

Abstract
X‐adrenoleukodystrophy (X‐ALD) is a complex disease where inactivation of ABCD1 gene results in clinically diverse phenotypes, the fatal disorder of cerebral ALD (cALD) or a milder disorder of adrenomyeloneuropathy (AMN). Loss of ABCD1 function results in defective beta oxidation of very long chain fatty acids (VLCFA) resulting in excessive accumulation of VLCFA, the biochemical “hall mark” of X‐ALD. At present, the ABCD1‐mediated mechanisms that determine the different phenotype of X‐ALD are not well understood. The studies reviewed here suggest for a “three‐hit hypothesis” for neuropathology of cALD. An improved understanding of the molecular mechanisms associated with these three phases of cALD disease should facilitate the development of effective pharmacological therapeutics for X‐ALD.
Keywords: axonal degeneration, demyelination, neuroinflammation, oxidative stress, peroxisomes, plasmalogens, VLCFA, X‐adrenoleukodystrophy
INTRODUCTION
With an incidence of 1 in 17 000 males, X‐linked adrenoleukodystrophy (X‐ALD, OMIM number 300100) is the most common monogenic leukodystrophy and peroxisomal disorder. It is characterized by axonopathy in the spinal cord, demyelination in the cerebral hemispheres and adrenal insufficiency. X‐ALD patients accumulate saturated, and to a lesser extent, monounsaturated, very long chain fatty acids (VLCFA) in plasma and tissues, most notably in the brain and adrenal cortex 14, 40, 59.
All X‐ALD patients present with mutations in the ABCD1 gene, which encodes a peroxisomal adenosin triphosphate (ATP)‐binding cassette transporter protein (ABCD1, ALDP) (41). ABCD1 has been proposed to play a crucial role in transport of VLCFA, or their Coenzyme A (CoA) derivatives, into peroxisomes. This is based on the VLCFA accumulation and their defective beta oxidation of VLCFA and on the role of the ABCD transporter orthologs in the peroxisomal import of fatty acids in yeast 26, 54. Recent studies support this hypothesis as the overexpression of human ABCD1 in Saccaromices cerevisiaeallows the import of several acyl‐CoA esters of long chain and VLCFA, such as C18:1ω9, C16:0, C22:0, and C24:6ω3, into peroxisomes (60).
X‐ALD is a complex inherited disease where the same mutation in the ALD gene (ABCD1) can lead to clinically very distinct phenotypes, a fatal childhood cerebral ALD (cALD) or the relatively benign adult disease of adrenomyeloneuropathy (AMN) 40, 59.
cALD is characterized by progressive cerebral demyelination with a strong inflammatory response in the white matter leading to neurodegeneration and death often before the patient reaches adolescence.
AMN affects adults (second to fourth decade) and is characterized by a pure myelopathy and peripheral neuropathy. The neuropathological hallmark of AMN is an axonopathy (resembling spastic paraparesis or spastic paraplegia) without significant myelin degeneration or neuroinflammation. However, about 35% of AMN patients subsequently develop cerebral demyelination: these patients share the same poor prognosis as children with cerebral ALD.
Frequently, the different X‐ALD phenotypes occur within the same family as a result of an identical ABCD1 mutation. No phenotype–genotype correlations have been established so far, which suggests that of modifier genes or environmental/epigenetic/stochastic factors modulate the clinical outcome of the disease. The molecular events that trigger the transition from the metabolic phase, which is the accumulation of VLCFA that is already found in utero, to neuroinflammation and demyelination in the brain in cALD or to axonal degeneration in spinal cords in AMN are largely unknown. Moreover, many AMN patients affected by axonal degeneration later develop inflammatory demyelination leading to death for reasons that remain obscure.
Until recently, the generally held view on the pathogenesis of X‐ALD was that the loss of function of ALD protein (ALDP) causes the accumulation of VLCFA, which would be incorporated in an anomalous manner into complex lipids such as gangliosides or phosphatidilcholine 20, 57. These structural alterations in the myelin lipidic components would cause destabilization of myelin sheath and onset of demyelinating pathology. Axonal degeneration could respond to the same cause because integrity of the cellular membrane is a prerequisite for myelin–axonal interaction.
However, the X‐ALD mouse model, a classical knockout of the ABCD1 gene, do not exhibit signs of demyelination in spite of accumulation of VLCFA in the brain and the nervous tissue 16, 37. Instead, ABCD1 null mice presents with a late onset neurodegenerative phenotype with defective motor coordination, locomotor activities and slower nerve conduction velocity at around 20 months of age. First immunohistochemical evidence of axonal degeneration and myelin pathology was detectable in the spinal cord after 16 months of age but not in the brain. Later in life, it was accompanied by microgliosis and astrocytosis 11, 52, 53.
