

Abstract
Vascular endothelial growth factor-A (VEGF) typically induces abnormal angiogenesis in the adult, thereby aggravating disease pathology and limiting utility of VEGF for therapeutic angiogenesis. To identify strategies for rectifying defects in pathological VEGF neovessels, we investigated consequences of modulating the Rho GTPase Cdc42. In a mouse skin model of VEGF-driven pathological angiogenesis, transduction with active Cdc42 (L28Cdc42) markedly improved VEGF neovessels, as measured by increased lumen formation, enlarged vessel diameter, and enhanced perfusion of macromolecular tracers. Conversely, transduction with dominant-negative Cdc42 (N17Cdc42) impaired endothelial cell (EC) assembly into lumenized blood vessels and reduced neovessel diameter and tracer perfusion. In vitro, active Cdc42 improved coordination between actin filaments and microtubules and enhanced formation of vascular cords, suggesting that active Cdc42 rectifies defects in angiogenesis by improving cytoskeletal dynamics and capillary morphogenesis. Analyses of Cdc42 signaling in microvascular ECs indicated that active Cdc42 also inhibits glycogen synthase kinase-3β (GSK-3β), a multi-functional serine/threonine protein kinase. Pharmacological inhibition of GSK-3β improved vascular cord formation in vitro and promoted proper neovessel formation in vivo comparably to active Cdc42, thus linking GSK-3β inhibition to the mechanism by which active Cdc42 rectifies pathological neovascularization. These studies identify activation of Cdc42 and inhibition of GSK-3β as novel strategies for correcting abnormalities associated with VEGF-driven angiogenesis, and they suggest new approaches for achieving improved therapeutic neovascularization with VEGF.
Keywords: Vascular endothelial growth factor (VEGF), pathological angiogenesis, lumens, endothelial cells, cytoskeleton, Cdc42, glycogen synthase kinase-3β (GSK-3β), capillary morphogenesis
Introduction
In contrast to the hierarchical vascular network formed during normal angiogenesis, pathological angiogenesis is characterized by abnormal angio-architecture and vessel leakiness, resulting in poor vascular perfusion (Fukumura and Jain, 2007; Jain, 2003; Nagy et al., 2009; Nagy and Senger, 2006; Pierce et al., 1995). These vascular abnormalities are likely attributable to comparatively high expression of VEGF in pathological settings (Ozawa et al., 2004) and an imbalance between VEGF and other important factors (Jain, 2003), including angiopoietin-1 (Chae et al., 2000). Still, the specific mechanisms responsible for the aberrant neovascular architecture that distinguishes pathological angiogenesis remain largely unexplored.
In this study, we designed experiments to investigate the consequences of modulating Cdc42 (Nobes and Hall, 1999) for pathological angiogenesis. We viewed this key member of the Rho family of small GTPases (Nobes and Hall, 1999) as an attractive candidate for such investigations because it regulates cytoskeletal dynamics, cell shape, and numerous other cellular processes including polarity, migration, cell cycle progression and cell fate determination (Cau and Hall, 2005; Etienne-Manneville, 2004b; Heasman and Ridley, 2008; Schlessinger et al., 2007; Wedlich-Soldner et al., 2003). Moreover, these critical functions specifically suggest an important role for Cdc42 in neovascularization and vascular biology. Indeed, siRNA-mediated suppression of Cdc42 in vitro blocks lumen formation by endothelial cells (ECs) in vitro (Koh et al., 2008). Cdc42 also regulates the EC cytoskeleton in response to flow-mediated shear stress (Tzima et al., 2003) and restores EC barrier function following disruption by thrombin (Kouklis et al., 2004). Nevertheless, possible relationships between Cdc42 activity and pathological angiogenesis in vivo had not been examined. Therefore, we designed experiments with a model of VEGF-driven angiogenesis that recapitulates pathological neovascularization. Our findings illustrate that active Cdc42 markedly improves the angio-architecture and lumenization of neovessels induced by VEGF. In addition, these studies identify inhibition of glycogen synthase kinase-3β (GSK-3β) as a key mechanism by which active Cdc42 improves the organization of endothelial cells (ECs) into new blood vessels and thereby enhances overall angio-architecture. Finally, we illustrate a pharmacological strategy involving a GSK-3β inhibitor that rectifies defects in pathological VEGF neovessels similarly to active Cdc42.
Materials and methods
Reagents
Purified recombinant human VEGF165, expressed in Sf21 cells, was obtained from the NCI Preclinical Repository, Biological Resources Branch, Frederick, MD. GSK-3β and serine-9-phosphorylated GSK-3β antibodies were from Cell Signaling Technology Inc. (Danvers, MA); antibodies to α–tubulin (Clone DM 1A) and acetylated α–tubulin were from Sigma-Aldrich (St. Louis, Missouri). ECL Western Blotting Substrate kit was from Pierce (Rockford, IL). Texas-Red secondary antibody for microtubule staining was from Jackson Immunoresearch (West Grove, PA). CD31 (PECAM-1) antibody was from BD Biosciences Pharmingen (San Diego, CA). DAB substrate Kit was from Zymed Laboratory Inc. (Carlsbad, CA). Matrigel and collagen-1 were from BD Biosciences (Bedford, MA). Oregon Green-conjugated phalloidin was from Invitrogen. GSK-3β inhibitor-I (TDZD-8) was purchased from Calbiochem (San Diego, CA). Lysine-fixable Texas-red dextran (MW 70 kD) for perfusion studies was from Invitrogen (Carlsbad, CA).
Preparation of packaging cells expressing retroviruses encoding Cdc42 mutants
Human fast cycling constitutive active L28Cdc42 cDNA (Lin et al., 1999; Tu et al., 2002) was a generous gift from Richard A. Cerione (Cornell University, Ithaca, NY); DN N17Cdc42 cDNA was generated from wild-type human Cdc42 by PCR. The Cdc42 mutant cDNAs were subcloned into BamHI sites in retrovirus vector pLCNX2/IRES-EGFP (Hoang and Senger, 2005). This vector expresses GFP as a separate protein through an independent ribosome entry site (IRES). The fidelity of both clones was verified by sequencing. PT67 retroviral packaging cells (Clontech, Palo Alto, CA), which express the 10A1 viral envelope for production of amphotropic virus, were transfected with pLCNX2/IRES-EGFP vector containing DN N17Cdc42, active L28Cdc42 or vector without insert (empty vector). Transfectants were cloned, and clones expressing retrovirus at 1×105 c.f.u./ml were selected for subsequent experiments.
