

Abstract
Graft versus host disease (GVHD), mediated by donor T cells, is a significant source of morbidity and mortality following allogeneic stem cell transplantation. Mesenchymal stem cells (MSC) can successfully treat ongoing graft versus host disease, presumably due to their ability to suppress donor T cell proliferation. Little is known about the potential of MSC to prevent GVHD. Here we show that bone marrow-isolated MSC can suppress the development of GVHD if given after donor T cell recognition of antigen. IFN-γ was required to initiate MSC efficacy. Recipients of IFN-γ−/− T cells did not respond to MSC treatment and succumbed to GVHD. MSC, pre-treated with IFN-γ, became immediately active and could suppress GVHD more efficiently than a fivefold-greater number of MSC that were not activated. When given at the time of bone marrow transplantation, activated MSC could prevent GVHD mortality (100% survival, p=0.006). MSC activation was dependent on the magnitude of IFN-γ exposure, with increased IFN-γ exposure leading to increased MSC suppression of GVHD. Activated MSC present a new strategy for preventing GVHD using fewer MSC.
Keywords: Mesenchymal stem cell, GVH disease, IFN-γ
Introduction
Allogeneic hematopoietic stem cell transplants have the potential to play a significant curative role in the treatment of malignant and non-malignant hematopoietic disorders, autoimmune diseases, and immunological deficiencies, and in the induction of transplantation tolerance [1–10]. Widespread application of this therapeutic modality is limited due to the morbidity and mortality of graft versus host disease (GVHD), which affects 50% of stem cell transplant recipients [11–16]. While grafts highly matched to the recipient, young donors, donor/recipient sex match, and post-transplant immunosuppression are strategies used to reduce the risk of GVHD [17], thus far, the greatest preventative measure has been intentional underutilization of stem cell transplantation. Theoretically, strategies aimed at preventing GVHD would target early initiating factors either during the inflammatory milieu created in the wake of tissue damage from conditioning regimens [18, 19] or during T cell antigen recognition and proliferation [20, 21]. Once the efferent effector phase occurs, donor Tcell-mediated destruction of host tissues occurs and preventive strategies are replaced with treatment regimens [19].
Mesenchymal stem cells (MSC) have been used in the efferent phase of GVHD to successfully treat ongoing, acute, steroid-resistant GVHD [22, 23]. In contrast, when given at the time of BM transplant, for the prevention of GVHD, the incidence of grade III/IV GVHD was not significantly improved [24], suggesting the absence of necessary initiating factors for MSC activation and subsequent efficient suppression of donor-derived T cells. MSC may require activating signals from robustly proliferating T cells to induce their suppressive effects. MSC reliably suppress large scale T cell proliferation in response to polyclonal stimulation in vitro [25–28]. In contrast, with allogeneic mixed lymphocyte cultures of variable stimulation, MSC suppression is also variable; MSC do not completely abrogate lymphocyte proliferative responses between all donor and recipient pairs [26, 27, 29, 30]. In addition, MSC do not suppress the modest T cell proliferative response to recall antigens [31]. These findings suggest MSC may exert their optimal effects during the events surrounding larger scale T cell activation and proliferation, such as that encountered during steroid-resistant GVHD. Identification and simulation of the events that stimulate MSC to suppress GVHD might aid in the development of a preventive GVHD strategy.
Murine experimental models used to dissect the mechanism of MSC effects in the course of GVHD have yielded mixed results, with some studies showing MSC efficacy and others finding no effect [32–35]. Several factors are likely to contribute to the variable results. MSC tissue source, (i.e., BM, cord blood, adipose tissue), method of isolation to remove myeloid precursors (several weeks [36] vs. rapid immunodepletion [37]) and timing of MSC administration are potential variables that could explain these differences. Notably, such variation has not been observed clinically, with MSC treatment of ongoing GVHD reported to have significant efficacy. Interestingly, human MSC isolation can significantly differ from murine MSC isolation. Human MSC can undergo culture for as few as 14 days prior to administration [38]. It is possible that murine MSC might also be more consistently efficacious if similar methods of isolation and culture were used.
In these studies, we used a rapid immunodepletion method to isolate murine MSC, resulting in dramatically shortened culture times and low passage cells. We tested the effect of these MSC first for efficacy of GVHD treatment and observed their effect to be similar to clinical observations, with lack of efficacy in prevention and improved survival when given during ongoing GVHD. To further dissect the factors that might initiate MSC suppression of GVHD, we localized their earliest time point of efficacy to occur following antigen presentation. Since IFN-γ is produced by donor T cells in response to antigen recognition, we tested whether it could initiate MSC efficacy in vivo and observed a dose-response effect, with higher doses of IFN-γ being more effective than lower doses. These observations suggest that IFN-γ serves as an initiating stimulus for MSC immunosuppressive activity in vivo. MSC response to this pro-inflammatory cytokine is differential, with three-log increases in IFN-γ required for maximal T cell suppression in vivo. These observations indicate MSC exposure to concentrated amounts of IFN-γ can stimulate MSC to prevent GVHD and provide the basis for a new potential strategy in prevention of GVHD.
Results
MSC treatment of GVHD
Murine MSC were isolated using rapid immunodepletion. This method was observed to provide CD29+CD44+Sca1+ MSC in 14 days (Fig. 1), a time course which is similar to isolation of human MSC [38]. By the third passage, 0.15 × 106–0.30 × 106 MSC per donor mouse could be retrieved for transplantation experiments. This number of MSC exceeds the numbers obtained from the classic method in which MSC are not devoid of myeloid cells until passage six. This technique also avoided long-term exposure of primitive MSC to mature myeloid lineages, which may enable murine MSC to become more immunostimulatory than immunosuppressive in vivo [33]. Using previously reported methods, these cells were observed to be capable of differentiating into adipogenic and osteogenic tissues (data not shown) [27, 39].
Figure 1.
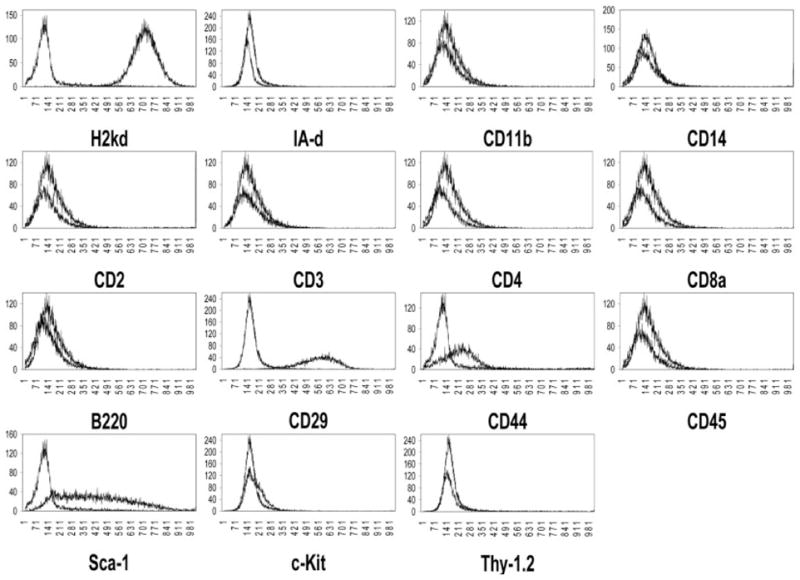
MSC phenotype following rapid immunodepletion. When compared to isotype control, MSC stained positive for MHC class I (H-2kd), the VLA complex marker CD29, the cell adhesion receptor CD44, and the hematopoietic stem cell marker Sca-1. MSC stained negative for MHC class II (I-Ad), macrophage cell surface markers (CD11b, CD14), B cell marker (B220), lymphocytes (CD2, CD3, CD4, and CD8a), and the hematopoietic stem cell markers Thy-1 and c-kit. Histograms represent consistent findings of more than 30 independent experiments.
