

Abstract
Background
To date, many genomic and pathway-related tools and databases have been developed to analyze microarray data. In published web-based applications to date, however, complex pathways have been displayed with static image files that may not be up-to-date or are time-consuming to rebuild. In addition, gene expression analyses focus on individual probes and genes with little or no consideration of pathways. These approaches reveal little information about pathways that are key to a full understanding of the building blocks of biological systems. Therefore, there is a need to provide useful tools that can generate pathways without manually building images and allow gene expression data to be integrated and analyzed at pathway levels for such experimental organisms as Arabidopsis.
Results
We have developed PathMAPA, a web-based application written in Java that can be easily accessed over the Internet. An Oracle database is used to store, query, and manipulate the large amounts of data that are involved. PathMAPA allows its users to (i) upload and populate microarray data into a database; (ii) integrate gene expression with enzymes of the pathways; (iii) generate pathway diagrams without building image files manually; (iv) visualize gene expressions for each pathway at enzyme, locus, and probe levels; and (v) perform statistical tests at pathway, enzyme and gene levels. PathMAPA can be used to examine Arabidopsis thaliana gene expression patterns associated with metabolic pathways.
Conclusion
PathMAPA provides two unique features for the gene expression analysis of Arabidopsis thaliana: (i) automatic generation of pathways associated with gene expression and (ii) statistical tests at pathway level. The first feature allows for the periodical updating of genomic data for pathways, while the second feature can provide insight into how treatments affect relevant pathways for the selected experiment(s).
Background
Novel technologies in genome research, such as Affymetrix GeneChips and DNA microarrays, have generated large amounts of data. These data have led to a more thorough understanding of gene function, regulation, interaction, and pathways. However, analysis of microarray data at the individual gene level is not sufficient for a thorough understanding of how a pathway/network is disturbed in the discovery of new drugs or in the modification of the existing plant variety. It is important to develop user-friendly tools that allow biologists to associate gene expression with pathways, test the treatment effects at the pathway level, and extract a comprehensive overview of experimental effects from the data.
Despite the importance of understanding biological pathways through microarray data, most microarray databases lack information on pathways. For example, SMD [1], a well-known microarray database for gene expression analysis in multi-organisms, does not feature pathway components. The Kyoto Encyclopedia of Genes and Genomes (KEGG) [2] and the National Center for Genome Research (NCGR, http://www.ncgr.org/) have pathway databases but they do not have microarray data analysis or statistical test components associated with pathways. A number of software packages for microarray data analysis or integration of pathway information does exist [3-7]. Their functions include querying pathway information, with some databases having the capability to overlay gene expression data on pathways. These packages focus on E. coli, yeast, mouse, human, or other organisms instead of plants, though. For example, GenMAPP 1.0 [6] provides useful tools to display and test gene expression, and interactively modify pathways. However, the pathways in GenMAPP are not relevant for plants. In addition, GenMAPP statistically tests gene expression at the individual enzyme level rather than the pathway level, and it uses local data files instead of a large database. Because plants and animals have evolved independently from unicellular eukaryotes and represent highly contrasting life forms, the analysis of plant genomes can elucidate fundamental principles of biology relevant to a variety of species, including humans as well as principles unique to plants.
Several plant-specific databases have been developed. For Arabidopsis thaliana, a model organism extensively studied by plant biologists [8], TAIR [9] provides a comprehensive database for many types of information on Arabidopsis. TAIR recently introduced a pathway component that can overlay expression patterns on known biological pathways. There are other Arabidopsis databases with more specific emphases, e.g. CATMA [10].
Despite the availability of web-based pathway databases, one current limitation of these databases is their reliance on static pathway image files. With the exception of NCGR, which provides partially automatic pathway graph generation features, the pathway graphs of these databases are built manually with other tools [2-5,9]. As our knowledge of genes evolves, gene information will need to be updated and older pathway images will become outdated. Static images cannot reflect any change and it is time-consuming to rebuild such detailed new data into pathways. Another limitation of the existing databases is that the analysis of gene expression data focuses on individual genes instead of pathways. An exception is Pathway Processor [7] that offers limited test for yeast on the basis of fold change only. PathMAPA transcends these limitations. It serves as a database to upload and retrieve Arobidopsis microarray data, automatically generates pathway graphs and visualizes pathways associated with gene expression. It displays and statistically tests gene expressions at the probe, locus and enzyme levels respectively for nearly 100 pathways. PathMAPA can also be used to compare gene expressions for individual experiments or across multiple experiments.