VEGF-driven angiogenesis in mouse skin, retroviral transduction and drug administration in vivo, and analyses of vascular parameters
Neovascularization was assayed in vivo according to a previously established method that includes both VEGF165-transfected cells and retroviral packaging cells, thereby providing a constant source of VEGF and retrovirus (Hoang and Senger, 2005; Hoang et al., 2004). Seven week old female athymic nude mice were injected subcutaneously on right and left flanks with 0.3 ml of 9 mg/ml Matrigel (BD Biosciences) containing 1×106 SK-MEL2 human melanoma cells stably transfected for CMV promoter-driven expression of human VEGF165 (210 ng VEGF per 1×106 cells per 24 h) together with 1×106 of retroviral packaging cells, as indicated. Untransfected parental SK-MEL2 cells do not provoke angiogenesis detectably, and therefore the VEGF-SK-MEL2 transfectants employed here allow for specific investigation of VEGF-driven angiogenesis (Senger et al., 1997). At times indicated, the animals were euthanized, dissected, and photographed. In addition, to analyze perfusion of new blood vessels, representative animals from each group received tail vein injections of 0.2 ml of 0.5% (w/v) Evan’s Blue dye or 70 kD Texas Red dextran (25 mg/ml) in sterile saline. For whole mount immunofluorescence analysis of tracer-filled vasculature, skin samples were fixed in 4% paraformaldehyde for 4 hr, mounted in immersion oil and viewed with a Bio-Rad MRC-1024 Confocal Microscope equipped with an Argon-Krypton Laser. Four-six fields per sample were visualized using x10 objective. For histology, implants together with associated skin were fixed for 1 hour in 10% buffered formalin and embedded in paraffin. Immunohistochemical staining of ECs with CD31 antibody was performed as described (Hoang and Senger, 2005). Total neovascular density and lumen area were traced through freehand selections on digital images and measured with NIH ImageJ software. The numbers of individual neovessels and numbers of ECs per unit area were measured through freehand point selections and internal vessel diameters were measured with line selections using NIH ImageJ software.
Vascular architecture was also analyzed with Microfil perfusion. The entire vascular tree of the mouse was filled with Microfil MV-122 (Flow Tech; Carver, MA) (Nagy et al., 1995). The Microfil was allowed to polymerize for 24 h at 4°C. Specimens of flank skin were cleared by dehydration in a graded series of glycerin solutions and photographed with a Wild M400 stereo-photomicroscope and a SPOT Insight digital camera.
Finally, for experiments with GSK-3β inhibitor-I (TDZD-8), angiogenesis assays were performed as above but without retroviral packaging cells. GSK-3β inhibitor-I was administered twice daily s.c. at 1 mg/kg in saline unless indicated otherwise.
MVEC isolation, cell culture, and retroviral transduction of microvascular ECs
Human dermal microvascular ECs were isolated from neonatal foreskins (Richard et al., 1998) and cultured (Hoang and Senger, 2005) in the continuous presence of 20 ng/ml VEGF165. All experiments were performed with cells at the fourth to seventh passage. MVECs at passage 5 or less were transduced with retroviruses according to a previously established, efficient method (Le Doux et al., 2001). The transduction procedure was repeated three times on consecutive days before subjecting cells to selection with 300 micrograms/ml G418. This method yields 100% transduction as indicated with GFP vectors (Hoang and Senger, 2005; Hoang et al., 2004). Cells were used within one week for experiments.
In vitro capillary morphogenesis assays with MVECs and cytoskeletal analyses
Capillary morphogenesis assays were performed as indicated by either “overlaying” or “sandwiching” confluent cell monolayers with rat tail collagen-I (BD Biosciences (Hoang and Senger, 2005; Hoang et al., 2004). The sandwich-type assay was performed in 12-well plates with 1.0 mg/ml collagen-I in full medium. VEGF (20 ng/ml) was included in all assays. Where indicated, GSK-3β inhibitor-1 was added for overnight incubation prior to adding the upper layer of collagen-I that also contained GSK-3β inhibitor-1. Capillary morphogenesis was allowed to proceed for 16h; the assay plates were fixed with 10% formalin for one hour and stained for F-actin with fluorescent Oregon Green-conjugated phalloidin (Invitrogen, final concentration 0.5 units/ml) and/or tubulin antibodies and subsequently photographed. For the “overlay” assay, GSK-3β inhibitor-1 was added and incubated with cells in 24-well plates overnight. The next day, each well was overlaid with 300 microliters of collagen-I at a concentration of 0.5 mg/ml in serum-free medium containing VEGF (20 ng/ml) together with inhibitor, as indicated. Capillary morphogenesis was allowed to proceed for 4h; cells were fixed in 10% formalin for 10 minutes, permeabilized for one minute with 0.02% Triton-X100 in PBS and stained for F-actin (as above) and/or tubulin followed by fluorescent secondary antibody. Cells were photographed with a Nikon inverted fluorescent microscope and digital camera. In all cases, cord length, blind ends, and polygons were quantified using NIH ImageJ software. Cord length was traced and measured through freehand line selections. Blind ends were determined with point selections. Measured parameters correspond to actual areas of 0.4 mm2.
Immunoblotting analyses of GSK-β, Cdc42 activity, and VEGF expression GSK-β Ser9 phosphorylation
MVECs transduced with active Cdc42, DN Cdc42, or empty vector were grown to confluence in full medium and switched to medium containing 2% serum for 12 hours. ECs were harvested in lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 10% Glycerol, 1% Nonidet P-40, 3 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 25 mM NaF, 5 mM Na3VO4, 150 μM sodium pyrophosphate, and a cocktail of protease inhibitors containing 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), pepstatin A, E-64, bestatin, leupeptin, and aprotinin. Lysates (20 μg protein) were subjected to electrophoresis with SDS PAGE on 4–20% gradient gel. Gels were electrophoretically transferred to PVDF Membrane (BIO-RAD) and stained with GSK-β Ser9 phospho-specific antibody (Cell Signaling, cat. #9336). Protein bands were detected with ECL Western blotting substrate. Next, the membrane was stripped and re-stained with total GSK-3β antibody (Cell Signaling, cat. #9315). Cdc42 activity: Active GTP-Cdc42 (1×107 ECs/sample) was measured with a Pak1/Cdc42 “pull-down” assay from Upstate Cell Signaling Solutions (Temecula, CA), followed by immuno-blotting. Active Cdc42 was quantified with a digital scanner. VEGF expression: To assay for any effects of retroviral transduction or GSK-3β inhbitor-1 on VEGF expression, VEGF165-SK-MEL2 cells were co-cultured with the various Cdc42 retroviral packaging cells (described above) in the same proportions used in vivo, or treated continuously with GSK-3β inhbitor-1 at the same dose used to improve cord formation (3μM). Medium was harvested daily for 8 days, and VEGF165 was concentrated with heparin-Sepharose chromatography (Senger et al., 1990) followed by electrophoresis and immunoblotting with VEGF-specific antibody (Sioussat et al., 1993) and quantification with a digital scanner.
Statistical analyses
All findings are presented as mean ± S.E.M. Statistical analyses were performed with InStat 3 software for Macintosh, employing the two-tail Mann-Whitney test and assuming unequal variances between the two groups under comparison. In all cases, a single experimental group was compared with the corresponding control group; and calculated p-values are based on direct comparisons between the two groups.