To establish whether MSC isolated in this manner proved efficacious in the treatment of GVHD, 0.10 × 106 MSC were administered during various phases of GVHD. First, MSC were administered on day 0 along with the BM graft and supplementary T cells to induce GVHD (Fig. 2A). For this administration, MSC were co-cultured with the BM graft and splenocytes for 2 h prior to administration. We hypothesized that cell contact between MSC and GVHD-producing T cells prior to donor antigen recognition could suppress Tcell activity and subsequent GVHD mortality. Preemptive cell contact of MSC with T cells did not prove to be effective; there was no statistical improvement in GVHD-related mortality when compared to control animals that received BM grafts and supplementary T cells.
Figure 2.
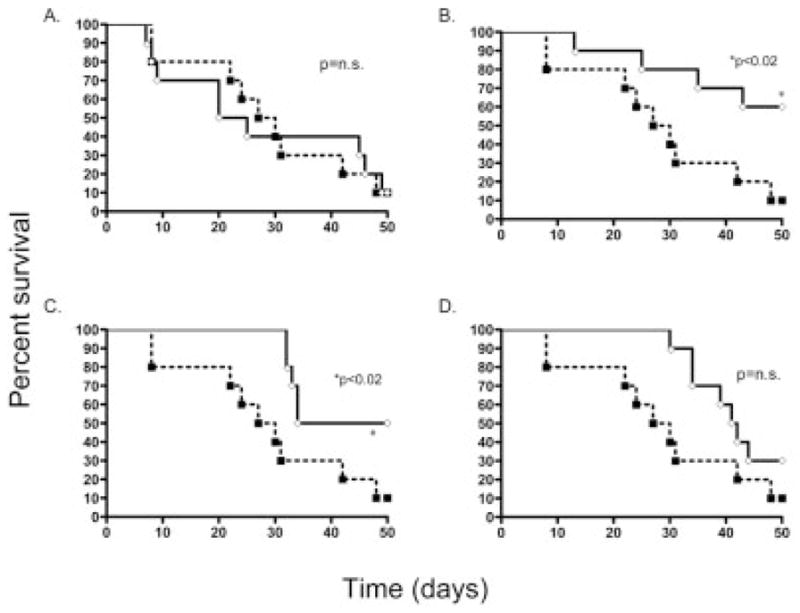
The effect of timing of MSC administration on 40-day survival following induction of GVHD. Following lethal irradiation, B6 recipients underwent transplantation with allogeneic BALB BMC and splenocytes on day 0 (control, dashed line) resulting in 30% survival, (experiment repeated six times). In experimental groups (solid line), BALB MSC were administered on days 0 (A), 2 (B), 20 (C) or 30 (D). MSC administered on days 2 and 20 significantly increased survival by 10–60% and 10–50%, respectively (p<0.05). n=10 for each experimental group.
During phase II of GVHD, donor T cells are exposed to host antigen and become active, serving to both proliferate and recruit additional Tcells [40]. We tested whether MSC, administered after donor antigen recognition, could mitigate GVHD mortality (Fig. 2B). Antigen recognition and/or subsequent activation of T cells appeared to be required for MSC efficacy as MSC given on day 2 increased survival from 10% to 60% (p<0.02). Following MSC infusion, some of the animals that had developed signs of GVHD, such as ruffled fur and alopecia, had improvement of these physical findings with many surviving animals experiencing a complete reversal to normal-appearing fur.
MSC were tested for their ability to treat ongoing GVHD (administered on day 20) or treatment of severe, pre-morbid GVHD (given on day 30). MSC administration increased survival from 10% to 50% when given on day 20 (p<0.02, Fig. 2C), and to 20% for day 30 treated animals (p=0.08, n.s, Fig. 2D). These data show MSC isolated with rapid immunodepletion are effective in preventing GVHD as well as treating ongoing GVHD. It is important to note that MSC contaminated with >3% CD45+ cells and MSC of late passage (greater than 6), had no significant effect on GVHD-related mortality (data not shown), indicating that early passage and significant immunodepletion were required for MSC suppression of GVHD.
MSC treatment is dose dependent
To further define the limits of MSC efficacy, we tested the effect of MSC dose on survival. Following transplantation, either 0.1 × 106 MSC or a fivefold greater number (0.5 × 106) were administered on either day 2 or day 20 (Fig. 3) and compared to transplanted animals that did not receive MSC. There was no dose-response effect when a higher dose was administered on day 2. Both 0.1 × 106 and 0.5 × 106 MSC significantly increased survival (p=0.004), and the two survival curves were indistinguishable from each other (Fig. 3A). These data indicated that higher doses, when given as a preventative measure, did not appear to change the course of mortality. For animals receiving MSC on day 20, survival following 0.5 × 106 MSC significantly increased from 10% to 85%, (p=0.0006, Fig. 3B). Statistical comparison between low and high dose revealed a strong trend, suggesting a difference between the two groups (p=0.07). Based on these data, it appeared that MSC behaved differently when given for treatment than when given as a preventative measure. Since higher numbers of T cells are likely to have undergone antigen recognition and proliferation on day 20 when compared to day 2 [41], we hypothesized that the difference in MSC behavior was due an increase in the magnitude of activating signals generated from the increased antigen presentation and/or T cell proliferative activity.
Figure 3.
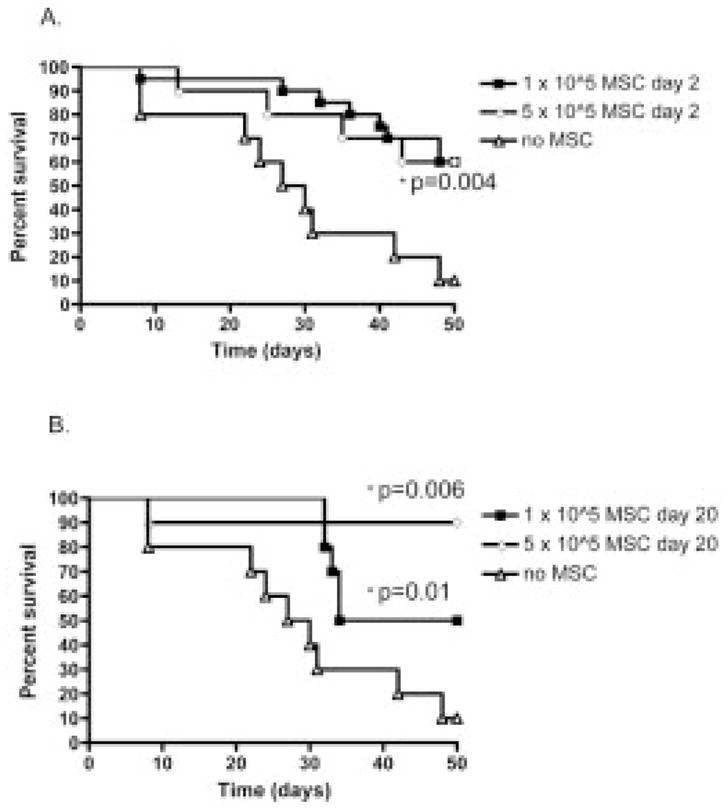
Effect of MSC dose escalation. Low (0.1 × 106) and high (0.5 × 106) doses of MSC were administered on day 2 (A, n=10) or day 20 (B, n=10). Both low and high doses led to a significant improved survival when given on either days 2 or 20. High dose MSC given on day 20 significantly increased survival to 85% (p=0.006).
Tissues were analyzed from recipients who underwent treatment with MSC. On histological examination of lung, spleen, colon, and skin, MSC treatment improved the severity of GVHD scoring. Spleen and lung displayed the greatest findings, with both tissues observed to appear normal after receiving 0.5 × 106 MSC (p<0.0007, and 0.03, respectively, Fig. 4).
Figure 4.