Implementation
System Architecture
PathMAPA uses a combination of Apache and Tomcat as its web server and is accessible through IE 5.0 or higher and Netscape 7 or higher from multiple platforms. The Java Plug-In can be easily installed for most platforms with the link provided. PathMAPA uses Oracle as its database management system, and Java/JSP as its programming language. Statistical components are called from R-project through an API interface except for the normalization component that is coded in C++. The system is periodically updated. Architecturally, the system is a three-tier application (Figure 1). Clients access the web site through the intranet/internet. The web server runs JSP, Java Servlets, and Java Beans. The Oracle database is accessed through JDBC. SQL Plus and SQL Loader are also used to optimize the performance of data processes. Functionally, PathMAPA consists of two components: Database and Pathway. The database part will not be discussed in this paper; the pathway part provides statistical and visualization tools to integrate microarray data with metabolic pathways (Figure 2).
Figure 1.
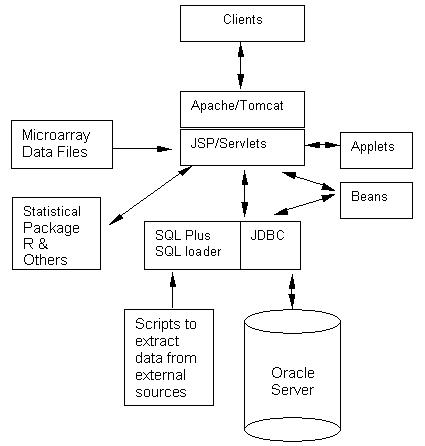
The system architecture.
Figure 2.
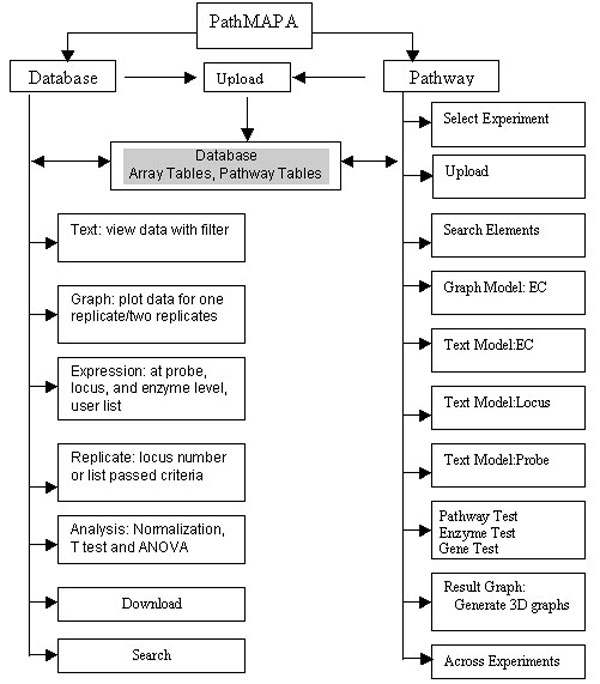
Flow chart of major functions of PathMAPA.
Microarray Data
Microarray data files generated through GenePix http://axon.com/GN_GenePixSoftware.html, exported Affymetrix http://www.affymetrix.com/ data and other types of exported microarray files can be populated into the PathMAPA database for analysis. The owners of these files have the option to modify the experimental information after the data have been uploaded into the server. PathMAPA provides data normalization through the "Upload" link in the Pathway component, with normalization taking place after uploading but before populating into the database. For GenePix data, PathMAPA normalizes the median-of-ratios column. Meanwhile, the user has the option to populate the original data into the database without normalization. An additional copy of the original text file is available for users to download.
The pathway details, including structures, enzymes, substrates, loci, probes, and gene ontology, have been collected mainly from the following sources: http://www.ncbi.nlm.nih.gov/; http://us.expasy.org/enzyme/; http://www.genome.ad.jp/; http://www.arabidopsis.org/; http://www.geneontology.org/; and other publications. A number of scripts have been written to fetch external files from ftp sites, parse data, and populate data into the database.