Results
Active Cdc42 improves neovessel architecture and lumen formation
We employed a mouse skin model of VEGF-driven pathological neovascularization that utilizes a constant source of VEGF together with packaging cells expressing retroviruses encoding different Cdc42 mutants. This model offers the advantage of high retroviral transduction efficiency because transduction is favored in proliferating cells, and endothelial cells (ECs) actively divide in response to continuous VEGF-stimulation (Hoang and Senger, 2005; Hoang et al., 2004). Moreover, the inclusion of packaging cells provides a constant source of freshly produced retrovirus throughout the entire experiment. Previously, we validated the efficiency of this model with packaging cells expressing retrovirus encoding GFP (Hoang and Senger, 2005), RhoA mutants (Hoang et al., 2004), and the transcription factor Nur77 (Zeng et al., 2006).
To initiate angiogenesis, transfected cells engineered for continuous expression of VEGF165 under the direction of a constitutively active cytomegalovirus immediate-early gene (CMV) promoter, were mixed with equal numbers of retroviral packaging cells in basement membrane Matrigel and injected sub-dermally. The retroviral packaging cells expressed retrovirus encoding either dominant negative Cdc42 (N17Cdc42) (Nobes and Hall, 1999), the “fast-cycling” active Cdc42 mutant (L28Cdc42) (Lin et al., 1999; Tu et al., 2002), or no insert (empty vector). Normally, Cdc42 is activated by guanine nucleotide exchange factors in response to cytokine stimulation and integrin-mediated cell adhesion (Price et al., 1998; Puls et al., 1999). Following activation, Cdc42 is inactivated by GTPase-activating proteins and thereby cycles between active GTP-bound and inactive GDP-bound configurations (Heasman and Ridley, 2008). The active fast-cycling mutant L28Cdc42 employed here possesses an enhanced intrinsic GTP/GDP exchange rate but maintains normal GTP hydrolytic activity. Thus, this fast-cycling form of Cdc42 is activated spontaneously but is also subject to normal inactivation and therefore closely reflects normal regulation (Lin et al., 1999; Tu et al., 2002).
Animals were analyzed on day 7 following induction of angiogenesis. As illustrated by intravital perfusion with Evans blue dye, which binds to serum albumin (Fig. 1A), and independently by perfusion with high molecular weight Texas Red-dextran (Fig. 1B), neovascularization of the over-lying dermis was extensive and well perfused in the active Cdc42 group, intermediate in the control empty vector group, and sharply inhibited in the dominant negative (DN) Cdc42 group. Moreover, as illustrated by tracer perfusion (Fig. 1A, B), and by vascular casting with Microfil (Fig. 1B), new blood vessels in the active Cdc42 group were larger and better defined than in the empty vector controls. Quantification from gross images indicated that active Cdc42 increased functional (i.e. perfusable) neovasculature by nearly 40% relative to empty vector controls; conversely, DN Cdc42 sharply decreased functional neovascularization by >80% (Fig. 1C). Also, as shown with CD31-staining of ECs in cross-section (Fig. 1A), neovessels with well-defined lumens were most abundant in the active Cdc42 group (red arrowheads) but nearly absent in the DN Cdc42 group. Active Cdc42 increased cumulative lumen area in cross-section >50% relative to empty vector controls, and this correlated with the increase in average neovessel diameter (Fig. 1C). Conversely DN Cdc42 decreased cumulative lumen area >70% and this correlated with reduction in average neovessel diameter (Fig. 1C). These findings are particularly intriguing given that both EC density and neovessel density in the DN Cdc42 group were slightly greater than for the empty vector control group (Fig. 1C). We interpret these results as indicating that the negative consequences associated with DN Cdc42 involve reductions in neovessel diameter and lumen formation rather than alterations in EC or neovessel density.
Figure 1. Active Cdc42 improves neovessel architecture and lumen formation in vivo.

(A) VEGF165-transfectants were mixed with packaging cells expressing retrovirus encoding active L28Cdc42, DN N17Cdc42, or empty vector (control), and injected together with Matrigel sub-dermally. After 7d, animals were injected with the indicated tracer (10 min) and harvested. Evans blue dye: Gross images of dermis overlying the Matrigel implants show that active L28Cdc42 improved formation of perfused blood vessels relative to control, whereas DN Cdc42 was inhibitory (Bar = 500 microns). CD31 Stain: ECs in cross section stained with CD31 antibody (brown color) illustrating that active Cdc42 improved lumen formation (arrows) relative to control, whereas DN Cdc42 abolished lumen formation. Bar = 40 microns. (B) Tx-Red dextran: Perfusion of vessels with 70kD, lysine-fixable, Texas-Red dextran and viewed with confocal microscopy (Bar = 200 microns) confirming that active L28Cdc42 promoted formation of perfused blood vessels. Microfil Vascular Cast: the entire vasculature was perfused with Microfil, illustrating improvement in neovessel diameter and architecture mediated by active Cdc42 (Bar = 375 microns). (C) Quantification of vascular parameters; n≥20 for all groups. From gross images: relative neovascularization (relative area of gross images occupied by perfused neovessels, p<0.01). From CD31 stained cross-sections: quantification of ECs per 0.01mm2, relative total lumen area (p<0.01), numbers of neovessels per 0.01mm2 (active Cdc42 vs. empty vector p=0.02), and average internal neovessel diameter (p<0.03). Parameters are shown also for control skin, i.e. without VEGF stimulation and therefore without neovascularization (clear bars).
Consistent with our observations that active Cdc42 and DN Cdc42 did not alter EC density significantly relative to empty vector control, co-culture of the various retroviral packaging cells with the VEGF-transfected cells, in the same proportions used in vivo, did not effect VEGF production (see Methods). This finding was not surprising, because VEGF expression was directed constitutively by a CMV promoter. Thus, the marked differences in neovessel architecture among the different experimental groups are best explained by differences in capillary morphogenesis and lumenization rather than by changes in VEGF expression.