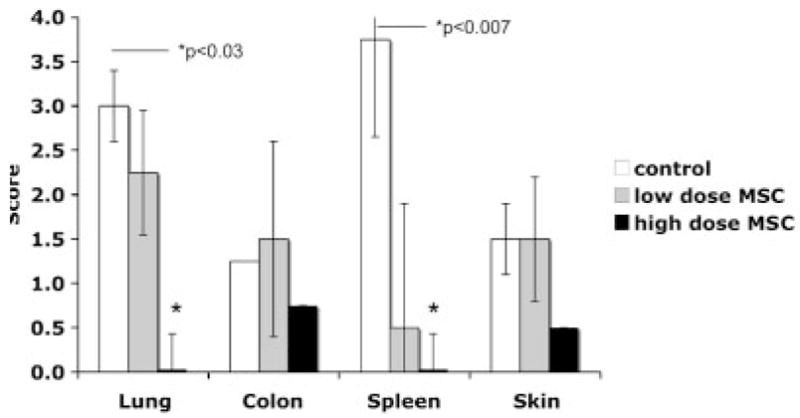
GVHD scores of lung, colon, spleen, and skin. Recipients of 0.5 × 106 (high) or 0.1 × 106 (low) MSC and control recipients were killed for histological examination. Sections taken from lung, colon, and spleen were scored on a scale of 1–4 with 4 being the most severe changes consisting of tissue destruction observed in GVHD. Skin was scored on a scale of 1–3, with 3 being the most severe. This experiment was repeated three times (n=3 per group).
IFN-γ is required for MSC reduction of GVHD mortality
Based on the hypothesis that antigen recognition and, potentially, Tcell proliferation were required to initiate MSC activity, we tested the regulatory protein IFN-γ for its ability to initiate suppressive activity in MSC. This protein was chosen for several reasons. First, IFN-γ can be produced by both donor dendritic cells following antigen recognition and donor T cells upon activation [42]. In response to high concentrations of IFN-γ, MSC are induced to produce indoleamine 2,3-dioxygenase (IDO), the enzyme known to promote the immunosuppressive barrier at the maternal-fetal interface [43]. Also, MSC treatment with IFN-γ in vitro has been observed to enhance MSC production of several immunosuppressive cytokines such as TGF-β [44]. We hypothesized that MSC would respond to the presence of this immunoregulatory protein by being stimulated to suppress GVHD.
To determine whether IFN-γ played a role in MSC suppression of GVHD in vivo, donor splenocytes, incapable of producing IFN-γ, were infused to induce GVHD. In this system, the sources of IFN-γ were limited to the low numbers of antigen-presenting cells (APC) within the BM and host hematopoietic cells. GHVD-related mortality was rapid and severe; 100% mortality occurred prior to day 30 (Fig. 5). Addition of MSC had no effect. These data indicate that the absence of donor IFN-γ led to accelerated GVHD, which could not be controlled by donor MSC.
Figure 5.
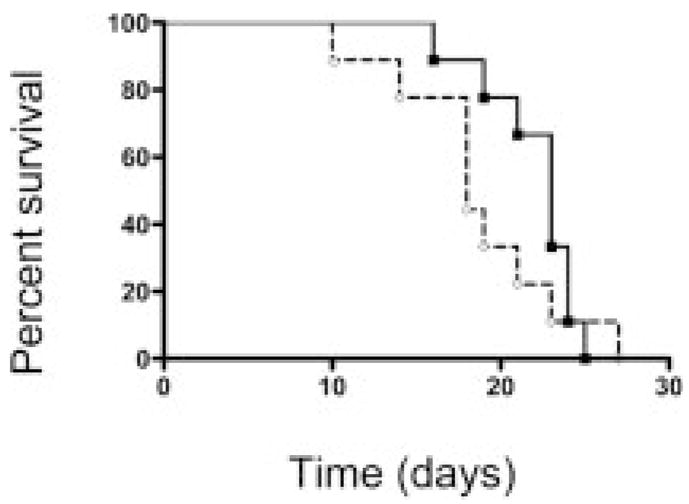
Requirement of IFN-γ for MSC mitigation of GVHD. Splenocytes from IFN-γ knockout mice were used to induce GVHD (solid line, n=10). Addition of 0.1 × 106 MSC on day 2 failed to affect survival (dashed line), indicating MSC required IFN-γ to initiate their suppressive effects.
MSC activation with IFN-γ
To further examine the ability of IFN-γ to initiate MSC suppression, we treated MSC with three concentrations of IFN-γ (5, 50, or 500 U) prior to their administration on day 0. We hypothesized that MSC were not effective on day 0 because they failed to receive a sufficient IFN-γ stimulus. By pre-treating the MSC with IFN-γ, we believed it could be possible to activate them for more efficient suppression of GVHD. When treated with either of the lower doses of IFN-γ, MSC were ineffective in significantly preventing GVHD mortality when compared to untreated MSC (Fig. 6A). In contrast, 500 U IFN-γ increased survival to 100% (p=0.02) and this treatment was significantly better than MSC pre-treatment with 5 U (p=0.006) or 50 U (p=0.005). These data suggested that MSC suppression could be initiated with high dose but not low dose IFN-γ, thereby identifying a threshold of MSC activation in response to the immunoregulatory protein.
Figure 6.
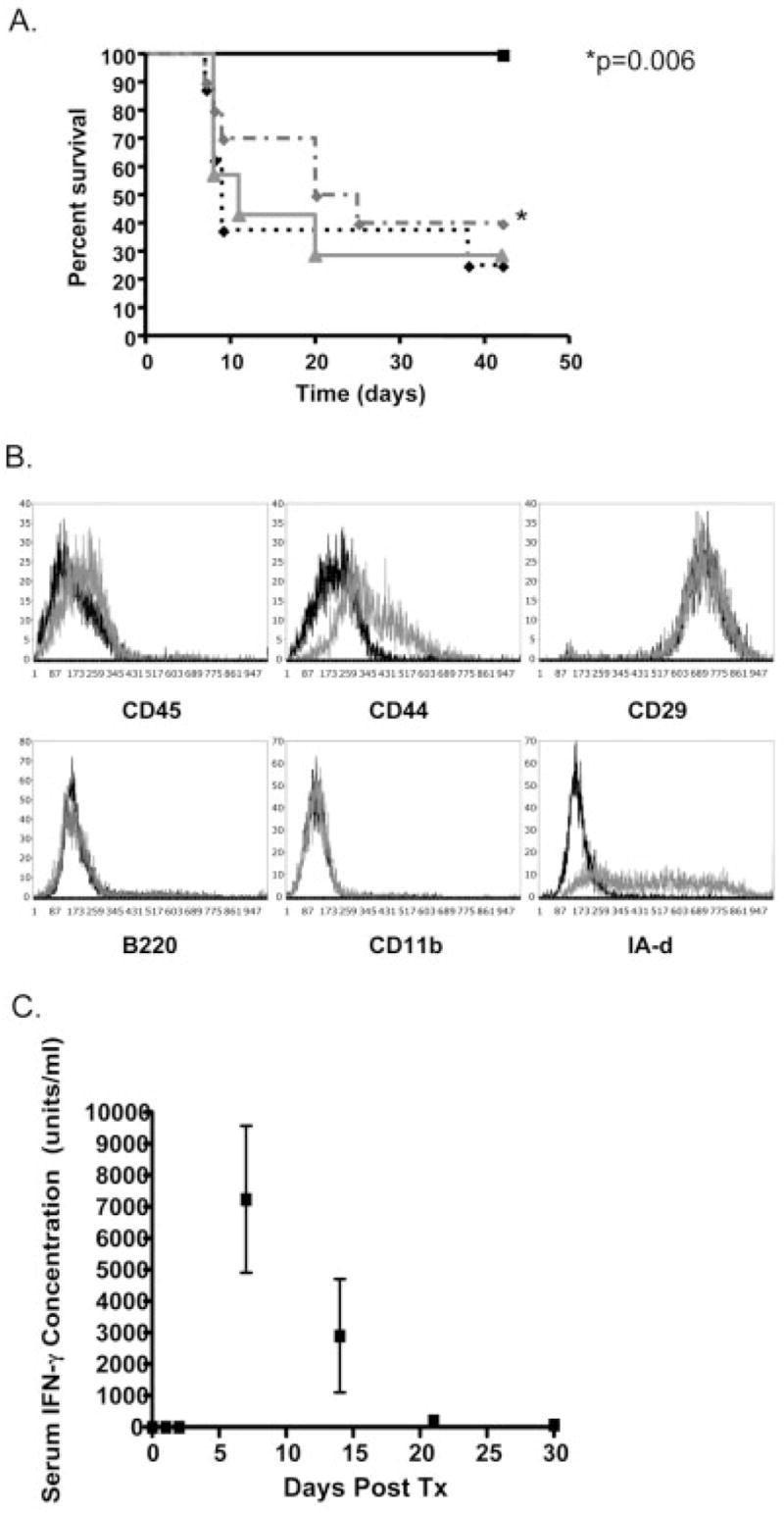
Activation of MSC with IFN-γ. Either untreated (A, dashed gray line, n=10) or IFN-γ-treated BALB MSC were administered on day 0. MSC treated with 5 U (solid gray line, n=10) or 50 U (dashed black, n=10) IFN-γ showed no effect on GVHD-related mortality when compared to untreated MSC. MSC treated with 500 U (solid black line, n=10) were significantly more effective than untreated MSC (p=0.006) and MSC treated with lower doses of IFN-γ. IFN-γ treatment appeared to have a direct effect on MSC (B), increasing expression of MHC class II. No detection of CD45 or CD11b populations were noted after IFN-γ treatment (gray line) when compared to pre-treatment (black line), indicating that IFN-γ treatment did not expand an immunoregulatory dendritic cell population, (experiment performed >10 times prior to each transplant). Following transplantation and the development of GVHD, circulating IFN-γ measured in the serum by ELISA (C) surpassed 500 U by day 7 with a gradual drop below 500 by days 21 and 30, suggesting there was sufficient circulating IFN-γ to activate MSC by day 7, but not after day 30, (each time print represents analysis of three to five recipients measured in duplicate).