Pathway visualization with gene expression
Several methods have been used to build pathway graphs in the literature. Most of the existing pathway software uses static images [2-4,9,11]. Static visualization cannot reflect any updated pathway information unless the static image is rebuilt [12]. Another approach is the semi-dynamic method. For example, Pathway Processor [7] puts several generated gene expression values at the same position on a corresponding KEGG static image when it visualizes pathways. This method is limited by pre-built image space. A third approach is automatic generation through standard graph layout algorithms. Examples have used algorithms for circular, orthogonal or planar drawing, and force-directed layout heuristics [13-15]. Becker et al. [16] used a combination of circular, hierarchical, and force-directed graph layout algorithms to compute the position of the graph elements representing main compounds and reactions. The resulting graphs are satisfactory when this method is applied for relatively regular pathways, such as circular or hierarchical pathways. However, a large number of irregular pathways exist. In addition, there is the added complexity of variations in enzyme number and gene expression for each step of a biochemical reaction. As for pathway analysis, pathway scores and distance functions have been proposed to analyze gene expression data in the context of pathways [17,18].
PathMAPA uses JSP/JavaBeans to retrieve database information for the automatic construction of pathways (Figure 3). It uses computational methods only for a small number of regular pathways, such as the circular Citrate cycle. The difference between our method and the automatic generation methods discussed above is that we draw elements for most of the complicated pathways with data stored in database tables instead of using a computational approach. The information retrieved from the database is obtained by joining a group of tables/views (Figure 4).
Figure 3.
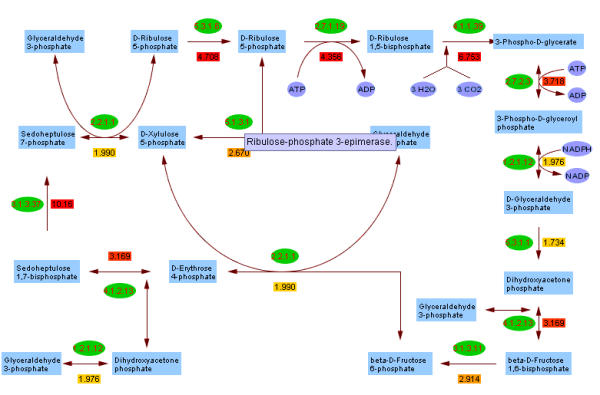
Calvin Cycle Pathway associated with gene expressions. The green ellipse is EC number, colored rectangular box is gene expression computed as the mean value of the probes that belong to the EC number. The long text box near EC 5.1.3.1 is the tool tip showing the enzyme name of EC 5.1.3.1. The user can click EC to open a new page to explore the enzyme details.
Figure 4.
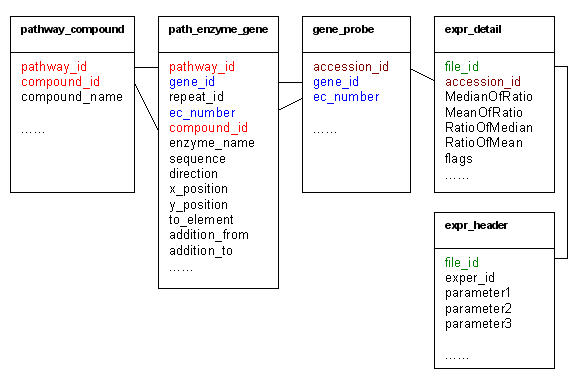
The major tables used to compute gene expression and generate pathway graphs. The table names are at the top of boxes in the bold font. The field names are at the bottom of boxes and the join columns between two tables are in the same color with link line.
Each pathway has a unique identifier. One pathway includes a list of enzymes with each enzyme including a list of genes (or loci). Each gene (or locus) may have a list of probes (accession ids) in the experiment. This relationship excludes any unrelated genes in the computation of gene expressions or test of pathways. There is a many-to-many relationship between EC_numbers. The combination of the EC_number, gene_id and accession_id columns is unique for the gene_probe table. The combination of the pathway_id, ec_number, gene_id and repeat_id columns is unique for the path_enzyme_gene table. In the query to compute the mean value of gene expression, a pathway_id is used to limit the EC_number into a particular pathway. Then a column repeat_id is used to group the repeated EC_numbers within a pathway. In each step of a pathway, such as from Ribulose 5-phosphate to Ribulose 1,5 bisphosphate of Figure 3, we treat Ribulose 5-phosphate, enzyme 2.7.1.19, the reaction arrow, the gene expression value and ATP-ADP as one unit. The path_enzyme_gene table (Figure 4) includes the pathway column (pathway_id), the relative order number of the unit (sequence), the reaction direction (direction), the x and y positions of Ribulose 5-phosphate (x_position, y_position), the next unit order number (to_element), the EC number (ec_number), the enzyme name (enzyme_name), and additional energy/chemicals like ATP-ADP involved in the reaction (addition_from, addition_to). The information for one unit is filled once for each EC_number appearance in a pathway. The compound name (Ribulose 5-phosphate) is obtained from the compound_name column by joining the path_enzyme_gene table with the pathway_compound table through the pathway_id and compound_id columns. Chemicals that are input to or output from a pathway are handled by filling in the addition_from and addition_to columns. If there is only input like H2O, or output like CO2, the addition_to column will be filled in with "input" or "output". The program can detect this and draw an up or down arrow. Additional code to handle very specific items for each pathway is very limited. The data storage for constructing pathway graphs is less than 100 M(G)B. Most of the data can be integrated from public sources, and it is simple to implement. The modules are scalable, and consist of classes that are repeatedly used for generation or visualization of different components of pathways.