Active Cdc42 coordinates microtubules and actin filaments and improves capillary morphogenesis in vitro
To identify cellular mechanisms through which active Cdc42 improves VEGF-driven angiogenesis, we transduced dermal MVECs with the same active Cdc42 and DN Cdc42 mutants employed in vivo. MVECs transduced with empty vector served as controls. MVECs transduced with active L28Cdc42 exhibited ~ 6x the Cdc42 activity of controls, and MVECs transduced with DN N17 Cdc42 exhibited a ~80% reduction in activity (Fig. 2A). Subsequently, these cells were overlaid with three-dimensional collagen-I matrix, thereby inducing formation of vascular cords (Davis and Camarillo, 1996; Whelan and Senger, 2003), corresponding to the pre-capillary cords observed in vivo prior to the appearance of mature tubes with lumens (Drake and Fleming, 2000; Vernon and Sage, 1995). During cord formation, MVECs were stained for microtubules (Fig. 2B, top panels – tubulin red color) and F-actin (Fig. 2B, middle panels – actin green color) to view cytoskeletal re-organization. Merged images of the microtubule and actin cytoskeletons illustrated striking differences among the different cell populations (Fig. 2B, bottom panels). In MVECs transduced with active Cdc42, microtubules (red color) and F-actin (green color) exhibited a well-organized and coordinated distribution consistent with cooperative organization and function. In particular, active Cdc42 organized actin filaments at the cell periphery in a cortical pattern that fully circumscribed the microtubules. In marked contrast, MVECs transduced with control vector showed comparatively poor coordination between F-actin and microtubules; and MVECs transduced with DNCdc42 exhibited a poorly developed actin cytoskeleton that, in many cases, failed to circumscribe or balance microtubules. In addition, microtubule staining of MVECs in monolayer culture showed that active Cdc42 enhanced microtubule polymerization whereas DN Cdc42 had the opposite effect (Supplementary Fig. 1A). Furthermore, staining of vascular cords for acetylated tubulin, an indicator of microtubule stabilization (Piperno et al., 1987), indicated that active Cdc42 enhanced microtubule stabilization whereas DN Cdc42 reduced microtubule stability (Supplementary Fig. 1B).
Figure 2. Active Cdc42 coordinates actin filaments and microtubules during capillary morphogenesis.

(A) MVECs transduced with active L28Cdc42 exhibited ~ 6x increase in Cdc42 activity (GTP-bound form) relative to controls, whereas MVECs transduced with DN Cdc42 exhibited ~80% reduction. (B) MVECs transduced with Cdc42 mutants and cultured in the presence of VEGF were overlaid with three-dimensional collagen-I matrix, thereby inducing reorganization into vascular cords; 4h later MVECs were stained for microtubules (red color) and F-actin (green color). Merged images of microtubule and actin cytoskeletons (bottom panels) illustrated that active Cdc42 organized actin filaments at the cell periphery in a cortical pattern that fully circumscribed the microtubules, consistent with coordinated balance between these two cytoskeletal elements. In marked contrast, MVECs transduced with control vector showed comparatively poor coordination between F-actin and microtubules. MVECs transduced with DN Cdc42 exhibited a poorly developed actin cytoskeleton and failed to re-organize.
Next, the same transduced MVEC populations were sandwiched between two layers of collagen I to induce formation of vascular cords. In this model system, MVECs are completely surrounded by flexible matrix, thereby providing a uniformly malleable environment permissive for capillary morphogenesis. Consistent with the cytoskeletal analyses described above, active L28Cdc42 enhanced the formation of vascular cords relative to controls, as measured by increased cord length and improved integration, i.e. reduction in blind ends (Fig. 3A, B). In contrast, DN Cdc42 significantly reduced cord length and increased the number of blind ends (Fig. 3A, B). Neither active Cdc42 nor DN Cdc42 significantly altered MVEC density relative to that of empty vector control (Fig. 3B) indicating that alterations in MVEC morphogenesis were solely responsible for the observed changes in vascular network formation. Thus, to summarize, active Cdc42 promotes coordinated dynamics between actin and microtubules and also promotes the formation of integrated vascular cord networks in vitro; and these findings are consistent with our observations that active Cdc42 improves VEGF-driven angiogenesis in vivo.
Figure 3. Active Cdc42 improves and DN Cdc42 disrupts formation of vascular cords in vitro.
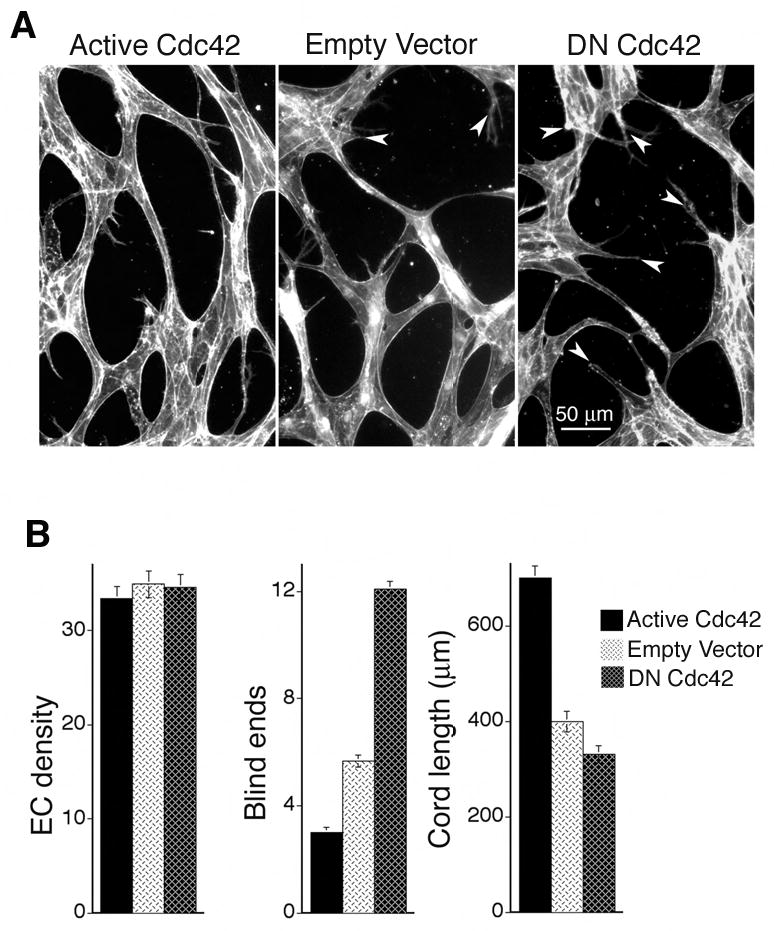
(A) Dermal MVECs, transduced with Cdc42 mutants or empty vector control were induced to undergo capillary morphogenesis by “sandwiching” between two layers of collagen I in the presence of VEGF and next stained for F-actin. Note presence of blind ends (arrows) in control and DN N17Cdc42 specimens that are absent in the active L28Cdc42 specimen. (B) Quantification of cord parameters; n ≥ 25 for all groups. Active Cdc42 significantly reduced blind ends (p < 0.001) and increased cord length (p < 0.001), relative to controls whereas DN N17Cdc42 increased blind ends (p < 0.001) and reduced cord length (p < 0.01).
Active Cdc42 improves capillary morphogenesis by inhibiting GSK-3β
Next, we sought to identify downstream molecular targets through which active Cdc42 improves capillary morphogenesis. In particular, we considered GSK-3β because Cdc42 signaling has been linked previously to inhibition of GSK-3β (Etienne-Manneville and Hall, 2003) and also because inhibition of GSK-3β is associated with improved microtubule dynamics (reviewed (Zhou and Snider, 2005)). Transduction of MVECs with active Cdc42 resulted in robust inhibition of GSK-3β as indicated by increased phosphorylation of GSK-3β at the Ser9 residue (Fig. 4A). Scanning densitometry indicated that active L28Cdc42 increased phosphorylation of GSK-3β Ser9 > 10-fold relative to controls. Conversely, transduction of MVECs with DN N17Cdc42 reduced GSK-3β Ser9 phosphorylation ~ 50% (Fig. 4A). The marked inhibition of GSK-3β by active Cdc42, as indicated by the large increase in GSK-3β Ser9 phosphorylation, suggested inhibition of GSK-3β as a potentially important mechanism by which active Cdc42 facilitates capillary morphogenesis.