Since suppression of T cells has also been observed by APC [45], we wished to exclude the possibility that IFN-γ preferentially expanded an APC subpopulation (found to be less than 3%) within the MSC preparation. If this were the case, a phenotypic analysis of the MSC following treatment with IFN-γ would show a distinct increase in the CD11b+, B220+ or CD45+ population. MSC phenotype analyses were performed prior to each transplant experiment with IFN-γ-treated MSC (>10 times). No significant increases in CD45, CD11b, CD44, CD29 or B220 were noted, indicating there was no enrichment of an additional hematopoietic cell type to account for this effect (Fig. 6B). In response to IFN-γ treatment, MSC increased expression of MHC class II, as has been previously described [46]. These findings suggest MSC phenotype and function are modified in response to IFN-γ concentration.
With the identification of a critical level of IFN-γ required for MSC activation, we next analyzed the serum of transplant recipients to determine when such levels were observed in vivo. Serum IFN-γ was measured by ELISA (in picograms) on days 0, 1, 2, 7, 14, 21, 30 and was converted to units (conversion factor =10 U/pg, Fig. 6C). No serum IFN-γ was detected on days 0 (at the time of transplant) or 1. A small increase on day 2 (2.7 ± 2.7 U) was followed by a dramatic increase on day 7 (7232 ± 2340 U). By day 21, levels had decreased to 207 ± 149 U, with 63 ± 63 U noted on day 30. These data suggest, following transplantation, circulating IFN-γ at the time of transplant are not sufficient to activate MSC. By day 30 there is also insufficient circulating IFN-γ for MSC activation, providing a possible explanation for the lack of MSC efficacy when administered on day 30. To test whether IFN-γ alone had an effect on GVHD, we treated three animals with 500 U IFN-γ i.v. on day 0 in the absence of MSC. All three animals survived to day 50 (data not shown), indicating a beneficial effect of this treatment on mortality; however, all three developed severe GVHD with alopecia, weight loss, and scabbing, requiring euthanasia. These data suggest that, while IFN-γ has a beneficial effect on GVHD mortality, activated MSC appear to have superior efficacy, since treatment with activated MSC on day 0 prevented the development of GVHD.
Discussion
The purpose of these experiments was to identify potential factors that initiated MSC suppressive behavior in vivo to develop a preventative strategy for GVHD. First, an experimental model of MSC treatment of GVHD was established to closely mimic clinical observations. Next, this model was applied in studies to identify factors that initiated and optimized the efficacy of MSC. The model selected was based on a different and more rapid method of MSC isolation, used primarily to closely match the conditions used during human MSC isolation and culture. Notable differences with this method when compared to the classic method of isolation described by Friedenstein [36] include the use of low-passage MSC, diminished culture time prior to administration, and lack of prolonged MSC exposure to myeloid cells. Using cells isolated in this manner, we observed that MSC significantly reduced the mortality of GVHD as evidenced by survival data and supportive histological analyses. These data correlate to findings reported in clinical studies [22, 24, 47]. MSC had no significant efficacy when given at the time of transplant, on day 0, but significantly improved GVHD mortality when given during ongoing GVHD, on day 20. Due to the similarities between this model and clinical observations, we believe this model is useful in studying clinical variables that cannot be easily or quickly undertaken with the clinical trials currently underway.
The observations on the effect of timing of MSC administration led us to conclude that factors required for initiation of MSC suppressive activity were not present prior to antigen presentation. The pro-inflammatory milieu, which is induced as early as 4 h following total body irradiation, is comprised of high serum levels of TNF-α, IL-1α, and IL-6 [48]. TNF-α has been observed to induce immunosuppressive activity from MSC through the production of COX-2 and PGE2. Comparison of TNF-α and IFN-γ treatment of MSC revealed that IFN-γ treatment induced IDO as well as COX-2 and PGE2 production [49]. Heme-oxygenase 1 is also observed in the pro-inflammatory milieu, and can be elicited by MSC in conditions of hypoxia, also leading to T cell suppression [50]. TGF-β, also secreted by MSC, could also be instrumental in suppression [51]. Since MSC were not effective when given during this pro-inflammatory milieu, it is possible that the amount of circulating TNF-α was not sufficient to initiate MSC-induced suppression. Alternatively, it is possible that TGF-β, IDO, or hemoxygenase-1, produced as a consequence of IFN-γ-stimulated MSC, may be a critical component of the MSC effect and can only be elicited through sufficient IFN-γ stimulation.
Following analysis of recipient sera, we observed that serum levels may be sufficient to stimulate MSC on day 7. This suggests that, in the mouse model, this time period is likely to be effective for MSC activation. Correlation with IFN-γ serum levels in patients might be used to guide the timing of MSC therapy. A lack of stimulating levels of IFN-γ was observed on days 0–2. This could explain why there was no efficacy on day 0, but may not fully explain the efficacy observed when MSC were administered on day 2. We speculate that threshold levels were present in areas where newly activated dendritic and T cells were producing high levels of IFN-γ locally, such as the spleen and lymph nodes. It has been reported that following administration, T cells migrate to secondary lymphoid organs and target organs that predominantly express certain chemokines and chemokine receptors, such as MIP-1α, MIP-2, MCP-1, and MCP-3 [52]. MSC migration can also be enhanced by these chemokines [53] and MSC chemokine receptor expression can be regulated by IFN-γ [54]. Taken together, it is possible that, following infusion, MSC migrated in response to tissue chemokine expression, where local production of IFN-γ by activated dendritic cells and donor T cells, during early GVHD (day 2), was sufficiently concentrated to provide MSC activation. Following activation, MSC have the capability to inhibit both dendritic cell and early T cell responses [55–58]. It is possible that part of the efficacy of day 2 administration is due to the ability of MSC to dampen the escalation of GVHD by local control within both the target organs as well as the lymphoid organs. Of all the tissues examined, splenic tissue appeared to have some of the most significant improvement following MSC treatment. The powerful and seemingly preferential effect of MSC observed on this lymphoid tissue, may reflect the greater levels of locally produced IFN-γ during GVHD.
MSC administered on day 30 had no efficacy when compared to day 2 or day 20. This observation may be due to two factors: the overwhelming increase in the number of donor T cells for which the number of MSC were insufficient and/or the corresponding drop in the levels of IFN-β. Despite such increased numbers of T cells, T cell production of IFN-γ has been observed to decrease during ongoing GVHD [59]. We also observed low serum levels of IFN-γ at this time. It is possible that MSC administered on day 30 failed to receive sufficient IFN-γ, either through the circulation or through local production. The lack of available IFN-γ for MSC may have limited their ability to produce significant amounts of immunosuppressive molecules such as IDO, IL-10, TGF-β. All of these have been observed to have a dose-response relationship with IFN-γ treatment of MSC [43, 44]. Our in vivo observations are very similar to those observed in vitro; using this model, our data show MSC-induced suppression of GVHD is dependent on the magnitude of IFN-γ stimulus.
