

Abstract
Background
Olfactory loss is a debilitating symptom of chronic rhinosinusitis (CRS). Although olfactory sensory neurons (OSNs) are normally regenerated constantly in the olfactory epithelium (OE), a transgenic model of CRS-associated olfactory loss (inducible olfactory inflammation [IOI] mouse) shows that inflammation causes widespread OSN loss without progenitor cell proliferation. In this study, we further examine whether the inflammatory cytokine tumor necrosis factor alpha (TNF-alpha) inhibits olfactory regeneration.
Methods
IOI mice underwent either unilateral bulbectomy or sham surgery and then were induced to express TNF-alpha in the OE for 1 week. After death, the mice were assessed histologically and with bromodeoxyuridine staining to determine the effect of TNF-alpha on olfactory regeneration.
Results
In the absence of TNF-alpha, bulbectomy was associated with death of OSNs, followed by robust proliferation of neural progenitors and regrowth of the OE. At 12 days postbulbectomy, OE thickness on the operated side had recovered to >80% of the unoperated side. In mice in which TNF-alpha expression was induced, significantly reduced proliferation was observed, associated with failure of normal reconstitution of OE thickness.
Conclusion
The mechanism of olfactory dysfunction in CRS remains incompletely understood. Previous studies with a transgenic mouse model suggested that inflammation inhibits progenitor cell proliferation and olfactory regeneration. Here, the role of the CRS-associated cytokine TNF-alpha was investigated using surgical ablation of the olfactory bulb to stimulate synchronous OSN turnover. We find that TNF-alpha expression prevents normal OE recovery, supporting the role of suppressed olfactory regeneration in the pathophysiology of CRS-associated olfactory loss.
Olfactory dysfunction is a common and often debilitating symptom in patients with chronic rhinosinusitis (CRS).1 Nonetheless, there are few effective treatments that specifically target the olfactory loss observed in this patient population. The ability of corticosteroids to partially reverse the loss of smell in these individuals has long suggested that the decreased olfactory function in CRS patients may be due largely to local inflammation. This has been partially confirmed by histological evaluation of olfactory epithelium (OE) from CRS patients, which typically shows an influx of inflammatory cells as well as production of substantial quantities of inflammatory mediators.2–4 Despite this, the mechanism of sinusitis-associated olfactory loss and olfactory neuron regeneration remains poorly understood.
The mammalian OE is comprised of three primary cell types: olfactory receptor neurons, supporting sustentacular cells, and a population of basal progenitor cells. In humans and most other mammals, olfactory neurons are continuously regenerated from basal cells with an average life span of ~30–90 days.5,6 Replacement of olfactory neurons occurs relatively rapidly after neuroepithelial injury, but the mechanism and regulation of olfactory neuronal regeneration is currently unclear.
Olfactory bulbectomy is a commonly used experimental model to investigate the regulation of olfactory neuron degeneration and regeneration.7 Axons originating from newly differentiated olfactory receptor neurons project to the main olfactory bulb where they synapse with secondary nerves and, ultimately, project axons to the primary olfactory cortex. The removal of the olfactory bulb results in a surge in olfactory neuron cell death followed by a period of basal progenitor cell division and olfactory neuroepithelial regeneration.8 This sequence of events can therefore be used as a model for both traumatic and inflammatory olfactory loss, as well as olfactory neuron regeneration.
We previously developed a transgenic mouse model for sinusitis-associated olfactory loss.9 This inducible system replicates CRS-associated inflammation via a temporally controlled, tissue-specific expression of individual inflammatory mediators within the OE. Our data suggest that the inflammatory cytokine, tumor necrosis factor (TNF) α, causes olfactory dysfunction through a combination of three possible mechanisms, including (1) An initial desensitization of odorant-initiated signaling pathways, (2) loss of olfactory neurons due to direct neuronal and/or axonal injury, and (3) Inhibition of regeneration and proliferation of immature neurons from progenitor cells. Most recently, we showed that TNF-α causes olfactory neuron loss and reduces expression of growth-associated protein (GAP) 43, a neuronal marker protein associated with the growth and development of immature neurons.10 The effect of TNF-α on olfactory regeneration otherwise remains unclear.