The phenotype of ABCD1‐deficient mice mimics features of human AMN, thus providing a model for investigating the pathogenesis of this disease and for evaluation of efficacy of pharmacological or gene therapeutic approaches (53). The lack of disease progression from the metabolic abnormalities and axonal degeneration to cerebral demyelination and neuroinflammation suggest that in addition to loss of ALDP function and VLCFA excess, it is likely that other factors play a critical role for X‐ALD disease expression that differ in a mouse and a man.
THE CYTOTOXICITY OF VLCFA
The exact mechanism that links the VLCFA excess to axonal degeneration in AMN or to inflammation and demyelination in cALD remains elusive.
VLCFA are usually found as constituents of complex lipids, such as ganglioside, phosphatidilcholine and cholesterol ester fractions of brain myelin, and are also found in the proteolipid protein. In particular, gangliosides and phosphatidilcholine have been proposed as trigger of the immune response in cerebral ALD. Theda and colleagues had observed greatest VLCFA excess (16‐fold higher) accumulated in the phosphatidilcholine fraction in intact regions of brain white matter (57). Recently, highest concentration of VLCFA were also observed in lysophosphatidilcholine (lysoPC) in cALD brain (28). The gangliosides of an X‐ALD brain contain 28% to 50% of fatty acids with chains exceeding 22 carbons (20). Such VLCFA are absent from gangliosides of control individuals. Of note, immunologic properties of gangliosides vary with their fatty acid composition, and they have been implicated in a variety of immunological brain diseases (44).
Studies of VLCFA cytotoxicity using free fatty acids might bring in a limitation for interpretation of data as the largest pool of fatty acids in the cells are not found free but complexed to phospholipids and other lipidic species. Secondly, in addition to eliciting an immune response, this incorporation of VLCFA to complex lipids might destabilize cell membranes. It is well established that hexacosanoic acid (C26:0) alters the physiological properties of membranes. Microcalorimetric studies revealed that inclusion of C26:0 in a model membrane would disrupt its structure (27). Moreover, turnover of C26:0 from phospholipid membranes is 10 000 times slower than that of long and medium‐chain fatty acids (27). Indeed, the microviscosity of erythrocyte membranes was reported to be increased in X‐ALD patients (35), an abnormality that was replicated in adrenocortical cell membranes exhibiting increased microviscosity and a decreased Adrenocorticotropic hormon (ACTH)‐stimulated cortisol secretion as a proof of impaired membrane function (63). Therefore, it is tempting to speculate that similar alterations might be found in the cellular membranes of neural cells and even of membrane of organelles, thus impairing essential functions that would contribute to the pathogenesis of AMN and cALD (reviewed in Powers et al (47)).
Toxicity mechanisms of VLCFA excess were also investigated using various rat primary neural cell cultures reporting changes in the cellular calcium levels, alterations in mitochondrial functions and cell death (25). At 20 mmol/L and higher, VLCFA (C22:0. C24:0 and C26:0) induce cell death of oligodendrocytes and astrocytes. VLCFA induced increase in intracellular calcium concentrations in oligodendrocytes, being this cell type more sensitive than astrocytes and neurons. In a similar setting, the mitochondrial membrane potential was markedly decreased, in particular by C22:0. Mitochondria respiration, in absence of ADP (state 4) or presence of ADP (active respiration, state 3) was also inhibited. Consequently, the authors concluded that VLCFA caused cell death by disturbing mitochondria metabolism and calcium homeostasis. Production of reactive oxygen species (ROS) was not detected in this system, which used normal cells instead of ABCD1 deficient cells.
In contrast, a parallel study using human fibroblasts demonstrated that hexacosanoic acid excess (doses up to 100 uM) generates ROS together with depletion of reduced glutathione and a decrease in mitochondrial membrane potential in absence of cell death (17). Disturbance in mitochondria membrane potential seemed common to both types of cultured cells, neural and fibroblasts. Production of free radicals might lead to oxidative stress when antioxidant defenses are overwhelmed. As an outcome, oxidation damage of macromolecules such as DNA, RNA or proteins lead to detrimental consequences for cell and organisms. Indeed, incubation of fibroblasts with C26:0 excess generated oxidative damage to proteins, arising from metal‐catalyzed oxidation and glycoxidation/lipoxidation, as detected with mass spectrometry techniques. This oxidative damage to proteins occurs in X‐ALD fibroblasts but not in control fibroblasts upon C26:0 excess. Moreover, the loss of ABCD1 gene function also sensitizes fibroblasts to death upon glutathione depletion, in their culture medium, thus indicating that ABCD1 dysfunction alters oxidative stress homeostasis (17). The reversal of oxidative lesions caused by VLCFA excess reversed in vitro by vitamin E offers therapeutic hope as discussed in this symposium (4).