Figure 4. Active Cdc42 improves capillary morphogenesis by inhibiting GSK-3β.
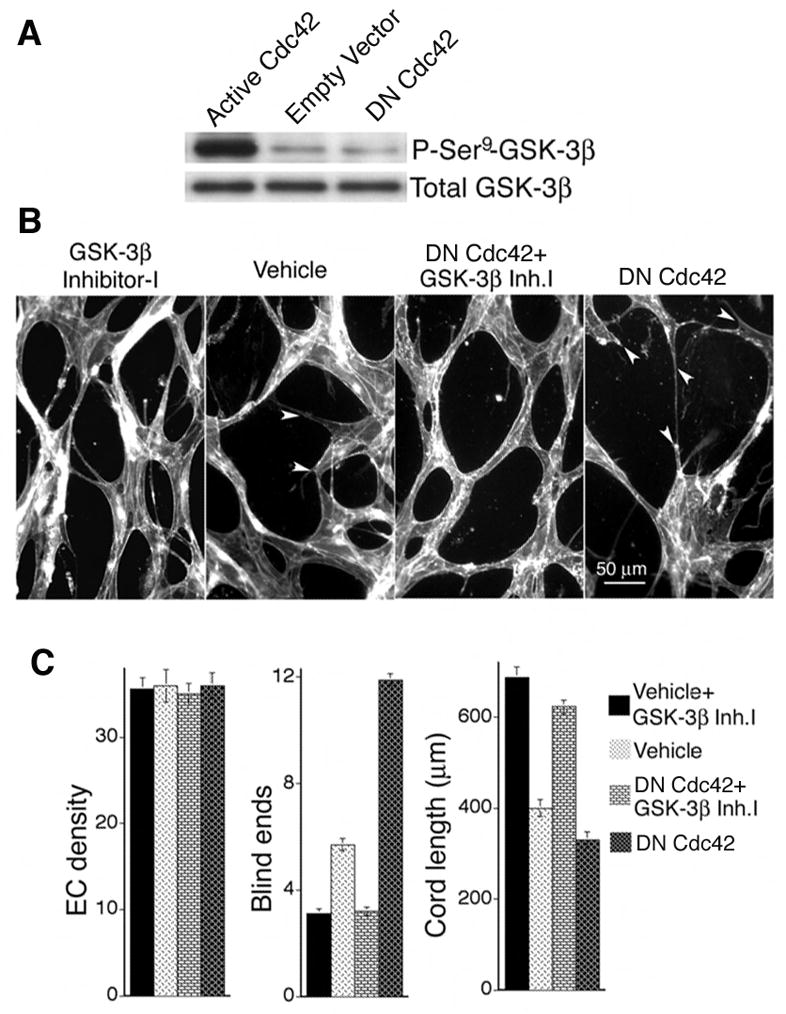
(A) Active L28Cdc42 inhibits GSK-3β in MVECs, as indicated by Ser9 phosphorylation. By scanning densitometry, active L28Cdc42 increased phosphorylation of GSK-3β Ser9 > 10-fold relative to controls in three separate experiments. Conversely, DN Cdc42 reduced GSK-3β Ser9 phosphorylation ~ 50%. (B) GSK-3β inhibitor-I (3μM) was added to control and DN N17Cdc42 MVECs “sandwiched” between two layers of collagen-I in the presence of VEGF. Note absence of blind ends (arrows) in the presence of GSK-3β inhibitor-I. (C) Quantification of cord parameters; n ≥ 22 for all groups. GSK-3β inhibitor-I significantly improved formation of capillary cords, as measured by increased cord length and reduction in cord blind ends (p < 0.001 for all paired comparisons between GSK-3β inhibitor-I and the corresponding control).
To test this hypothesis directly, we employed GSK-3β inhibitor-I (TDZD-8), a cell-permeable thiadiazolidinone analog that blocks GSK-3β independently of ATP binding with high specificity (Martinez et al., 2002). In preliminary analyses of capillary morphogenesis, four concentrations of TDZD-8 were tested (0.5, 2.0, 3.0, and 5.0 μM); the 3.0 μM dose (~1.5x IC50) best improved cord formation and was chosen for more extensive analyses. At a concentration of 3.0 μM, GSK-3β inhibitor-I strongly improved formation of capillary cords by MVECs as measured by increased cord length and reduction in the number of blind ends (Fig. 4B, C). Thus, GSK-3β inhibitor-I improved formation and integration of capillary cords very similarly to active Cdc42. In addition, GSK-3β inhibitor-I fully rectified the negative effects of DN Cdc42 on cord formation, as measured by increased cord length and reduction of blind ends (Fig. 4C). Moreover, microtubule staining of MVECs in monolayer culture showed that GSK-3β inhibitor-I enhanced microtubule polymerization similarly to active Cdc42 and rescued microtubule polymerization in cells transduced with DN Cdc42 (Supplementary Fig. 2A). Furthermore, staining of vascular cords for stabilized microtubules with antibody to acetylated tubulin indicated that GSK-3β inhibitor-I also improved microtubule stability (Supplementary Fig. 2B).
Pharmacological inhibition of GSK-3β improves VEGF-driven angiogenesis in vivo similarly to active Cdc42
The in vitro experiments described above suggested that inhibition of GSK-3β is likely the key mechanism through which active Cdc42 improves blood vessel formation during VEGF-driven angiogenesis in vivo. To test this possibility directly, we employed the same mouse skin angiogenesis model used for the Cdc42 experiments but without retroviral packaging cells. Instead, animals were treated systemically with GSK-3β inhibitor-I (TDZD-8). This compound is a highly selective, non-ATP competitive GSK-3β inhibitor that is active in the micromolar range and not known to inhibit other kinases (Martinez et al., 2002). In initial experiments, GSK-3β inhibitor-I (TDZD-8) was administered twice daily at different doses (0.3 mg/kg, and 1 mg/kg s.c.) beginning on day two following sub-dermal implantation of the VEGF-SK-MEL2 cells in Matrigel. These doses have been employed successfully in rats for reducing experimental colitis and also for reducing general organ dysfunction caused by endotoxemia (Dugo et al., 2005; Whittle et al., 2006); and, we observed no evidence of toxicity with either of these doses, as indicated by changes in animal appearance or behavior. As determined grossly on day 7 after initiation of angiogenesis (i.e., 6 days after beginning treatment with TDZD-8), the twice-daily 1 mg/kg dosing regimen most improved the formation of VEGF neovessels; and therefore more extensive experiments and analyses were performed with this dose. As quantified from gross flat mounts of tissues taken following Evans Blue dye perfusion, and with specimens viewed in cross-section, twice daily administration of 1 mg/kg GSK-3β inhibitor-I markedly increased perfused neovessel density, neovessel diameter, and lumen area (Fig. 5A, B). Quantitatively, these improvements were all comparable or better than the improvements observed in experiments involving transduction with active Cdc42 (compare Fig. 5B with Fig. 1C). Also, similarly to active Cdc42, GSK-3β inhibitor-I improved blood vessel formation without increasing EC density (Fig. 5B). Moreover, GSK-3β inhibitor-I, at the same concentrations used to improve cord formation in vitro, had no effect on VEGF production by the VEGF-transfected cells used to drive angiogenesis (see Methods), consistent with the fact that the VEGF expression vector was designed to express VEGF constitutively through a CMV promoter. Thus improved angiogenesis in vivo mediated by GSK-3β inhibitor-I (Fig. 5) is best explained by enhanced assembly of ECs into new blood vessels, consistent with our observations that GSK-3β inhibitor-I improved capillary morphogenesis in vitro (Fig. 4).