Although our previous studies examined the effects of TNF-α on the structure and characteristics of the mouse OE, the current study seeks to specifically detail the role of this inflammatory cytokine in olfactory neuronal regeneration. Using the transgenic mouse model, we assessed the effect of TNF-α on regrowth of the olfactory neuroepithelium after bulbectomy. Expression of TNF-α resulted in reduced proliferation of neuronal progenitor cells and failed reconstitution of the OE. This study shows that TNF-α–associated inflammation inhibits differentiation of progenitor cells into immature olfactory neurons and suggests a prominent role for local inflammation in CRS-associated olfactory dysfunction.
METHODS
Inducible Olfactory Inflammation (IOI) Mouse
The IOI mouse line was created, as previously described, by introduction of the Teton genetic system into the mouse genome under the control of the olfactory-specific cyp2g1 promoter.9 Briefly, the reverse tetracycline transactivator gene was knocked into the cyp2g1 coding region in mouse ES cells. The tetracycline responsive element (TRE)–TNF-α construct containing the murine TNF-α gene under control of the tetracycline-responsive element was generated by cloning the murine TNF-α gene11 into the pTRE vector (Clontech, Mountain View, CA). The TRE–TNF-α vector was injected into fertilized mouse eggs to generate transgenic mice. Transgenic lines were maintained by mating to BL/6 mice. The cyp2g1-rtTA mice were bred to wild-type BL/6 mice to establish germline transmission. True-breeding strains of TRE–TNF-α and cyp2g1-rtTA were housed under specific pathogen-free conditions. The two lines were crossed to create the IOI mouse, with the presence of both constructs determined by polymerase chain reaction.
Olfactory Bulbectomy
Targeted removal of the olfactory bulb was performed as described previously by Carr and Farbman.7 Mice were anesthetized with ketamine (Sigma, St Louis, MO) (80 mg/kg). A midline incision was made to expose the frontal bones overlying the olfactory bulbs. The right olfactory bulb was then exposed by drilling through the frontal bone. The entire olfactory bulb was then removed by gentle aspiration with a curved glass pipette. The cavity was then filled with sterile Gelfoam (Pfizer, New York) and the incision was reapproximated with absorbable sutures. Mice were allowed to recover from anesthesia and then returned to their enclosures. After bulbectomy, mice were housed under standard conditions for 5 days and then with drinking water supplemented with or without 0.5 mg/mL of doxycycline for 7 days before death by CO2 inhalation.
Histological Analyses
For histology, mouse nasal cavities were embedded in paraffin or were processed for frozen sections. After death by CO2 inhalation, mice were decapitated and the heads were fixed and decalcified by immersion in TBD2 solution (Shandon, Pittsburgh, PA) for 24 hours. The heads were then embedded in paraffin, and 12-μm sections were obtained and collected on glass slides for hematoxylin and eosin staining. For frozen section analysis, mice were anesthetized by i.p. injection of 100 mg/kg of xylaket (Sigma, St. Louis, MO), before intracardiac perfusion with 4% paraformaldehyde. The olfactory tissue was then dissected, postfixed in 4% paraformaldehyde, and transferred to a solution of 30% sucrose and 250 mM of EDTA for 48 hours. The decalcified heads were then infiltrated with OCT tissue-tek compound (Miles, Elkhart, IN) and frozen on dry ice into a plastic mold. Sections of mouse olfactory tissue in OCT were cut on a cryostat (12 μM), placed on Super-frost plus slides (Fisher Scientific, Pittsburgh, PA), and dried 60 minutes before use. Images shown are representative of at least three separate experiments for a minimum of three individual mice.
Thickness Measurements
The thickness of the OE was measured on hematoxylin and eosin–stained tissue sections. Images were taken using a Labophot-2 microscope with a DXM1200 Digital Camera (Nikon Corp., Melville, NY) at 400× magnification. Thickness measurements were made along the nasal septum and turbinates in increments of 200 μm over the entire length of the OE for each section. The measured thickness of the epithelium was from the basement membrane to the top of the olfactory knobs. All measurements were made on at least two sections from zone 3 of the OE for at least three mice from each data group. Values for each individual animal were averaged and data are represented as the mean ± SE for each group. Statistical comparisons were made using the students t-test.