Addition of a fivefold higher number of MSC (0.5 × 106) improved survival to 85% when MSC were administered on day 20. When MSC were administered on day 2, survival remained at 60% despite the higher dose. We hypothesized that MSC given on day 2 failed to receive sufficient amounts of IFN-γ to become activated. Following activation of MSC, 0.1 × 106 MSC administered on day 2 improved survival to 100%. This observation suggests that the efficacy of MSC can be manipulated by IFN-γ activation. Since IFN-γ production can vary during the course of GVHD, the efficacy of MSC may also vary unreliably, with some treatments not attaining full immunosuppressive potential. Depending on the number of activated T cells, the ratio of T cells to MSC, and the available IFN-γ, it is possible that lack of optimization of these three factors could result in MSC therapy which is only marginally beneficial. In clinical trial, some patients, who received MSC treatment of ongoing GVHD, had no beneficial effect [23]. It is possible that activated MSC may provide a more uniform control of GVHD, since initiation of MSC-induced suppression in vitro can provide both an immediate and a consistent effect. The potential of activated MSC might also provide potential savings in both cost and ease in obtaining sufficient numbers of MSC for patients, since high efficacy can be achieved without dose escalation.
One of the strategies cited for control and/or prevention of GVHD has been to induce a shift from Th1 to Th2 cytokines [60]. As IFN-γ is a known stimulant of Th1 cytokine production, a potential harmful side effect from IFN-γ-stimulated MSC treatment might be the release Th1 cytokines, IL-2, GM-CSF, and TNF-α [61]. In our preliminary studies, analysis of day 1 and day 6 supernatants from MSC exposed or not exposed to 500 U/mL IFN-γ (Biosource, 20-plex cytokine detection kit, Invitrogen) showed undetectable amounts of TNF-α and GM-CSF and only modest increases in IL-2 (50–80 pg/mL; manuscript in preparation) in IFN-γ-activated MSC. Untreated MSC had undetectable amounts of all three Th1 cytokines. Interestingly, IFN-γ had no effect on inducing IL-10 production but significantly increased TGF-β (p=0.001; manuscript in preparation). TGF-β has been implicated in T cell suppression by MSC [51]. These early data suggest that in response to IFN-γ, MSC increase suppression and limit Th1 responses, possibly due to their role in dampening destructive pro-inflammatory states in preparation for healing and regeneration [62].
With the incidence of GVHD exceeding 50%, and success in GVHD prevention being limited, new strategies in preventing GVHD and its significant morbidity and mortality are needed. Prevention of GVHD with activated MSC may play a role in broadening the therapeutic potential of allogeneic stem cell transplantation.
Animals
Male BALB/c (H-2Kd) and female C57BL/6 (H-2Kb) mice were purchased from Fredericks NCI (Frederick, MD) or Charles River (Wilmington, MA). Male C.129S7(B6)-Ifngtm1Ts/J (IFN-γ-deficient) mice were purchased from Jackson Laboratories (Bar Harbor, ME). All mice were housed in an AAALAC-accredited animal facility in microisolator cages equipped with autoclaved food and acidified water and were treated under conditions approved by the Animal Care Committee at the University of Illinois at Chicago (UIC).
Conditioning
Recipient mice were age matched (10–12 weeks) for each set of experiments and were exposed to lethal radiation 24 h prior to administration of donor BM and splenocytes. Irradiation was performed at the Department of Radiation Oncology at UIC after recipients were placed in a Lucite retainer for immobilization. The retainer was placed in a water equivalent phantom (box) 30 × 30 × 14.5 cc to ensure dose homogeneity during irradiation. Radiation was delivered via two portals (left and right) using a 6 MV photon beam from a Clinac 2100EX (Varian Medical Systems, Palo Alto, CA) linear accelerator. A total of 1000 cGy at a dose rate of 100 cGy/min was delivered to the prescription point situated in the middle of the box. Across the box the delivered dose was homogeneous within ±10%. The dose delivered to the animal was verified using ThermoLuminiscent Dosemeters (Harshaw/Bicron, Solon, OH) with agreement between measured and expected dose within ±3.0%.
BM, MSC, and splenocyte preparation
Following euthanasia, the BM contents of the femurs and tibia of donor BALB/c mice were flushed through a 40-μm filter (Becton Dickinson, Franklin Lakes, NJ) into a 50-mL tube (Corning, Corning, NY) containing MSC media (40% alpha Modified Eagle Medium (αMEM, Invitrogen, Rockville, MD), 40% F-12 nutrient mixture (Invitrogen), 10% FBS (Valley Biomedical, Winchester, VA), and 1% antibiotic-anti-mycotic solution (Invitrogen). Splenocytes were flushed from spleens and filtered through a 40-μm filter (BD) into a 50-mL tube (Corning). BM cells and splenocytes were counted and resuspended in HBSS (Invitrogen) to the appropriate dose and administered to recipient mice retro orbitally on day 0 (day after irradiation) in a total volume of 200 μL per recipient.
To obtain MSC, BM cells were plated at a density of 20 × 106/9.6 cm2 in MSC media at 37°C in 5% C02 as previously described [63–65]. The nonadherent population was removed after 72 h and the adherent cells were washed with fresh media and cultured for 7 additional days. The resulting adherent cells were harvested by incubating with 0.25% trypsin (Invitrogen) for 6 min at 37°C followed by gentle scraping. Cells were then incubated with biotinylated antibodies to mouse CD11b (10 μg/mL, e-biosciences, San Diego, CA) and CD45 (10 μg/mL, e-biosciences) for 30 min at 4°C. Positive cells were discarded after binding with MACS anti-biotin beads and cohering to a magnetic column (Miltenyi Biotec, Auburn, CA). Negative cells were placed back into culture in Nunclon SoLo 185-cm2 flasks (Nalge Nunc International, Rochester, NY) at a density of 1 × 106 cells/flask. A homogenous cell population was obtained immediately following immunodepletion. The uniform phenotype was confirmed, based on the expression of CD29, CD44, and Sca1, and the absence of hematopoietic (CD45, CD14, CD11b) markers. All antibodies were purchased from ebiosciences. The proportion of CD45+ cells in the MSC preparations used in the various experiments never exceeded 3% CD45+ cells. Prior to transplantation, cells had been passaged one to four times. MSC primed with IFN-γ were plated at a density of 0.116 × 106 per 9.6 cm2 well in 6-well plates. This density is the equivalent to 1 × 106/185-cm2 flask. The 4 mL of MSC media in each well was supplemented with 500 U/mL recombinant murine IFN-γ (PeproTech, Rocky Hill, NJ). On the day of transplantation (day 0), MSC were counted and resuspended at the appropriate dose in 100 μL HBSS per recipient in a 1-mL syringe (BD). MSC were injected retro-orbitally on either day 2, 20 or 30 post irradiation. When co-administered with BM and splenocytes on day 0, the total volume remained 200 μL.
Flow cytometry
MSC were characterized by flow cytometry (Cytomics FC 500, Beckman Coulter, Miami, FL). Briefly, MSC were resuspended at 1 × 106 cells/mL in FACS buffer (PBS (Invitrogen) with 2% FBS (Valley Biomedical, Winchester, VA) and 0.1% sodium azide (Sigma, St. Louis, MO)). Following Fc block (BD Pharmingen, San Jose, CA) at 1 μg/106 cells for 15 min at 4°C, cells were stained with the following PE- or FITC-conjugated antibodies: H2kd (SF1–1,1, BD), I-Ad (AMS-32.1, BD), CD2 (RM2–5, BD), CD3 (17A2, BD), CD4 (GK1.5, BD), CD8a (53–6.7, ebiosciences), CD11b (M1/70, ebiosciences), CD14 (rmC5–3, BD), CD44 (IM7, ebiosciences), CD45 (30-F11, BD), B220 (RA3–6B2, BD), Sca-1 (E13–161.7, BD), c-Kit (2B8, ebiosciences), Thy-1 (30-H12, BD), IFN-γ beta receptor (Abcam, Cambridge, MA), and appropriate isotype controls (ebiosciences or Abcam). Cells were also stained with a primary purified anti-CD29 antibody (BD) at a concentration of 1 μg/106cells, washed with FACS buffer, and then stained with a secondary PE F(ab′)2 fragment donkey anti-rat IgG (Jackson ImmunoResearch, West Grove, PA) at 0.5 μg/106 cells. Flow analysis was performed following the acquisition of 10 000 events. MSC purity was verified within 2 days of transplantation. MSC were stained with FITC-conjugated CD11b and CD45 as above.