Figure 5. Pharmacological inhibition of GSK-3β improves VEGF-driven angiogenesis in vivo.

Mice were treated systemically with GSK-3β inhibitor-I, beginning on day two following implantation of the VEGF165-transfected cells in Matrigel. (A) As shown grossly at day 7 (upper panels) and as shown following perfusion for 10 min with Evan’s blue tracer (middle panels), GSK-3β inhibitor-1 increased formation of readily perfused neovessels. Bars = 400 microns. Also, as viewed in cross-sections stained for CD31 (bottom panels), neovessels with well-developed lumens were most abundant in the GSK-3β inhibitor-1 group. Bar = 30 microns. (B) Quantification of vascular parameters (n ≥ 17 for all groups). From gross images: relative neovascularization, i.e. relative area of gross images occupied by perfused neovessels (p<0.001). From CD31 stained cross-sections: quantification of ECs per 0.01mm2, numbers of neovessels per 0.01mm2 (p=0.005), average internal neovessel diameter (p<0.002) and relative total lumen area (p<0.001). No effects of GSK-β inhibitor-1 were observed in the vasculature of skin away from the site containing VEGF165-transfected cells in Matrigel (not shown).
Discussion
Experiments described here demonstrate that active L28Cdc42 significantly improved VEGF-driven pathological angiogenesis in vivo as measured by an increase in functional neovascularization, neovessel diameter, and lumen formation. In contrast, DN N17Cdc42 blocked formation of neovessels with prominent lumens despite the abundance of ECs in the host skin adjacent to the angiogenic stimulus. Our findings with DN Cdc42 are consistent with previous observations that Cdc42 siRNA blocked formation of lumens by ECs embedded in three-dimensional collagen I in vitro (Koh et al., 2008). Nonetheless, data presented here are the first to demonstrate that interference with Cdc42 signaling suppresses lumen formation in vivo and, most importantly, that active Cdc42 can improve and augment lumen formation in vivo. Interestingly, in vitro studies described here also demonstrated that active Cdc42 improved formation of pre-capillary cords that are the precursors to tubes with lumens (Drake and Fleming, 2000; Vernon and Sage, 1995). In contrast, DN Cdc42 robustly disrupted formation of pre-capillary cords. Cytoskeletal analyses indicated that active Cdc42 distinctly facilitated coordination between actin filaments and microtubules during cord formation, and improved microtubule stability, suggesting a cytoskeletal mechanism by which active Cdc42 improves capillary morphogenesis. Taken together, these findings indicate that Cdc42 is essential for proper cytoskeletal dynamics during capillary morphogenesis, and they suggest that active Cdc42 rectifies VEGF-driven angiogenesis by improving coordinated interplay between actin filaments and microtubules.
Actin-microtubule interactions provide essential mechanical stability for dynamic changes in cell shape and direction, and microtubules are required for proper guidance of actin-based contraction (reviewed (Etienne-Manneville, 2004a; Rodriguez et al., 2003)). Formation of pre-capillary cords in vitro and new blood vessels in vivo is strongly driven by actin-based contraction (Hoang et al., 2004; Whelan and Senger, 2003), and therefore it is not surprising that actin-microtubule coordination would be important for normal angiogenesis. Of particular relevance, Cdc42 regulates crosstalk between actin and microtubules in developing neurons, and actin-microtubule interactions are critical for axon guidance and elongation (reviewed (Conde and Caceres, 2009; Georges et al., 2008)). Although molecular mechanisms for coordinating actin and microtubules are highly complex and not yet well defined, they likely involve cortical microtubule receptors, microtubule tip proteins, bridging proteins, and/or other microtubule associated proteins (i.e. MAPs) (reviewed (Gundersen et al., 2004)).
In searching for downstream signaling molecules through which active Cdc42 improves VEGF-driven angiogenesis, we found that active Cdc42 strongly inhibited GSK-3β, a serine-threonine kinase that was initially identified as a phosphorylating inactivator of glycogen synthase. GSK-3β serves many functions including regulation of energy metabolism, neuronal cell development, and body pattern formation (reviewed (Doble and Woodgett, 2003)). Of likely relevance to our findings that Cdc42 improves coordination of actin and microtubules and also improves microtubule stability in MVECs, active GSK-3β has been shown to negatively regulate microtubule polymerization and stability (reviewed (Zhou and Snider, 2005)). Moreover, active GSK-3β induces neurite retraction, and suppression of GSK-3β activity is essential for stability of neurite extensions (Sanchez et al., 2001).
GSK-3β is inhibited through phosphorylation of the Ser9 residue (Doble and Woodgett, 2003) in response to a variety of external stimuli. Atypical PKC is one of the signaling intermediates that phosphorylate this residue; and, in astrocytes, cell polarity is controlled through Cdc42-mediated GSK-3β inhibition by a complex consisting of Par6 and atypical PKC (Etienne-Manneville and Hall, 2003). Importantly, EC lumen formation in vitro also has been linked to the Cdc42-Par3-Par6-atypical PKC complex (Koh et al., 2008). GSK-3β is also inhibited by the PI-3′-kinase pathway through PKB/Akt, and also by the MEK/ERK and p38 MAPK pathways, PKA, p90RSK/MAPKAP kinase-1, and S6 kinase (Forde and Dale, 2007; Liu et al., 2002). Therefore, one or more of these kinases may also be involved. We did not investigate intermediate kinases responsible for inhibiting GSK-3β downstream of Cdc42 but instead focused on investigating the significance of GSK-3β inhibition for Cdc42-mediated improvement of VEGF-driven angiogenesis. We found that, at appropriate doses, GSK-3β inhibitor-I improved all vascular parameters comparably to or better than active Cdc42. Moreover, improvements were not attributable to increases in EC density but rather improved neovessel formation. Thus, it is most likely that active Cdc42 rectifies neovascular defects associated with pathological VEGF-driven angiogenesis through inhibition of GSK-3β.