Bromodeoxyuridine Labeling
Mice were injected i.p. with bromodeoxyuridine (BrdU) (Sigma), 50 μg/g of body weight, 60 minutes before death. Paraformaldehydefixed sections were then incubated with 3 N HCl for 30 minutes before immunostaining with rat anti-BrdU antibody (Abcam, Cambridge MA) as previously described.9 BrdU+ cells were counted along the nasal septum in zone 3 of the OE. Data were expressed graphically as the mean ± SE for each group. Statistical comparisons were made using the students t-test.
RESULTS
Bulbectomy as a Model for Olfactory Neurogenesis
The IOI mouse models chronic sinusitis–associated olfactory dysfunction via the spatially and temporally controlled expression of individual inflammatory cytokines. In the current study we sought to evaluate the effect of TNF-α on neuronal regeneration after olfactory bulbectomy. Targeted removal of the olfactory bulb results in a massive loss of olfactory receptor neurons within 72 hours of injury.8 This retrograde neuronal degeneration stimulates mitosis of progenitor cells, and the ultimate regeneration of millions of olfactory neurons within 2–3 weeks after bulbectomy. Figure 1 shows a hematoxylin and eosin–stained section of a wild-type bulbectomized mouse 7 days after surgery. As shown, the right olfactory bulb has been surgically ablated. The right OE is atrophic secondary to rapid loss of olfactory neurons.
Figure 1.
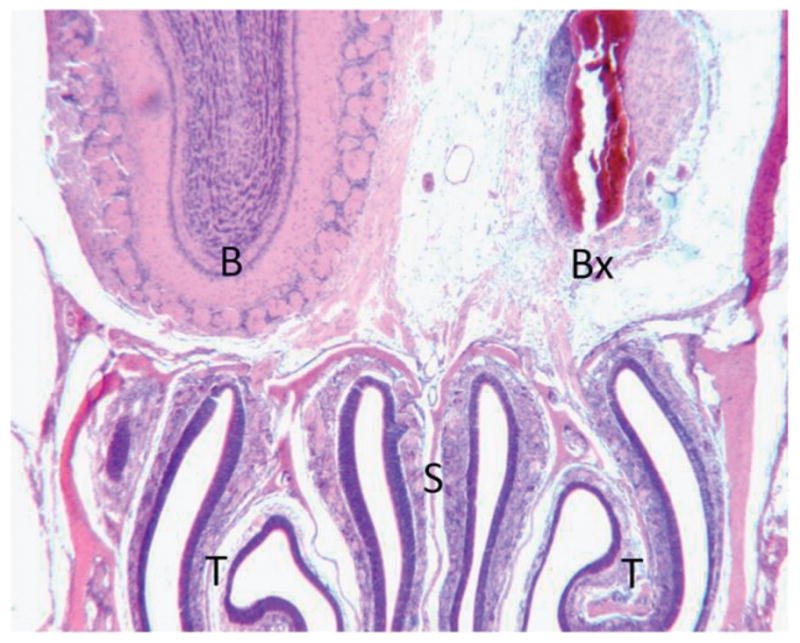
Olfactory bulbectomy is a model for olfactory neuronal regeneration. Wild-type and inducible olfactory inflammation (IOI) mice were bulbectomized and killed, and their nasal cavities were then embedded in paraffin and processed as described in the Methods section. Shown is a cross-section of a wild-type mouse nasal cavity at the level of the olfactory bulb, processed 7 days after bulbectomy. Identified is the normal left olfactory bulb (B), right olfactory bulb after bulbectomy (BX), nasal septum (NS), and turbinates (T).
TNF-α Expression Inhibits Olfactory Neurogenesis after Bulbectomy
In previous work we showed that expression of TNF-α in the IOI mouse results in olfactory neuron loss, defects in olfactory function, and thinning of the olfactory neuroepithelium. Besides an apparent ability to stimulate olfactory neuron death, our data suggest that TNF-α may also impede growth and differentiation of neuronal progenitor cells and immature olfactory neurons. To further evaluate the role of TNF-α on olfactory regeneration, we examined the effect of TNF-α expression on the olfactory neuroepithelium in IOI mice. Wild-type and IOI mice were bulbectomized and then reared for 12 days. In IOI mice, TNF-α expression was induced on day 5. Twelve days after bulbectomy in wild-type mice, the olfactory neuroepithelium on the bulbectomy side had largely recovered and resembled that of the nonbulbectomy side (Fig. 2). In contrast, the OE is thinned in bulbectomized IOI mice and the axon bundles are atrophic. These data are represented graphically in Fig. 3. The OE in bulbectomized wild-type mice recovered to almost 90% of the unoperated side (51.46 μm versus 59.31 μm). The OE in bulbectomized IOI mice had recovered to <70% of the unoperated side (41.69 μm versus 60.85 μm). These results suggest that TNF-α inhibits the regeneration of olfactory neurons after bulbectomy.