Results and Discussion
Display of gene expressions on pathways at different levels for an individual experiment and across experiments
PathMAPA displays gene expressions for a given pathway and experiment at the enzyme, locus, and probe levels, respectively. The gene expressions at different levels are generated automatically from a database upon a web client's request. The value is computed by joining the table expr_header, expr_detail, gene_probe and path_enzyme_gene (Figure 4). The microarray files are selected and joined for computation once the user has selected an experiment. Gene expressions are computed as the average of the MedianOfRatio column by grouping at a user-selected gene expression level (probe, gene or enzyme). Pathways overlaid with gene expression levels provide investigators with an overview on whether gene expressions are up or down regulated, and how the gene expressions are distributed with each enzyme. Gene expressions are displayed on the pathway graph only at the enzyme level because a pathway describes biochemical reactions with enzymes. Since multiple loci may correspond to the same enzyme in many cases, expressions of these loci allow investigators to compare multiple loci within one enzyme. Furthermore, since more than one probe may exist on a microarray that corresponds to the same locus, gene expressions from multiple probes corresponding to the same locus may provide variation across these probes. In addition to displaying gene expression data for individual experiments, PathMAPA allows investigators to view expression data for a specific pathway across a group of experiments. Currently, gene expressions for multiple experiments are displayed in text format because of the limited space of the pathway graph. Gene expression comparisons across multiple experiments provide the investigator with an overview of whether gene expression patterns are related to experimental conditions. This is especially useful for investigators with time-course or multiple treatment level experiments. The computation of gene expressions with current microarray technology is limited in that it is currently impossible to separate gene expressions from two different reactions catalyzed by the same enzyme that has the same probes of the microarray experiments because the RNA from the sample is a mixture. The RNA from different transcriptions cannot be separated from the mixed sample either. However, as there are a limited number of genes for which this is the case, microarray technology plays an important role in biological research.
Display of automatic generated pathway images
PathMAPA automatically generates pathway image upon the web client's request in contrast with KEGG, MetaCyc, EcoCyc and TAIR [2-5,9] that rely on a large number of static images (gif files). The web servers of these databases respond to a client's request to view a pathway by sending a gif image file to client computer. PathMAPA uses Java code to automatically generate the images displayed at the users' computer rather than manually built image files. The algorithm involved can be summarized as: (1) query database to compute gene expression; (2) query database to retrieve selected pathway elements and coordinate information; (3) use Java code to draw the graph. More details in solving the complexity of pathway graphs have been described in the section "Pathway visualization with gene expression". The method used by PathMAPA is quite efficient in building images and reflects updated information through easy modification of data in database. As of yet the user cannot change image on the client side, but this feature will be added soon. On the machines we have tested, the generation of a pathway graph usually takes around 5 seconds. The pathway image provides two additional features: (1) it displays the full enzyme name as a tool-tip when the user's mouse is over any Enzyme Commission (EC) number and (2) if the user clicks on an EC-number spot, a new page is opened, from which the user can explore more details. Any change of the enzyme-gene-probe, substrate-reaction-compound or experiment-parameter-data will reflect on the pathway graph once the data are updated in the database table. It would take more time to re-build static images. Because biologists periodically modify some of the pathway gene lists, it is essential to be able to update pathway data when a pathway is studied in detail. The pathway construction method used in PathMAPA combines the flexibility of reusable code with the simplicity of database storage.