Interestingly, others have shown that hepatocyte growth factor (HGF) enhances EC barrier function in pulmonary artery ECs through multiple signaling pathways that converge on inhibition of GSK-3β (Liu et al., 2002). Although we did not investigate barrier function in this study, the relationship between GSK-3β inhibition and barrier function clearly warrants further investigation in the context of VEGF and pathological neovascularization. Also, interestingly, others have shown that adenoviral transduction with an active mutant of GSK-3β inhibited angiogenesis and that local adenoviral transduction with a kinase-dead GSK-3β mutant promoted angiogenesis in Matrigel plugs that included basic fibroblast growth factor (bFGF) as the driver of angiogenesis (Kim et al., 2002). Analyses were confined to quantification of ECs within the implanted Matrigel plugs, in contrast to our current analyses of neovascularization in the overlying host dermis that monitored neovascular architecture, perfusion, neovessel diameter, and lumen formation, in addition to EC density. Given the differences in angiogenic stimuli (i.e. bFGF vs. VEGF) and considerable differences in experimental design and analyses, direct comparisons between these different studies are not practical. Nonetheless, they are in agreement regarding the conclusion that inhibition of GSK-3β can support angiogenesis. Moreover, promotion of angiogenesis by GSK-3β inhibition in the Matrigel model using bFGF as the driver of angiogenesis was linked to increased stability of β-catenin (Skurk et al., 2005). HGF-mediated inhibition of GSK-3β in ECs also improves β-catenin and VE-cadherin linkage to the cytoskeleton (Liu et al., 2002). Thus, the relationship between GSK-3β inhibition and stability of β-catenin also warrants further investigation in the context of VEGF and pathological neovascularization.
Although a possible connection between β–catenin stability and the mechanism by which GSK-3β inhibition rectifies defects in VEGF-driven pathological angiogenesis remains to be determined, our findings specifically broaden understanding of the importance of GSK-3β by demonstrating that GSK-3β inhibition stabilizes microtubules in MVECs cultured in the presence of VEGF (Supplementary Fig. 2), improves formation of precapillary cords in collagen gels containing VEGF (Figure 4), and improves VEGF neovessel lumenization in vivo (Figure 5). They also suggest the more general hypothesis that impaired regulation of the EC cytoskeleton is responsible for vascular defects associated with pathological angiogenesis (Nagy and Senger, 2006). However, it is important to emphasize that our findings do not establish that defects in Cdc42 or GSK-3β signaling are directly responsible for vascular defects associated with pathological angiogenesis. Rather our findings identify a Cdc42/GSK-3β pathway that can be manipulated to rectify such defects. Moreover, if defective GSK-3β signaling does prove to be integral to the mechanism by which VEGF induces pathological neovascularization, it need not be attributable to defective Cdc42 signaling. As summarized above, numerous kinases inhibit GSK-3β activity and defects associated with VEGF pathological neovessels may relate to reduced activities of any one or more of these upstream kinases. Also, because multiple signaling pathways in addition to Cdc42/GSK-3β regulate the cytoskeleton (Nobes and Hall, 1999), there are likely numerous other cytoskeletal-targeting strategies that may prove suitable for rectifying defects in VEGF neovasculature (Nagy and Senger, 2006). Indeed, active RhoA, a key regulator of cytoskeletal dynamics, can also markedly improve VEGF-driven angiogenesis in vivo (Hoang et al., 2004). Targeting of yet additional pathways may also prove beneficial. For example, lumen size can be increased by a constitutively active form of Notch (reviewed (Iruela-Arispe and Davis, 2009)). Thus, multiple targeting strategies, possibly used in combination, may eventually provide optimal rectification of pathological neovessels.
From a more immediate translational standpoint, our experiments identify inhibition of GSK-3β as a “stand alone” pharmacological strategy for rectifying neovascular defects associated with VEGF. Abnormal blood vessel formation, resulting in poor perfusion, seriously reduces the utility of VEGF as a therapeutic agent for tissue revascularization (Jain, 2003), and abnormal neovessels resulting from hypoxia-induced VEGF expression contribute to the pathology of disease. For example, in ischemic retinopathies, abnormal neovascularization driven by VEGF damages retina and ultimately leads to blindness (Pierce et al., 1995). Importantly, GSK-3β inhibitors are already under consideration for other clinical applications (Eldar-Finkelman, 2002; Meijer et al., 2004) including diabetes and a variety of neurodegenerative conditions (Medina and Castro, 2008). Thus, pharmacological targeting GSK-3 activity may prove useful in treating a variety of important pathologies, including abnormal neovascularization.
Supplementary Material
Acknowledgments
We thank Dr. Richard Cerione, Cornell University, for generously providing the L28Cdc42 construct, and we thank Mary Whelan for isolation of microvascular endothelial cells. This research was supported by NIH grant CA129339 from the National Cancer Institute (D.R.S.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cau J, Hall A. Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J Cell Sci. 2005;118:2579–87. doi: 10.1242/jcs.02385. [DOI] [PubMed] [Google Scholar]
- Chae JK, et al. Coadministration of angiopoietin-1 and vascular endothelial growth factor enhances collateral vascularization. Arterioscler Thromb Vasc Biol. 2000;20:2573–8. doi: 10.1161/01.atv.20.12.2573. [DOI] [PubMed] [Google Scholar]
- Conde C, Caceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–32. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Davis GE, Camarillo CW. An alpha 2 beta 1 integrin-dependent pinocytic mechanism involving intracellular vacuole formation and coalescence regulates capillary lumen and tube formation in three-dimensional collagen matrix. Exp Cell Res. 1996;224:39–51. doi: 10.1006/excr.1996.0109. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CJ, Fleming PA. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood. 2000;95:1671–9. [PubMed] [Google Scholar]
- Dugo L, et al. GSK-3beta inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit Care Med. 2005;33:1903–12. doi: 10.1097/01.ccm.0000178350.21839.44. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8:126–32. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S. Actin and microtubules in cell motility: which one is in control? Traffic. 2004a;5:470–7. doi: 10.1111/j.1600-0854.2004.00196.x. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S. Cdc42--the centre of polarity. J Cell Sci. 2004b;117:1291–300. doi: 10.1242/jcs.01115. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature. 2003;421:753–6. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- Forde JE, Dale TC. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci. 2007;64:1930–44. doi: 10.1007/s00018-007-7045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges PC, et al. The yin-yang of dendrite morphology: unity of actin and microtubules. Mol Neurobiol. 2008;38:270–84. doi: 10.1007/s12035-008-8046-8. [DOI] [PubMed] [Google Scholar]