Figure 2.

Tumor necrosis factor (TNF) α inhibits repopulation of olfactory neurons after bulbectomy in the inducible olfactory inflammation (IOI) mouse. Wild-type and IOI mice were bulbectomized (OBX) or underwent Sham surgery (Sham) as described in the Methods section. Shown are images of the nasal septum at (A–D) 10× and (E–H) 20× magnification with the unoperated side on the left and the operated side on the right. (A and E) Wild-type, sham-operated mouse with multiple layers of olfactory neurons and an overlying layer of sustentacular cells on both sides of the nasal cavity. (B and F) Wild-type, bulbectomized mouse; the olfactory epithelium has largely recovered and is similar in appearance to the contralateral side. (C and G) Sham-operated IOI mouse olfactory epithelium is histologically identical to that of wild-type mouse. (D and H) Bulbectomized IOI mouse with substantially thinned olfactory epithelium and disorganized axon bundles. Data are representative of at least three different mice for each condition. Scale bars: panels A–D, 100 μm; panels E–H, 50 μm.
Figure 3.

Tumor necrosis factor (TNF) α inhibits repopulation of the olfactory epithelium after bulbectomy in inducible olfactory inflammation (IOI) mice. Thickness of the olfactory epithelium was measured on each side of the nasal septum for both sham-operated and bulbectomized mice. Data are shown as the mean thickness ± SE for at least three different mice. *p < 0.05 versus unoperated side; **p < 0.001 versus unoperated side; #p < 0.005 versus wild type.
Basal Cell Mitosis after Bulbectomy Is Inhibited by Expression of TNF-α
Regenerating olfactory neurons are produced from a pluripotent line of progenitor cells that reside along the basal epithelium. To further assess neurogenesis after bulbectomy, we measured the incorporation of BrdU into developing neurons. In wild-type mice, bulbectomy resulted in a significant mitotic response, as assessed by BrdU incorporation ipsilateral to the lesion (Fig. 4, A and B). These dividing cells reside largely along the basal cell layer. This mitotic response is substantially reduced in the IOI mouse (Fig. 4, C and D). Most mitotic cells are instead limited to the subepithelium, consistent with inflammatory cell infiltration (Andrew P. Lane, et al., unpublished data, 2010). Results are expressed graphically in Fig. 4 E. These results suggest that TNF-α inhibits basal cell mitosis and the resulting generation of immature olfactory neurons.
Figure 4.

Tumor necrosis (TNF) α inhibits mitosis of basal progenitor cells in the olfactory epithelium of inducible olfactory inflammation (IOI) mice. Wild-type and IOI mice were processed for BrdU staining as described in the Methods section. Representative images are shown for wild-type (WT) and IOI mice that underwent either (A and C) sham surgery or (B and D) bulbectomy. Proliferating basal progenitor cells (solid arrows) and infiltrating inflammatory cells (dashed arrows) are noted. (E). BrdU+ cells were counted along the entire length of the nasal septum on both the operated and unoperated side. Data are represented as the mean number of BrdU cells ± SE for at least three different mice. *p < 0.05 versus wild type; **p < 0.01 versus unoperated side. Scale bar, 100 μm.
DISCUSSION
Olfactory dysfunction is common in patients with CRS. Although the cause remains unclear, the use of topical or systemic corticosteroids can often partially or fully reverse the olfactory loss in these patients, suggesting a prominent role for inflammation. Using the IOI mouse, a transgenic model for CRS-associated olfactory loss, we have begun to explore the mechanism by which CRS produces olfactory dysfunction. In previous work, we showed that expression of the inflammatory cytokine TNF-α results in a loss of olfactory function and neurons. We also noted a decrease in expression of GAP-43, a marker of immature or developing neurons, suggesting that the olfactory loss in this model may be due, in part, to inhibition of olfactory neuron regeneration and maturation. Unlike most neuronal cell populations in the mammalian central nervous system, olfactory neurons have the ability to regenerate. This replacement occurs both as part of the normal turnover of the OE and as a response to widespread olfactory neuron death. In the current study, we therefore sought to specifically address the effect of TNF-α–mediated inflammation on olfactory neuron regeneration.