Statistical test for pathways
One unique advantage of PathMAPA over with TAIR is its ability to analyze pathways as a unit. For each statistical procedure, we use a Java Server Page (JSP) or Java Bean to generate data with JDBC and pass that data to the statistical component through an Application Programming Interface (API) call. The analysis result page is then displayed using JSP. Currently, Fisher's exact test [19,20] is used to test whether a given pathway is affected in a specific experiment. The enzymes, genes and probes involved in each pathway are restricted in their biological relationships as described in the implementation section. There are no unrelated genes involved in the statistical test. The Fisher's test considers the number of loci in each pathway, the number of loci with "altered" gene expression, and the total number of loci in the microarray. We have implemented a T-test across replicates to separate significant and non-significant probes. The cutoff value is determined by the P-value of the probe from the T-tests. The gene expression of a probe is considered to be significantly altered under the experimental condition if its P-value is less than the given threshold. We have also implemented a cutoff value method based on fold change, such as 2.0 fold [7], as another option. However, this method does not consider the variation in gene expressions because a mean value that is greater than 2 may not be statistically significant. The significance of gene expressions depends on both the mean value and the variance when sample size and significant level are given [21]. PathMAPA can identify up-regulated and down-regulated pathways depending on whether the mean gene expression in a significant pathway is greater or less than that of the loci that are significant but are not in the pathway. Each significant pathway is marked either up-regulated or down-regulated in the output and the resulting text file can be downloaded (Table 1). Note that if genes on a microarray are only a small portion of the whole genome or a small portion of the pathway, the results may not be meaningful even though the Fisher's exact test can still be performed.
Table 1.
Statistical test of gene expression at pathway level.
| Pathway Name | P-Value | Significant or not (NS) | Up or down regulation |
| ATP synthesis | 0.02358 | Significant | Up |
| Photosynthesis | 0.04368 | Significant | Up |
| Glutamate metabolism | 0.00569 | Significant | Up |
| Glyoxylate and dicarboxylate metabolism | 1.0E-4 | Significant | Up |
| Carbon fixation | 1.0E-4 | Significant | Up |
| Proteasome | 0.02033 | Significant | Up |
| Oxidative phosphorylation | 0.001279 | Significant | Down |
| Tyrosine metabolism | 0.02515 | Significant | Down |
| Glycolysis or Gluconeogenesis | 0.515 | NS | |
| Citrate cycle (TCA cycle) | 0.5626 | NS | |
| Pentose phosphate pathway | 0.5711 | NS | |
| Inositol metabolism | 1.0 | NS | |
| Pentose and glucuronate interconversions | 0.7458 | NS | |
| Fructose and mannose metabolism | 1.0 | NS | |
| Galactose metabolism | 0.3579 | NS | |
| Ascorbate and aldarate metabolism | 0.8625 | NS | |
| Fatty acid biosynthesis (path 2) | 1.0 | NS | |
| Fatty acid metabolism | 0.101 | NS | |
| Sterol biosynthesis | 0.4255 | NS | |
| Bile acid biosynthesis | 0.4304 | NS | |
| Ubiquinone biosynthesis | 0.08707 | NS | |
| Androgen and estrogen metabolism | 0.6917 | NS | |
| Urea cycle and metabolism of amino groups | 0.6471 | NS | |
| Purine metabolism | 0.8689 | NS |
This is an example to identify pathways that are affected by experimental treatment for one experiment with four replicates. NS: non-significant. Significance level is 0.05.
PathMAPA also plots a 3-D graph (Figure 5) for visualization of multiple experiments. The input data for plotting the graph are generated from the "Pathway Test" menu. The results show up-regulated (red), down-regulated (green) and non-regulated (grey) at different days (F1, F2, F3). The integrated graph provides the investigator with information on how related pathways are affected under different treatments across experiments. The resulting graph can be downloaded to the user's computer.
Figure 5.

The integrated pathway test graph showing the light effect on gene expressions across experiments. In this figure, F1, F2, and F3 mean data file 1, file 2 and file 3 that correspond to experiment 1, experiment 2 and experiment 3, respectively. The squares in the color of red, green and gray mean up-regulated, down-regulated and non-regulated, respectively.
Search of pathway elements across pathways
PathMAPA allows a biologist to quickly identify pathways to which a specific locus or probe belong. It also provides tools to search across pathways for EC-Number, GeneBank Accession ID, and Gene Ontology ID. Investigators can simply enter the item on the web page and click a button. Currently, PathMAPA contains around 100 plant pathways that can be used for queries.
