuroimmunol. 2002;130:78–85. doi: 10.1016/s0165-5728(02)00219-9. [DOI] [PubMed] [Google Scholar]
- Koeberle PD, Gauldie J, Ball AK. Effects of adenoviral-mediated gene transfer of interleukin-10, interleukin-4, and transforming growth factor-beta on the survival of axotomized retinal ganglion cells. Neuroscience. 2004;125:903–920. doi: 10.1016/S0306-4522(03)00398-1. [DOI] [PubMed] [Google Scholar]
- Kremlev SG, Palmer C. Interleukin-10 inhibits endotoxin-induced pro-inflammatory cytokines in microglial cell cultures. J Neuroimmunol. 2005;162:71–80. doi: 10.1016/j.jneuroim.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Marques CP, Hu S, Sheng W, Cheeran MC, Cox D, Lokensgard JR. Interleukin-10 attenuates production of HSV-induced inflammatory mediators by human microglia. Glia. 2004;47:358–366. doi: 10.1002/glia.20045. [DOI] [PubMed] [Google Scholar]
- McPhail LT, McBride CB, McGraw J, Steeves JD, Tetzlaff W. Axotomy abolishes NeuN expression in facial but not rubrospinal neurons. Exp Neurol. 2004;185:182–190. doi: 10.1016/j.expneurol.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Olsson T, Diener P, Ljungdahl A, Hojeberg B, van der Meide PH, Kristensson K. Facial nerve transection causes expansion of myelin autoreactive T cells in regional lymph nodes and T cell homing to the facial nucleus. Autoimmunity. 1992;13:117–126. doi: 10.3109/08916939209001912. [DOI] [PubMed] [Google Scholar]
- Qian L, Block ML, Wei SJ, Lin CF, Reece J, Pang H, Wilson B, Hong JS, Flood PM. Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of NADPH oxidase. J Pharmacol Exp Ther. 2006;319:44–52. doi: 10.1124/jpet.106.106351. [DOI] [PubMed] [Google Scholar]
- Radwanski E, Chakraborty A, Van Wart S, Huhn RD, Cutler DL, Affrime MB, Jusko WJ. Pharmacokinetics and leukocyte responses of recombinant human interleukin-10. Pharm Res. 1998;15:1895–1901. doi: 10.1023/a:1011918425629. [DOI] [PubMed] [Google Scholar]
- Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggieri M, Avolio C, Livrea P, Trojano M. Glatiramer acetate in multiple sclerosis: a review. CNS Drug Rev. 2007;13:178–191. doi: 10.1111/j.1527-3458.2007.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer U, Gunnersen J, Karch C, Wiese S, Holtmann B, Takeda K, Akira S, Sendtner M. Conditional gene ablation of Stat3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J Cell Biol. 2002;156:287–297. doi: 10.1083/jcb.200107009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Byram SC, Sanders VM, Jones KJ. Brain-derived neurotrophic factor supports facial motoneuron survival after facial nerve transection in immunodeficient mice. Brain Behav Immun. 2005;19:173–180. doi: 10.1016/j.bbi.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Coers S, Sanders VM, Jones KJ. CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain Behav Immun. 2003;17:393–402. doi: 10.1016/s0889-1591(03)00028-x. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J Neurosci. 1999;19:RC7. doi: 10.1523/JNEUROSCI.19-11-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Sanders VM, Jones KJ. Kinetics of facial motoneuron loss following facial nerve transection in severe combined immunodeficient mice. J Neurosci Res. 2000;62:273–278. doi: 10.1002/1097-4547(20001015)62:2<273::AID-JNR11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Steinman L. Elaborate interactions between the immune and nervous systems. Nat Immunol. 2004;5:575–581. doi: 10.1038/ni1078. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol. 2002;513:87–113. doi: 10.1007/978-1-4615-0123-7_3. [DOI] [PubMed] [Google Scholar]
- Strle K, Zhou JH, Shen WH, Broussard SR, Johnson RW, Freund GG, Dantzer R, Kelley KW. Interleukin-10 in the brain. Crit Rev Immunol. 2001;21:427–449. [PubMed] [Google Scholar]
- Taskinen HS, Olsson T, Bucht A, Khademi M, Svelander L, Roytta M. Peripheral nerve injury induces endoneurial expression of IFN-gamma, IL-10 and TNF-alpha mRNA. J Neuroimmunol. 2000;102:17–25. doi: 10.1016/s0165-5728(99)00154-x. [DOI] [PubMed] [Google Scholar]
- Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology. 2006;117:433–442. doi: 10.1111/j.1365-2567.2006.02321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, Wang X, Butowt R. Anterograde axonal transport, transcytosis, and recycling of neurotrophic factors: the concept of trophic currencies in neural networks. Mol Neurobiol. 2001;24:1–28. doi: 10.1385/MN:24:1-3:001. [DOI] [PubMed] [Google Scholar]
- Wilms H, Zecca L, Rosenstiel P, Sievers J, Deuschl G, Lucius R. Inflammation in Parkinson’s diseases and other neurodegenerative diseases: cause and therapeutic implications. Curr Pharm Des. 2007;13:1925–1928. doi: 10.2174/138161207780858429. [DOI] [PubMed] [Google Scholar]
- Xin J, Wainwright DA, Serpe CJ, Sanders VM, Jones KJ. Phenotype of CD4+ T cell subsets that develop following mouse facial nerve axotomy. Brain Behav Immun. 2008;22:528–537. doi: 10.1016/j.bbi.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]