A recent, study has addressed the cytotoxicity of VLCFA complexed to phospholipids in particular lysophosphatidilcholine (lysoPC C24:0) (12). In this study, Eichler and coworkers injected lysoPC C24:0 into brain parietal cortex of mice, causing microglial activation and apoptosis. This toxicity did not occur when lysoPC C16:0 was used, indicating a fatty chain length dependent cytotoxicity. Of note, the C26:0 content of lysoPC fraction in peripheral blood is being currently validated as biomarker for the newborn screening of X‐ALD (29).
THE FIRST HIT: METABOLIC DISEASE OF X‐ALD AND AXONAL DEGENERATION
Underscoring the described in vitro results, evidence of oxidative damage was also documented in human X‐ALD brain: oxidative lesions were found in the form of the lipid peroxidation product HNE (hidroxy‐nonenal) (50). Both AMN and cALD patient had increased lipid peroxidation (TBA‐RS) and decreased tissue nonenzymatic antioxidants (TAR) 7, 61. Recently, damage to proteins has also been reported in peripheral mononuclear cells from X‐ALD patients (19).
Because oxidative stress is involved in a wide variety of neurological disorders, it may be considered a common event in neurodegenerative pathway that occurs in late stages of the disease and is simply epiphenomenal or a consequence of tissue damage. Therefore, the use of antioxidants is being questioned as a valid therapeutic strategy for treating multifactorial diseases in which multiple and parallel pathways contribute to neurodegeneration, in Alzheimer disease, Parkinson disease or multiple sclerosis. In X‐ALD, however, we have recently reported evidence suggesting that oxidative stress is an early etiologic factor contributing to disease pathogenesis in X‐ALD mice do not exhibit an overt locomotor phenotype until 20 months of age in spite of early signs of axonal pathology of APP. Synaptophysin deposition in axonal swellings of spinal cord can be detected at 15 months of age along with ultrastructural signs of myelin and axonal damage in peripheral nerves (52).
Recently, we have also observed oxidative damage to proteins and lipids of the same nature, observed in lesions produced by excess of VLCFA in X‐ALD fibroblasts, as an early biochemical feature of the neurodegenerative cascade in the spinal cord of ABCD1 null mice at 12 months, prior to disease onset. Interestingly, as early as 3.5 months of age, the malonaldehyde‐lysine (MDAL) marker, a consequence of lipoxidative damage to proteins, is detected as the most predominant type of lesion to proteins derived from lipid oxidation adducts (17). Moreover, the degree of VLCFA accumulation parallel the levels of lipoxidative damage to proteins as recently seen in a double knockout mouse deficient for both ABCD1 and ABCD2 genes 19, 53. It correlates with an earlier disease onset of more severe neurodegenerative phenotype of the double mutant mice, with axonal damage, locomotor and behavioral abnormalities and peripheral nerve conduction impairment 13, 53. ABCD1 and ABCD2 share overlapping functions in the beta oxidation of hexacosanoic acid 13, 19 and only double mutants present accumulation of C26:0 in serum (18). However, ABCD2 seems to have higher affinity to other fatty acid species, such as C20:0, C22:1ω9 and the precursor of DHA (C24:6ω3) 18, 36. Of note, the double mutant ABCD1/ABCD2 presents occasionally and at very old ages with inflammatory infiltrates of T‐lymphocytes, a feature absent in ABCD1 or ABCD2 single mutants (53).
In addition to VLCFA abnormality, the functional loss of ABCD1 in X‐ALD results in an additional defective peroxisomal function, the synthesis of plasmalogens (vinyl ether lipids with antioxidant activity). Accordingly, decreased plasmalogens were reported in X‐ALD patient brains along with increased levels of VLCFA 33, 56. Plasmalogen biosynthesis utilizes acyl‐CoA generated by the peroxisomal beta oxidation pathway 23, 24. Presumably, ABCD1 inactivation results in lower amounts of plasmalogens because of lack of substrates (acyl‐CoA). Therefore, metabolic derangements in levels of VLCFA and plasmalogens might act synergistically in increasing oxidative stress in X‐ALD.
As oxidative stress was reported to be directly responsible for axonal degeneration in cultured primary neurons 15, 51 or in vivo in an experimental autoimmune encephalomyelitis (EAE) model (2), it seems reasonable to propose oxidative burden (direct consequence of peroxisomal metabolic impairment) as a first hit in the physiopathogenesis of axonal damage in X‐ALD (Figure 1).
Figure 1.