Examples of Using PathMAPA to Study Pathways through Gene Expression
Identification of pathways affected
To identify pathways affected by treatment perturbations, we considered gene expressions under the wild type/white light condition versus those under the wild type/dark condition on different days after planting as follows: (1) WildType/White versus WildType/Dark; (2) WildType/White versus WildType/Dark 1.5 d; and (3) WildType/White versus WildType/Dark 5 WeekLeaf. There were four replicates for each experiment.
By clicking on the "Select Experiment" and "Pathway Test" links in the Pathway component of PathMAPA once for each experiment, we obtained one result file of the pathway test for each experiment. The result file was similar to Table 1. Then, after we clicked on the "Result Graph" menu, uploaded the three result files and completed the processing, we obtained the result graph (Figure 5). When the plants were treated under white light condition versus dark condition, one of the most significant changes was the photosynthesis pathway, which can be designated as an up-regulated pathway. The graph shows that gene expression of photosynthesis was up-regulated at 1.5 days and 5 weeks after planting but not at the very young stage (the same day of planting). These results are consistent with biological knowledge that photosynthesis is activated under white light condition. On the other hand, the gene expressions of other pathways, such as nitrogen metabolism, are not on this significance list.
Identification of enzymes activated
The Calvin cycle pathway is used to demonstrate how PathMAPA can help investigators to identify individual enzymes in a pathway affected under an experimental condition. We can click on the "Select Experiment" and "Enzyme Test" links to conduct a T-test. All enzymes in the pathway are significant at the 0.05 level (Table 2). This is consistent with the pathway test discussed above. We can display the Calvin cycle graph by using the "Select Experiment" and "Graph Model: EC" menus and selecting "Carbon Fixation". The overview of the gene expression for the pathway is displayed (Figure 3). The enzymes with the two highest changes are Sedoheptulose-bisphosphatase (3.1.3.37) and Ribulose 1,5-bisphosphate carboxylase/oxygenase (4.1.1.39). The latter enzyme, also called Rubisco, is the most extensively studied enzyme in photosynthesis. Rubisco works either as a carboxylase or oxygenase in photosynthesis and photorespiration [22,23]. The reaction catalyzed by Rubisco is the key step that fixes carbon dioxide, and the high gene expression changes in Rubisco are consistent with its functional importance. An investigator can click on an enzyme commission number such as 1.2.1.12. The hyperlink for enzyme/locus in the new page allows for further examination of enzyme information.
Table 2.
Statistical test of gene expression at enzyme level.
| EC_ID | Enzyme_Name | Signficance | Regulation |
| 1.2.1.12 | Glyceraldehyde 3-phosphate dehydrogenase | S | Up |
| 2.2.1.1 | Transketolase. | S | Up |
| 2.6.1.2 | Alanine aminotransferase. | S | Up |
| 2.7.1.19 | Phosphoribulokinase. | S | Up |
| 2.7.2.3 | Phosphoglycerate kinase. | S | Up |
| 3.1.3.11 | Fructose-bisphosphatase. | S | Up |
| 3.1.3.37 | Sedoheptulose-bisphosphatase. | S | Up |
| 4.1.1.39 | Ribulose-bisphosphate carboxylase. | S | Up |
| 4.1.2.13 | Fructose-bisphosphate aldolase. | S | Up |
| 5.3.1.1 | Triosephosphate isomerase. | S | Up |
| 5.3.1.6 | Ribose-5-phosphate isomerase | S | Down |
This is an example to identify enzymes that are affected by experimental treatment for the Calvin cycle pathway. S means significant and NS means non-significant. Significance level is 0.05.
Identification of loci involved
An enzyme like Rubisco may consist of several small subunits. We can examine these subunits by using the "Select Experiment", "Text Model: Locus", and "Text Model: Probe" links to display pages containing the gene expressions of loci or probes. By clicking the "Select Experiment", "Gene Test" and "Carbon fixation" links, statistical test results for all genes in the Calvin cycle can be obtained (Table 3). Let us take an example to examine the loci at individual EC. All loci of EC 4.1.1.39 are significant. On the other hand, only 3 of the 6 loci from EC 4.1.2.13 are significant. The detailed information from each locus may yield information on how each locus works as part of an enzyme. Overall, identification of gene expression for loci involved could help the investigator to identify whether the putative gene function is correctly defined, and whether different loci from one enzyme have different functions like Rubisco's carboxylase and oxygenase described in the previous section. Such analyses might provide a clue as to how to understand and manipulate pathways at different levels.