GVHD scoring
Mice were weighed twice weekly and monitored daily for survival and clinical evidence of GVHD (ruffled fur, cachexia, alopecia, and diarrhea). Control mice receiving no MSC and recipients of either 105 or 5 × 105 MSC administered on day 2 were killed on day 20 for histological examination. Lung, colon, spleen, and skin were excised, sectioned, stained with hematoxylin and eosin, and examined and scored by two independent pathologists blinded to treatment groups. GVHD was scored on a scale from 0 (none) to 4.0 based on the scales reported by Ferrara (skin) [66], Grass (liver, spleen) [67], and Hill (colon) [68]. The scales for each tissue are defined as follows: for lung, 0=normal; 0.5=minimal perivascular cuffing; 1.0=perivascular cuffing, 1–2 cells in thickness, involving up to 15% of vessels; 1.5=perivascular cuffing, 1–2 cells in thickness, involving up to 15% of vessels and infiltration into parenchyma proper; 2.0=perivascular cuffing, 2–3 cells in thickness, involving up to 15% of vessels and infiltration into parenchyma proper; 2.5=perivascular cuffing, 2–3 cells in thickness, involving 25–50% of vessels and infiltration into parenchyma proper; 3.0=perivascular cuffing, 4–5 cells in thickness, involving 25–50% of vessels, and infiltration into parenchyma proper; 3.5=perivascular cuffing, 6–7 cells in thickness, involving greater than 50% of vessels, peribronchiolar cuffing (4–5 cells), and infiltration into parenchyma proper with severe disruption of structure, 4.0=perivascular cuffing, 6 to 7 cells in thickness, involving greater than 50% of vessels, peribronchiolar cuffing (>6 cells), and infiltration into parenchyma proper with severe disruption of structure; for colon, 0=normal; 0.5=occasional necrotic crypt cell, minimal infiltration in lamina propria and submucosa (colon); 1.0=necrotic cells in up to 15% of crypts, minor infiltration of up to 20% of lamina propria (1–2 cell thickness in intermucosal areas and submucosa; 1.5=necrotic cells in up to 15% of crypts, minor infiltration of less than or equal to one third of the lamina propria (1–2 cell thickness in intermucosal areas and submucosa); 2.0=necrotic cells in ≤25% of crypts, infiltration of less than or equal to one third of the lamina propria (3 cell thickness in intermucosal areas and submucosa); 2.5=necrotic cells in 25–50% of crypts, infiltration of less than or equal to one third of lamina propria (3–4 cell thickness in intermucosal areas and submucosa); 3.0=necrotic cells in greater than 50% of crypts, infiltration of lamina propria (5–6 cell thickness in intermucosal areas and submucosa) with loss of ≤25% of goblet cells; 3.5=necrotic cells in greater than 50% of crypts, infiltration of lamina propria resulting in displacement of ≤50% of mucosa with loss of 50% of goblet cells; 4.0=necrotic cells in greater than 50% of crypts, infiltration of lamina propria resulting in displacement of greater than 50% of mucosa with loss of 75–100% of goblet cells; for spleen, 0=normal; 1.0=necrotic/apoptotic cells, up to 10 cells/mm2 of tissue; 1.5=necrotic/apoptotic cells, up to 10 cells/mm2 of tissue and occasional hemolysis; 2.0=necrotic/apoptotic cells, ≤20 cells/mm2 of tissue, and occasional hemolysis with abnormal architecture; 2.5= necrotic/apoptotic cells, ≤20 cells/mm2 of tissue, and hemolysis in ≤25% of the tissue with abnormal architecture; 3.0=necrotic/apoptotic cells, ≤40 cells/mm2 of tissue, hemolysis in 25–50% of tissue with abnormal architecture and areas of leukopenia involving ≤25% of tissue, formation of fibrous bands; 3.5= necrotic/apoptotic cells, up to 40 cells/mm2 of tissue, hemolysis evident in greater than 50% of tissue with abnormal architecture and areas of leukopenia involving 25–50% of tissue, formation of fibrous bands; 4.0=large areas of necrosis and hemolysis evident in greater than 50% of tissue with abnormal architecture and large areas of leukopenia involving greater than 50% of tissue; for skin, 0=normal; 1.0=basal keratinocyte ballooning; 2.0=sebaceous and adnexal infiltrate; 3.0=loss of epidermis.
Quantification of serum IFN-γ
Twenty B6 recipients underwent irradiation followed BALB/c BM transplant with accessory splenocytes as described above. Three to five recipients underwent serum sampling for each time point, on days 0, 1, 2, 7, 14, 20, and 30. Sera were cryopreserved and batch analyzed using Quantikine Mouse IFN-y Immunoassasy by R&D Systems (Minneapolis, MN; Cat no. MIFOO) according to the manufacturer’s instructions. A Multiskan Ascent (Labsystems) plate reader provided OD read at 450 nm with 540 nm wavelength correction. Data were expressed as means with SD.
Statistical analysis
Treatment group sizes were designed based on an alpha of 0.05 and a power of 0.80. Each experiment was repeated at least three times with a minimum of n=10 per group, unless otherwise stated. Kaplan Meier curves (log rank test) were used to compare survival between treatment groups. ANOVA was used to compare GVHD scoring between groups. In all statistical analyses, a p value of 0.05 was deemed significant.
Acknowledgments
The authors gratefully acknowledge grant support received from NIAID, R01AI67506–01 (A.B.) and the Illinois Regenerative Medicine Institute, Project 5 (A.B.). The authors wish to thank Drs Tapas DasGupta and Ed Cohen for their insightful editorial comments.
Abbreviations
- GVHD
Graft versus host disease
- MSC
mesenchymal stem cell
Footnotes
See accompanying commentary by Dazzi and Marelli-Berg
See accompanying commentary http://dx.doi.org/10.1002/eji.200838433
Conflict of interest: The authors declare no financial of commercial conflict of interest.