Unilateral bulbectomy causes rapid degeneration of olfactory neurons, followed by regeneration over a period of weeks. Using this model, we showed that TNF-α expression inhibits olfactory neuron regeneration. This was evidenced by a failure in reconstitution of OE thickness, as well as reduced mitosis among basal progenitor cells. Although TNF-α has been shown to induce apoptosis in OE in vitro,12 the ability of TNF-α and other inflammatory cytokines to disrupt olfactory neuron regeneration is a previously unreported phenomenon. Inflammation does appear to inhibit functional recovery after injury in both the central and the peripheral nervous system, although few studies have directly investigated a role for TNF-α. In a report by Neumann et al., TNF-α was found to inhibit neurite outgrowth and branching of hippocampal neurons, important components in neuronal development and regeneration.13 Likewise, in an experimental model of peripheral nerve injury, TNF-α mediates injury-induced NF-κB activity and inhibits postinjury axonal sprouting.14 A role for TNF-α in tissue regeneration is more validated in nonneuronal cells and tissues. TNF-α inhibits chondrogenesis by human mesenchymal stem cells through a mechanism involving NF-κB15 and induces premature senescence in endothelial progenitor cells via a p38 mitogen-activated protein kinase–dependent pathway.16 TNF-α also blocks skeletal muscle regeneration via an effect on satellite and myoblast differentiation,17 and can inhibit liver regeneration after hepatectomy.18 It is hypothesized that TNF-α and other cytokines may alter expression of gene expression sequences necessary for regenerative responses.
In our previous studies we proposed three mechanisms through which TNF-α–induced inflammation can cause olfactory dysfunction, including (1) desensitization of odorant signaling pathways, (2) death of olfactory neurons, and (3) reduced regeneration of immature olfactory neurons from basal progenitor cells. Although previous studies have suggested that TNF-α may induce apoptosis in the OE, we still have to confirm this in our model (although we are confident that it does occur). There is, undoubtedly, a substantial loss of olfactory neurons in the presence of TNF-α, but this loss is delayed and protracted, suggesting that its effects may be mediated partially by a modulation of normal and stimulated olfactory neuron turnover. This hypothesis was initiated by our previous study, in which we showed that persistent expression of TNF-α substantially reduces expression of GAP-43, a marker for immature neurons, while having only a marginal effect on the expression of markers for mature neurons. Unilateral bulbectomy typically results in prolonged proliferation of immature olfactory neurons on the side of the lesion. In our model, TNF-α expression prevented olfactory neuron regeneration, in part, by affecting the mitotic activity of basal progenitor cells within the olfactory neuroepithelium. The division and differentiation of these pluripotent cells into immature olfactory neurons is the basis for the regenerative capacity of mammalian olfactory tissue. We therefore propose that chronic inflammation secondary to TNF-α and other cytokines may perturb precise gene transcription profiles during an important “check point,” at which basal progenitor cells transition into immature olfactory neurons.
The results of the current study are both substantial and exciting. Despite being a significant patient complaint, the mechanism of olfactory loss in sufferers of CRS has remained largely unknown. We have shown that local TNF-α–mediated inflammation impedes olfactory neuron turnover and regeneration. The remarkable regenerative ability of the olfactory neuroepithelium creates a scenario by which mediators of sinonasal inflammation may become worthy therapeutic targets in the treatment of CRS-associated olfactory dysfunction.