Table 3.
Statistical test of gene expression at gene level.
| EC_ID | Enzyme_Name | Locus | Signficance | Regulation |
| 1.2.1.12 | Glyceraldehyde 3-phosphate dehydrogenase | At1g13440 | S | Up |
| At3g26650 | S | Up | ||
| At3g04120 | S | Down | ||
| At1g79530 | NS | |||
| At1g16300 | S | Up | ||
| At1g12900 | S | Up | ||
| At1g42970 | NS | |||
| 2.2.1.1 | Transketolase. | At2g45290 | NS | |
| At3g60750 | S | Up | ||
| 2.6.1.2 | Alanine aminotransferase. | At1g23310 | S | Up |
| 2.7.1.19 | Phosphoribulokinase. | At1g32060 | S | Up |
| 2.7.2.3 | Phosphoglycerate kinase. | At1g56190 | S | Up |
| At1g79550 | S | Down | ||
| At3g12780 | S | Up | ||
| 3.1.3.11 | Fructose-bisphosphatase. | At1g43670 | NS | |
| At3g54050 | S | Up | ||
| 3.1.3.37 | Sedoheptulose-bisphosphatase. | At3g55800 | S | Up |
| 4.1.1.39 | Ribulose-bisphosphate carboxylase. | At1g67090 | S | Up |
| At5g38430 | S | Up | ||
| At5g38410 | S | Up | ||
| At5g38420 | S | Up | ||
| 4.1.2.13 | Fructose-bisphosphate aldolase. | At2g01140 | NS | |
| At2g21330 | S | Up | ||
| At2g36460 | NS | |||
| At3g52930 | S | Up | ||
| At4g26530 | NS | |||
| At4g38970 | S | Up | ||
| 5.3.1.1 | Triosephosphate isomerase. | At3g55440 | NS | |
| At2g21170 | S | Up |
This is an example to identify genes that are affected by experimental treatment for the Calvin cycle pathway. S means significant and NS means non-significant. Significance level is 0.05.
Conclusions
PathMAPA can automatically generate and display biological pathways integrated with gene expression data for Arabidopsis thaliana. It is a useful bioinformatics tool for performing statistical test at the pathway, enzyme and gene levels and studying enzyme, locus and probe details within a pathway. It can also be used to identify pathways to which a set of elements belongs as well as to compare gene expressions across experiments. PathMAPA facilitates the exploration of microarray data, the investigation of pathways, and the generatation of insights from microarray data.
Availability and requirements
The web site can be accessed from http://bioinformatics.med.yale.edu/pathmapa.htm through IE 5.0 or higher, or Netscape 7 or higher from multiple platforms. It needs to install Java Plug-In 1.3.1_02 or higher for some platforms.
Authors' contributions
DP carried the pathway data collection, designed and developed the database and web site. HZ and KC supervised the study. NS added to the pathway analysis method. ZG coded the statistical analysis component for microarray data. LM and XD provided microarray data and biological input. MH took part in coding the web pages. All authors have read and approved the final manuscript.
Acknowledgments
Acknowledgements
We thank two reviewers for their constructive comments. Research supported in part by NIH grants T15 LM07056, R01 GM59507, R01 GM-47850, ACS IRG 58-012-45, K25 HG02378, NSF grants 0241160 and 0135442.
Contributor Information
Deyun Pan, Email: deyun.pan@yale.edu.
Ning Sun, Email: ning.sun@yale.edu.
Kei-Hoi Cheung, Email: kei.cheung@yale.edu.
Zhong Guan, Email: zhong.guan@yale.edu.
Ligeng Ma, Email: ligeng.ma@yale.edu.
Matthew Holford, Email: matthew.holford@yale.edu.
Xingwang Deng, Email: xingwang.deng@yale.edu.
Hongyu Zhao, Email: hongyu.zhao@yale.edu.