References
- 1.European Group for Bone Marrow Transplantation. Allogeneic bone marrow transplantation for leukaemia in Europe. Report from the Working Party on Leukaemia, European Group for Bone Marrow Transplantation. Lancet. 1988;1:1379–1382. [PubMed] [Google Scholar]
- 2.Lucarelli G, Polchi P, Galimberti M, Izzi T, Delfini C, Manna M, Agostinelli F, et al. Marrow transplantation for thalassaemia following busulphan and cyclophosphamide. Lancet. 1985;1:1355–1357. doi: 10.1016/s0140-6736(85)91784-2. [DOI] [PubMed] [Google Scholar]
- 3.Boulad F, Gillio A, Small TN, George D, Prasad V, Torok-Castanza J, Regan AD, et al. Stem cell transplantation for the treatment of Fanconi anaemia using a fludarabine-based cytoreductive regimen and T-cell-depleted related HLA-mismatched peripheral blood stem cell grafts. Br J Haematol. 2000;111:1153–1157. doi: 10.1046/j.1365-2141.2000.02443.x. [DOI] [PubMed] [Google Scholar]
- 4.van Besien K, Bartholomew A, Stock W, Peace D, Devine S, Sher D, Sosman J, et al. Fludarabine-based conditioning for allogeneic transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2000;26:445–449. doi: 10.1038/sj.bmt.1702518. [DOI] [PubMed] [Google Scholar]
- 5.Reisner Y, Kapoor N, Kirkpatrick D, Pollack MS, Cunningham-Rundles S, Dupont B, Hodes MZ, et al. Transplantation for severe combined immunodeficiency with HLA-A,B,D,DR incompatible parental marrow cells fractionated by soybean agglutinin and sheep red blood cells. Blood. 1983;61:341–348. [PubMed] [Google Scholar]
- 6.Wulffraat N, van Royen A, Bierings M, Vossen J, Kuis W. Autologous haemopoietic stem-cell transplantation in four patients with refractory juvenile chronic arthritis. Lancet. 1999;353:550–553. doi: 10.1016/S0140-6736(98)05399-9. [DOI] [PubMed] [Google Scholar]
- 7.Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D, Dupuis-Girod S, et al. Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med. 2001;344:1758–1762. doi: 10.1056/NEJM200106073442304. [DOI] [PubMed] [Google Scholar]
- 8.Friedrich W, Goldmann SF, Vetter U, Fliedner TM, Heymer B, Peter HH, Reisner Y, Kleihauer E. Immunoreconstitution in severe combined immunodeficiency after transplantation of HLA-haploidentical, T-cell-depleted bone marrow. Lancet. 1984;1:761–764. doi: 10.1016/s0140-6736(84)91277-7. [DOI] [PubMed] [Google Scholar]
- 9.Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, Fasth A, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: Report of the European experience 1968–99. Lancet. 2003;361:553–560. doi: 10.1016/s0140-6736(03)12513-5. [DOI] [PubMed] [Google Scholar]
- 10.Spitzer TR, Delmonico F, Tolkoff-Rubin N, McAfee S, Sackstein R, Saidman S, Colby C, et al. Combined histocompatibility leukocyte antigen-matched donor bone marrow and renal transplantation for multiple myeloma with end stage renal disease: The induction of allograft tolerance through mixed lymphohematopoietic chimerism. Transplantation. 1999;68:480–484. doi: 10.1097/00007890-199908270-00006. [DOI] [PubMed] [Google Scholar]
- 11.Kernan NA, Bartsch G, Ash RC, Beatty PG, Champlin R, Filipovich A, Gajewski J, et al. Analysis of 462 transplantations from unrelated donors facilitated by the National Marrow Donor Program. N Engl J Med. 1993;328:593–602. doi: 10.1056/NEJM199303043280901. [DOI] [PubMed] [Google Scholar]
- 12.Powles RL, Morgenstern GR, Kay HE, McElwain TJ, Clink HM, Dady PJ, Barrett A, et al. Mismatched family donors for bone-marrow transplantation as treatment for acute leukaemia. Lancet. 1983;1:612–615. doi: 10.1016/s0140-6736(83)91793-2. [DOI] [PubMed] [Google Scholar]
- 13.Gale RP, Reisner Y. Graft rejection and graft-versus-host disease: Mirror images. Lancet. 1986;1:1468–1470. doi: 10.1016/s0140-6736(86)91503-5. [DOI] [PubMed] [Google Scholar]
- 14.Wagner JE, Thompson JS, Carter SL, Kernan NA. Effect of graft-versus-host disease prophylaxis on 3-year disease-free survival in recipients of unrelated donor bone marrow (T-cell Depletion Trial): A multicentre, randomised Phase II–III trial. Lancet. 2005;366:733–741. doi: 10.1016/S0140-6736(05)66996-6. [DOI] [PubMed] [Google Scholar]
- 15.Lee SJ. New approaches for preventing and treating chronic graft-versus-host disease. Blood. 2005;105:4200–4206. doi: 10.1182/blood-2004-10-4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bacigalupo A. Management of acute graft-versus-host disease. Br J Haematol. 2007;137:87–98. doi: 10.1111/j.1365-2141.2007.06533.x. [DOI] [PubMed] [Google Scholar]
- 17.Bacigalupo A, Palandri F. Management of acute graft versus host disease (GvHD) Hematol J. 2004;5:189–196. doi: 10.1038/sj.thj.6200399. [DOI] [PubMed] [Google Scholar]
- 18.Reddy P, Ferrara JL. Immunobiology of acute graft-versus-host disease. Blood Rev. 2003;17:187–194. doi: 10.1016/s0268-960x(03)00009-2. [DOI] [PubMed] [Google Scholar]
- 19.Ferrara JL, Yanik G. Acute graft versus host disease: Pathophysiology, risk factors, and prevention strategies. Clin Adv Hematol Oncol. 2005;3:415–419. 428. [PubMed] [Google Scholar]
- 20.Andre-Schmutz I, Le Deist F, Hacein-Bey-Abina S, Vitetta E, Schindler J, Chedeville G, Vilmer E, et al. Immune reconstitution without graft-versus-host disease after haemopoietic stem-cell transplantation: A phase 1/2 study. Lancet. 2002;360:130–137. doi: 10.1016/S0140-6736(02)09413-8. [DOI] [PubMed] [Google Scholar]
- 21.Michalek J, Collins RH, Hill BJ, Brenchley JM, Douek DC. Identification and monitoring of graft-versus-host specific T-cell clone in stem cell transplantation. Lancet. 2003;361:1183–1185. doi: 10.1016/S0140-6736(03)12917-0. [DOI] [PubMed] [Google Scholar]
- 22.Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, Uzunel M, Ringden O. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 23.Le Blanc K, Frassoni F, Ball L, Lanino E, Sundberg B, Lonnies L, Roelofs H, et al. Mesenchymal stem cells for treatment of severe acute graft-versus-host disease. Blood (ASH Annual Meeting Abstract) 2006;108:5304a. [Google Scholar]
- 24.Lazarus HM, Koc ON, Devine SM, Curtin P, Maziarz RT, Holland HK, Shpall EJ, et al. Cotransplantation of HLA-identical sibling culture-expanded mesenchymal stem cells and hematopoietic stem cells in hematologic malignancy patients. Biol Blood Marrow Transplant. 2005;11:389–398. doi: 10.1016/j.bbmt.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: Implications in transplantation. Transplantation. 2003;75:389–397. doi: 10.1097/01.TP.0000045055.63901.A9. [DOI] [PubMed] [Google Scholar]
- 26.Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, Grisanti S, Gianni AM. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 27.Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, Hardy W, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30:42–48. doi: 10.1016/s0301-472x(01)00769-x. [DOI] [PubMed] [Google Scholar]
- 28.Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, Dazzi F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–3729. doi: 10.1182/blood-2002-07-2104. [DOI] [PubMed] [Google Scholar]
- 29.Klyushnenkova EN, Shustova V, Mosca JD, Buyaner D, Hendricks JK, Moseley AB, McIntosh KR. Human mesenchymal stem cell-mediated suppression of allogeneic T cell responses: A cytokine analysis. FASEB J. 1999;13:A615. [Google Scholar]
- 30.Le Blanc K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy. 2003;5:485–489. doi: 10.1080/14653240310003611. [DOI] [PubMed] [Google Scholar]
- 31.Potian JA, Aviv H, Ponzio NM, Harrison JS, Rameshwar P. Veto-like activity of mesenchymal stem cells: Functional discrimination between cellular responses to alloantigens and recall antigens. J Immunol. 2003;171:3426–3434. doi: 10.4049/jimmunol.171.7.3426. [DOI] [PubMed] [Google Scholar]
- 32.Chung NG, Jeong DC, Park SJ, Choi BO, Cho B, Kim HK, Chun CS, et al. Cotransplantation of marrow stromal cells may prevent lethal graft-versus-host disease in major histocompatibility complex mismatched murine hematopoietic stem cell transplantation. Int J Hematol. 2004;80:370–376. doi: 10.1532/ijh97.a30409. [DOI] [PubMed] [Google Scholar]
- 33.Sudres M, Norol F, Trenado A, Gregoire S, Charlotte F, Levacher B, Lataillade JJ, et al. Bone marrow mesenchymal stem cells suppress lymphocyte proliferation in vitro but fail to prevent graft-versus-host disease in mice. J Immunol. 2006;176:7761–7767. doi: 10.4049/jimmunol.176.12.7761. [DOI] [PubMed] [Google Scholar]