Acknowledgments
A. Lane received funding by R01 DC009026
Footnotes
Presented at the 55th annual meeting of the American Rhinologic Society, San Diego, California, October 3, 2009
References
- 1.Loury KM. Chronic sinusitis and nasal polyposis. In: Getchell TV, Bartoshuk LM, Doty RL, Snow JB, editors. Smell and Taste in Health and Disease. New York: Raven Press; 1991. pp. 711–730. [Google Scholar]
- 2.Kern RC. Chronic sinusitis and anosmia: Pathologic changes in the olfactory mucosa. Laryngoscope. 2000;110:1071–1077. doi: 10.1097/00005537-200007000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Kuehnemund M, Ismail C, Brieger J, et al. Untreated chronic rhinosinusitis: A comparison of symptoms and mediator profiles. Laryngoscope. 2004;114:561–565. doi: 10.1097/00005537-200403000-00032. [DOI] [PubMed] [Google Scholar]
- 4.Lennard CM, Mann EA, Sun LL, et al. Interleukin-1 beta, interleukin-5, interleukin-6, interleukin-8, and tumor necrosis factor-alpha in chronic sinusitis: Response to systemic corticosteroids. Am J Rhinol. 2000;14:367–373. doi: 10.2500/105065800779954329. [DOI] [PubMed] [Google Scholar]
- 5.Samanen DW, Forbes WB. Replication and differentiation of olfactory receptor neurons following axotomy in the adult hamster: A morphometric analysis of postnatal neurogenesis. J Compar Neurol. 1984;225:201–211. doi: 10.1002/cne.902250206. [DOI] [PubMed] [Google Scholar]
- 6.Mackay-Sim A, Kittel PW. On the life span of olfactory receptor neurons. Eur J Neurosci. 1991;3:209–215. doi: 10.1111/j.1460-9568.1991.tb00081.x. [DOI] [PubMed] [Google Scholar]
- 7.Carr VM, Farbman AI. Ablation of the olfactory bulb up-regulates the rate of neurogenesis and induces precocious cell death in olfactory epithelium. Exp Neurol. 1992;115:55–59. doi: 10.1016/0014-4886(92)90221-b. [DOI] [PubMed] [Google Scholar]
- 8.Carter LA, MacDonald JL, Roskams AJ. Olfactory horizontal basal cells demonstrate a conserved multipotent progenitor phenotype. J Neurosci. 2004;24:5670–5683. doi: 10.1523/JNEUROSCI.0330-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane AP, Turner J, May L, Reed R. A genetic model of chronic rhinosinusitis-associated olfactory inflammation reveals reversible functional impairment and dramatic neuroepithelial reorganization. J Neurosci. 2010;30:2324–2329. doi: 10.1523/JNEUROSCI.4507-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner JH, May L, Reed RR, Lane AP. Reversible loss of neuronal marker protein expression in a transgenic mouse model for sinusitis-associated olfactory dysfunction. Am J Rhinol Allergy. 2010;24:192–196. doi: 10.2500/ajra.2010.24.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki Y, Farbman AI. Tumor necrosis factor-alpha-induced apoptosis in olfactory epithelium in vitro: Possible roles of caspase 1 (ICE), caspase 2 (ICH-1), and caspase 3 (CPP32) Exp Neurol. 2000;165:35–45. doi: 10.1006/exnr.2000.7465. [DOI] [PubMed] [Google Scholar]
- 13.Neumann H, Schweigreiter R, Yamashita T, et al. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J Neurosci. 2002;22:854–862. doi: 10.1523/JNEUROSCI.22-03-00854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith D, Tweed C, Fernyhough P, Glazner GW. Nuclear factor-kappaB activation in axons and Schwann cells in experimental sciatic nerve injury and its role in modulating axon regeneration: Studies with etanercept. J Neuropathol Exp Neurol. 2009;68:691–700. doi: 10.1097/NEN.0b013e3181a7c14e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Herbert BS, Rajashekhar G, et al. Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. FASEB J. 2009;23:1358–1365. doi: 10.1096/fj.08-110296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wehling N, et al. Interleukin-1beta and tumor necrosis factor alpha inhibit chondrogenesis by human mesenchymal stem cells through NF-kappaB-dependent pathways. Arthritis Rheum. 2009;60:801–812. doi: 10.1002/art.24352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alter J, Rozentzweig D, Bengal E. Inhibition of myoblast differentiation by tumor necrosis factor alpha is mediated by c-Jun N-terminal kinase 1 and leukemia inhibitory factor. J Biol Chem. 2008;283:23224–23234. doi: 10.1074/jbc.M801379200. [DOI] [PubMed] [Google Scholar]
- 18.Hou Z, Yanaga K, Kamohara Y, et al. A new suppressive agent against interleukin-1beta and tumor necrosis factor-alpha enhances liver regeneration after partial hepatectomy in rats. Hepatol Res. 2003;26:40–46. doi: 10.1016/s1386-6346(02)00334-0. [DOI] [PubMed] [Google Scholar]