References
- Gollub J, Ball CA, Binkley G, Demeter J, Finkelstein DB, Hebert JM, Hernandez-Boussard T, Jin H, Kaloper M, Matese JC, Schroeder M, Brown PO, Botstein D, Sherlock G. The Stanford Microarray Database: data access and quality assessment tools. Nucleic Acids Research. 2003;31:94–96. doi: 10.1093/nar/gkg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD, Riley M, Saier M, Paulsen IT, Collado-Vides J, Paley SM, Pellegrini-Toole A, Bonavides C, Gama-Castro S. The EcoCyc Database. Nucleic Acids Research. 2002;30:56–8. doi: 10.1093/nar/30.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD, Riley M, Paley SM, Pellegrini-Toole A. The MetaCyc Database. Nucleic Acids Research. 2002;30:59–61. doi: 10.1093/nar/30.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD, Paley S, Romero P. The Pathway Tools software. Bioinformatics. 2002;18:S225–S232. doi: 10.1093/bioinformatics/18.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nature Genetics. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- Grosu P, Townsend JP, Hartl DL, Cavalieri D. Pathway Processor: A Tool for Integrating Whole-Genome Expression Results with Metabolic Networks. Genome Research. 2002;12:1121–1126. doi: 10.1101/gr.226602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinke DW. Arabidopsis thaliana : A Model Plant for Genome Analysis. Science. 1998;282:679–682. doi: 10.1126/science.282.5389.662. [DOI] [PubMed] [Google Scholar]
- Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M, et al. The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Research. 2003;31:224–228. doi: 10.1093/nar/gkg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe ML, Serizet C, Thareau V, Aubourg S, Rouzé P, Hilson P, Beynon J, Weisbeek P, Hummelen P, Reymond P, Paz-Ares J, Nietfeld W, Trick M. CATMA: A complete Arabidopsis GST database. Nucleic Acids Research. 2003;31:156–158. doi: 10.1093/nar/gkg071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel R, Bairoch A, Hochstrasser D. A new generation of information retrieval tools for biologists: the example of the ExPASy WWW server. Trends Biochem Sci. 1994;19:258–260. doi: 10.1016/0968-0004(94)90153-8. [DOI] [PubMed] [Google Scholar]
- Brandenburg FJ, Gruber B, Himsolt M, Schreiber F. Automatische Visualisierung biochemischer Information. In Proceedings of the Workshop Molekulare Bioinformatik, GI Jahrestagung. 1998. pp. 24–38.
- Battista DG, Eades P, Tamassia R, Tollis IG. Annotated bibliography on graph drawing algorithms. Comput Geom-Theor Appl. 1994;4:235–282. doi: 10.1016/0925-7721(94)00014-X. [DOI] [Google Scholar]
- Battista DG, Eades P, Tamassia R, Tollis IG. Graph Drawing: Algorithms for the Visualization of Graphs. Prentice Hall, New Jersey. 1999.
- Brandenburg FJ, Junger M, Mutzel P. Algorithmen zum automatischen Zeichnen von Graphen. Informatik Spektrum. 1997;20:199–207. doi: 10.1007/s002870050066. [DOI] [Google Scholar]
- Becker MY, Rojas I. A graph layout algorithm for drawing metabolic pathways. Bioinformatics. 2001;17:461–467. doi: 10.1093/bioinformatics/17.5.461. [DOI] [PubMed] [Google Scholar]
- Hanisch D, Zien A, Zimmer R, Lengauer T. Co-clustering of biological networks and gene expression data. Bioinformatics. 2002;18:145S–154S. doi: 10.1093/bioinformatics/18.suppl_1.s145. [DOI] [PubMed] [Google Scholar]
- Zien A, Kueffner R, Zimmer R, Lengauer T. Analysis of Gene Expression Data with Pathway Scores. ISMB. 2000:407–417. [PubMed] [Google Scholar]
- Fisher RA. The logic of inductive inference. Journal of the Royal Statistical Society Series A. 1935;98:39–54. [Google Scholar]
- Fisher RA. Confidence limits for a cross-product ratio. Australian Journal of Statistics. 1962;4:41. [Google Scholar]
- Speed TP. Statistical analysis of gene expression microarray data. Chapman & Hall/CRC. 2003.
- Lorimer GH, Andrews TJ. Plant photorespiration. an inevitable consequence of the existence of atmospheric oxygen. Nature. 1973;243:359. [Google Scholar]
- Lorimer GH. The carboxylation and oxygenation of ribulose 1,5-bisphosphate: the primary events in photosynthesis and photorespiration. Annu Rev Plant Physiol. 1981;32:349–383. doi: 10.1146/annurev.pp.32.060181.002025. [DOI] [Google Scholar]