- 34.Tisato V, Naresh K, Girdlestone J, Navarrete C, Dazzi F. Mesenchymal stem cells of cord blood origin are effective at preventing but not treating graft-versus-host disease. Leukemia. 2007;21:1992–1999. doi: 10.1038/sj.leu.2404847. [DOI] [PubMed] [Google Scholar]
- 35.Min CK, Kim BG, Park G, Cho B, Oh IH. IL-10-transduced bone marrow mesenchymal stem cells can attenuate the severity of acute graft-versus-host disease after experimental allogeneic stem cell transplantation. Bone Marrow Transplant. 2007;39:637–645. doi: 10.1038/sj.bmt.1705644. [DOI] [PubMed] [Google Scholar]
- 36.Friedenstein A, Chailakhjan R, Lalykina K. Heterotopic Transplants of Bone Marrow: Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6:230–247. [PubMed] [Google Scholar]
- 37.Baddoo M, Hill K, Wilkinson R, Gaupp D, Hughes C, Kopen GC, Phinney DG. Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection. J Cell Biochem. 2003;89:1235–1249. doi: 10.1002/jcb.10594. [DOI] [PubMed] [Google Scholar]
- 38.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 39.Moadsiri A, Polchert D, Genrich K, Napoles P, Reina E, Turian J, Smith B, Bartholomew A. Mesenchymal stem cells enhance xenochimerism in NK-depleted hosts. Surgery. 2006;140:315–321. doi: 10.1016/j.surg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Andre-Schmutz I, Dal Cortivo L, Fischer A, Cavazzana-Calvo M. Improving immune reconstitution while preventing GvHD in allogeneic stem cell transplantation. Cytotherapy. 2005;7:102–108. doi: 10.1080/14653240510027118. [DOI] [PubMed] [Google Scholar]
- 41.Blazar BR, Lees CJ, Martin PJ, Noelle RJ, Kwon B, Murphy W, Taylor PA. Host T cells resist graft-versus-host disease mediated by donor leukocyte infusions. J Immunol. 2000;165:4901–4909. doi: 10.4049/jimmunol.165.9.4901. [DOI] [PubMed] [Google Scholar]
- 42.Hirayama M, Azuma E, Kumamoto T, Iwamoto S, Yamada H, Nashida Y, Araki M, et al. Prediction of acute graft-versus-host disease and detection of distinct end-organ targets by enumeration of peripheral blood cytokine spot-forming cells. Transplantation. 2005;80:58–65. doi: 10.1097/01.tp.0000163431.57482.8c. [DOI] [PubMed] [Google Scholar]
- 43.Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–4621. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- 44.Ryan JM, Barry F, Murphy JM, Mahon BP. Interferon-gamma does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin Exp Immunol. 2007;149:353–363. doi: 10.1111/j.1365-2249.2007.03422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato K, Yamashita N, Yamashita N, Baba M, Matsuyama T. Regulatory dendritic cells protect mice from murine acute graft-versus-host disease and leukemia relapse. Immunity. 2003;18:367–379. doi: 10.1016/s1074-7613(03)00055-4. [DOI] [PubMed] [Google Scholar]
- 46.Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890–896. doi: 10.1016/s0301-472x(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 47.Ringden O, Uzunel M, Rasmusson I, Remberger M, Sundberg B, Lonnies H, Marschall HU, et al. Mesenchymal stem cells for treatment of therapy-resistant graft-versus-host disease. Transplantation. 2006;81:1390–1397. doi: 10.1097/01.tp.0000214462.63943.14. [DOI] [PubMed] [Google Scholar]
- 48.Xun CQ, Thompson JS, Jennings CD, Brown SA, Widmer MB. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood. 1994;83:2360–2367. [PubMed] [Google Scholar]
- 49.English K, Barry FP, Field-Corbett CP, Mahon BP. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol Lett. 2007;110:91–100. doi: 10.1016/j.imlet.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Chabannes D, Hill M, Merieau E, Rossignol J, Brion R, Soulillou JP, Anegon I, Cuturi MC. A role for heme oxygenase-1 in the immunosuppressive effect of adult rat and human mesenchymal stem cells. Blood. 2007;110:3691–3694. doi: 10.1182/blood-2007-02-075481. [DOI] [PubMed] [Google Scholar]
- 51.Nasef A, Chapel A, Mazurier C, Bouchet S, Lopez M, Mathieu N, Sensebe L, et al. Identification of IL-10 and TGF-beta transcripts involved in the inhibition of T-lymphocyte proliferation during cell contact with human mesenchymal stem cells. Gene Expr. 2007;13:217–226. doi: 10.3727/000000006780666957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.New JY, Li B, Koh WP, Ng HK, Tan SY, Yap EH, Chan SH, Hu HZ. Tcell infiltration and chemokine expression: Relevance to the disease localization in murine graft-versus-host disease. Bone Marrow Transplant. 2002;29:979–986. doi: 10.1038/sj.bmt.1703563. [DOI] [PubMed] [Google Scholar]
- 53.Wang L, Li Y, Chen X, Chen J, Gautam SC, Xu Y, Chopp M. MCP-1, MIP-1, IL-8 and ischemic cerebral tissue enhance human bone marrow stromal cell migration in interface culture. Hematology. 2002;7:113–117. doi: 10.1080/10245330290028588. [DOI] [PubMed] [Google Scholar]
- 54.Croitoru-Lamoury J, Lamoury FM, Zaunders JJ, Veas LA, Brew BJ. Human mesenchymal stem cells constitutively express chemokines and chemokine receptors that can be upregulated by cytokines, IFN-beta, and Copaxone. J Interferon Cytokine Res. 2007;27:53–64. doi: 10.1089/jir.2006.0037. [DOI] [PubMed] [Google Scholar]
- 55.Zhang W, Ge W, Li C, You S, Liao L, Han Q, Deng W, Zhao RC. Effects of mesenchymal stem cells on differentiation, maturation, and function of human monocyte-derived dendritic cells. Stem Cells Dev. 2004;13:263–271. doi: 10.1089/154732804323099190. [DOI] [PubMed] [Google Scholar]
- 56.Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, Galun E, Rachmilewitz J. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105:2214–2219. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- 57.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 58.Gur-Wahnon D, Borovsky Z, Beyth S, Liebergall M, Rachmilewitz J. Contact-dependent induction of regulatory antigen-presenting cells by human mesenchymal stem cells is mediated via STAT3 signaling. Exp Hematol. 2007;35:426–433. doi: 10.1016/j.exphem.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 59.Szebeni J, Wang MG, Pearson DA, Szot GL, Sykes M. IL-2 inhibits early increases in serum gamma interferon levels associated with graft-versus-host-disease. Transplantation. 1994;58:1385–1393. [PubMed] [Google Scholar]
- 60.Blazar BR, Korngold R, Vallera DA. Recent advances in graft-versus-host disease (GVHD) prevention. Immunol Rev. 1997;157:79–109. doi: 10.1111/j.1600-065x.1997.tb00976.x. [DOI] [PubMed] [Google Scholar]
- 61.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 62.Phinney DG, Prockop DJ. Concise review: Mesenchymal stem/multipotent stromal cells: The state of transdifferentiation and modes of tissue repair–Current views. Stem Cells. 2007;25:2896–2902. doi: 10.1634/stemcells.2007-0637. [DOI] [PubMed] [Google Scholar]
- 63.Friedenstein A, Chailakhjan R, Lalykina K. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970;3:393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 64.Witte PL, Robinson M, Henley A, Low MG, Stiers DL, Perkins S, Fleischman RA, Kincade PW. Relationships between B-lineage lymphocytes and stromal cells in long- term bone marrow cultures. Eur J Immunol. 1987;17:1473–1484. doi: 10.1002/eji.1830171014. [DOI] [PubMed] [Google Scholar]
- 65.Kopen GC, Prockop DJ, Phinney DG. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc Natl Acad Sci USA. 1999;96:10711–10716. doi: 10.1073/pnas.96.19.10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferrara J, Guillen FJ, Sleckman B, Burakoff SJ, Murphy GF. Cutaneous acute graft-versus-host disease to minor histocompatibility antigens in a murine model: Histologic analysis and correlation to clinical disease. J Invest Dermatol. 1986;86:371–375. doi: 10.1111/1523-1747.ep12285612. [DOI] [PubMed] [Google Scholar]
- 67.Grass JA, Wafa T, Reames A, Wages D, Corash L, Ferrara JL, Lin L. Prevention of transfusion-associated graft-versus-host disease by photochemical treatment. Blood. 1999;93:3140–3147. [PubMed] [Google Scholar]
- 68.Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: The role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90:3204–3213. [PubMed] [Google Scholar]