- Gundersen GG, et al. Cortical control of microtubule stability and polarization. Curr Opin Cell Biol. 2004;16:106–12. doi: 10.1016/j.ceb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Hoang MV, Senger DR. In vivo and in vitro models of mammalian angiogenesis. Methods Mol Biol. 2005;294:269–85. doi: 10.1385/1-59259-860-9:269. [DOI] [PubMed] [Google Scholar]
- Hoang MV, et al. Rho activity critically and selectively regulates endothelial cell organization during angiogenesis. Proc Natl Acad Sci U S A. 2004;101:1874–9. doi: 10.1073/pnas.0308525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iruela-Arispe ML, Davis GE. Cellular and molecular mechanisms of vascular lumen formation. Dev Cell. 2009;16:222–31. doi: 10.1016/j.devcel.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- Kim HS, et al. Regulation of angiogenesis by glycogen synthase kinase-3beta. J Biol Chem. 2002;277:41888–96. doi: 10.1074/jbc.M206657200. [DOI] [PubMed] [Google Scholar]
- Koh W, et al. Cdc42- and Rac1-mediated endothelial lumen formation requires Pak2, Pak4 and Par3, and PKC-dependent signaling. J Cell Sci. 2008;121:989–1001. doi: 10.1242/jcs.020693. [DOI] [PubMed] [Google Scholar]
- Kouklis P, et al. Cdc42 regulates the restoration of endothelial barrier function. Circ Res. 2004;94:159–66. doi: 10.1161/01.RES.0000110418.38500.31. [DOI] [PubMed] [Google Scholar]
- Le Doux JM, et al. Complexation of retrovirus with cationic and anionic polymers increases the efficiency of gene transfer. Hum Gene Ther. 2001;12:1611–21. doi: 10.1089/10430340152528110. [DOI] [PubMed] [Google Scholar]
- Lin R, et al. Specific contributions of the small GTPases Rho, Rac, and Cdc42 to Dbl transformation. J Biol Chem. 1999;274:23633–41. doi: 10.1074/jbc.274.33.23633. [DOI] [PubMed] [Google Scholar]
- Liu F, et al. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. Faseb J. 2002;16:950–62. doi: 10.1096/fj.01-0870com. [DOI] [PubMed] [Google Scholar]
- Martinez A, et al. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J Med Chem. 2002;45:1292–9. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- Medina M, Castro A. Glycogen synthase kinase-3 (GSK-3) inhibitors reach the clinic. Curr Opin Drug Discov Devel. 2008;11:533–43. [PubMed] [Google Scholar]
- Meijer L, et al. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–80. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Nagy JA, et al. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer. 2009;100:865–9. doi: 10.1038/sj.bjc.6604929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JA, et al. Pathogenesis of ascites tumor growth: angiogenesis, vascular remodeling, and stroma formation in the peritoneal lining. Cancer Res. 1995;55:376–85. [PubMed] [Google Scholar]
- Nagy JA, Senger DR. VEGF-A, cytoskeletal dynamics, and the pathological vascular phenotype. Exp Cell Res. 2006;312:538–48. doi: 10.1016/j.yexcr.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–44. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa CR, et al. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J Clin Invest. 2004;113:516–27. doi: 10.1172/JCI18420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce EA, et al. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci U S A. 1995;92:905–9. doi: 10.1073/pnas.92.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno G, et al. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. 1987;104:289–302. doi: 10.1083/jcb.104.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price LS, et al. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell. 1998;9:1863–71. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls A, et al. Activation of the small GTPase Cdc42 by the inflammatory cytokines TNF(alpha) and IL-1, and by the Epstein-Barr virus transforming protein LMP1. J Cell Sci. 1999;112(Pt 17):2983–92. doi: 10.1242/jcs.112.17.2983. [DOI] [PubMed] [Google Scholar]
- Richard L, et al. A simple immunomagnetic protocol for the selective isolation and long-term culture of human dermal microvascular endothelial cells. Exp Cell Res. 1998;240:1–6. doi: 10.1006/excr.1998.3936. [DOI] [PubMed] [Google Scholar]
- Rodriguez OC, et al. Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat Cell Biol. 2003;5:599–609. doi: 10.1038/ncb0703-599. [DOI] [PubMed] [Google Scholar]
- Sanchez S, et al. The inhibition of phosphatidylinositol-3-kinase induces neurite retraction and activates GSK3. J Neurochem. 2001;78:468–81. doi: 10.1046/j.1471-4159.2001.00453.x. [DOI] [PubMed] [Google Scholar]
- Schlessinger K, et al. Cdc42 and noncanonical Wnt signal transduction pathways cooperate to promote cell polarity. J Cell Biol. 2007;178:355–61. doi: 10.1083/jcb.200701083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger DR, et al. Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc Natl Acad Sci U S A. 1997;94:13612–7. doi: 10.1073/pnas.94.25.13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger DR, et al. Purification and NH2-terminal amino acid sequence of guinea pig tumor-secreted vascular permeability factor. Cancer Res. 1990;50:1774–8. [PubMed] [Google Scholar]
- Sioussat TM, et al. Inhibition of vascular permeability factor (vascular endothelial growth factor) with antipeptide antibodies. Arch Biochem Biophys. 1993;301:15–20. doi: 10.1006/abbi.1993.1109. [DOI] [PubMed] [Google Scholar]
- Skurk C, et al. Glycogen-Synthase Kinase3beta/beta-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ Res. 2005;96:308–18. doi: 10.1161/01.RES.0000156273.30274.f7. [DOI] [PubMed] [Google Scholar]
- Tu SS, et al. Antiapoptotic Cdc42 mutants are potent activators of cellular transformation. Biochemistry. 2002;41:12350–8. doi: 10.1021/bi026167h. [DOI] [PubMed] [Google Scholar]
- Tzima E, et al. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J Biol Chem. 2003;278:31020–3. doi: 10.1074/jbc.M301179200. [DOI] [PubMed] [Google Scholar]
- Vernon RB, Sage EH. Between molecules and morphology. Extracellular matrix and creation of vascular form. Am J Pathol. 1995;147:873–83. [PMC free article] [PubMed] [Google Scholar]
- Wedlich-Soldner R, et al. Spontaneous cell polarization through actomyosin-based delivery of the Cdc42 GTPase. Science. 2003;299:1231–5. doi: 10.1126/science.1080944. [DOI] [PubMed] [Google Scholar]
- Whelan MC, Senger DR. Collagen I initiates endothelial cell morphogenesis by inducing actin polymerization through suppression of cyclic AMP and protein kinase A. J Biol Chem. 2003;278:327–34. doi: 10.1074/jbc.M207554200. [DOI] [PubMed] [Google Scholar]
- Whittle BJ, et al. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3beta. Br J Pharmacol. 2006;147:575–82. doi: 10.1038/sj.bjp.0706509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, et al. Orphan nuclear receptor TR3/Nur77 regulates VEGF-A-induced angiogenesis through its transcriptional activity. J Exp Med. 2006;203:719–29. doi: 10.1084/jem.20051523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Cell biology. GSK-3beta and microtubule assembly in axons. Science. 2005;308:211–4. doi: 10.1126/science.1110301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.