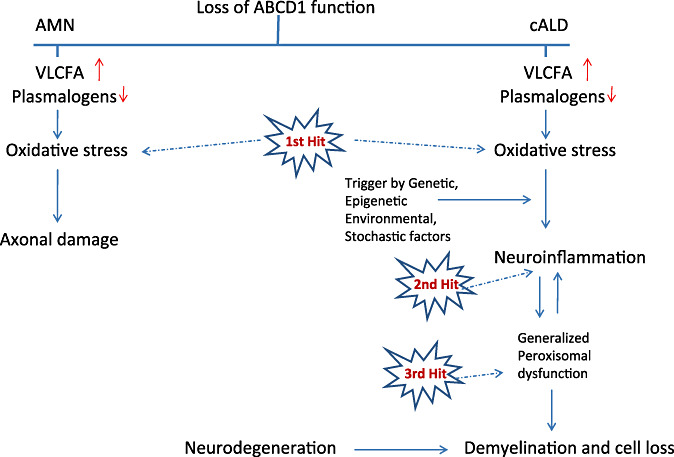
Three‐hit hypothesis on the physiopathogenesis of X‐ALD.
THE SECOND HIT: INDUCTION OF INFLAMMATORY PHENOTYPE OF cALD
The transition from the metabolic disturbance linked to oxidative stress to neuroinflammatory disease of cALD is driven by the “second hit.” The molecular events associated with the transition from a relatively benign metabolic disease to a fatal neuroinflammatory disease in cALD are not well understood at present.
A recent set of data provides some insights into the possible role of ABCD1 dysfunction in triggering of inflammatory response in glial (astrocytes and C6 glial cells) cells. Studies using rat primary astrocytes silenced for ABCD1 and/or ABCD2 reported accumulation of VLCFA, reduced plasmalogens, increased reactive oxygen species production, expression of enzymes of eicosanoid pathway (5‐lipooxygenase, cyclooxygenase‐2) and induction of inflammatory mediators such as cytokines (TNF‐alpha, IL‐1beta) and inducible nitric oxide synthase (iNOS) as well as alterations of peroxisomal metabolism (56). The validity of the relationship between VLCFA accumulation leading to inflammatory response was further established by down regulation of these changes by reducing the VLCFA load by treatment with Lorenzo's oil (mixture of oleic and erucic acids). Lorenzo's oil reduces the levels of VLCFA by competing for enzymes for fatty acid chain elongation. Another study reported that silencing of ABCD3 gene in C6 glial cell line also induces expression of inflammatory mediators (8). Astrocytes silenced for ABCD1 and/or ABCD2 and C6 glial cells silenced for ABCD3 accumulate VLCFA and express proinflammatory mediators suggesting that dysfunction of those transporters induces proinflammatory signaling pathways in these cell types. Further, macrophages (65) from ALD mouse and human X‐ALD‐derived lymphoblasts (58) were also reported to produce higher levels of inflammatory mediators as compared to the respective control cells. These studies document that cells induced to accumulate VLCFA become prone to higher expression and release of inflammatory mediators. But there are still very many unknowns as similar cell types defective for ABCD1 function, in ALD mouse brain, do no produce these inflammatory mediators.
In cALD, the inflammation affects the white matter and related cells 45, 49. In addition the neuropathology of cALD is associated with the infiltration of vascular inflammatory immune cells 22, 48 and NO‐mediated toxicity as evident from the detection of nitrotyrosine in the brain of X‐ALD patients (21). Myelin breakdown products, as a result of myelin instability, containing complex lipids and/or acylated proteins enriched in VLCFA were believed to play a role in this process possibly by inducing CNS resident immune cells to produce cytokines/chemokines that further upregulate the cellular events to induce myelin breakdown 1, 10. However, recent studies with ABC peroxisomal transporter (ABCD1 and/or ABCD2) silenced astrocytes and C6 glial cells silenced for ABCD3 in culture indicate that metabolic derangements, without myelin degradation products, are sufficient to induce inflammatory response (56). The observed expression of proinflammatory cytokines such as TNF‐α, IL‐1β, and IFN‐γ in X‐ALD brain indicates a role for these cytokines in inflammatory demyelination 38, 48. Studies using a combination of super gene array, RT‐PCR, and immunohistochemistry techniques have demonstrated the presence of cytokines (IL‐1beta, TNF‐alpha, IL‐6, IL‐2, IL‐3, and GM‐CF), chemokines (MCP‐1, MCP‐3, MIP‐1b, SDF‐2, fractalkine, eotaxin, MIP‐2, MDC, HCC‐4), chemokine receptors (CCR‐1, CCR‐4, CCR‐5) and iNOS in regions of cALD brains such as the demyelinating plaque and the active edge of the demyelinating plaque compared with histologically unaffected areas of X‐ALD brain and appropriate age‐matched control brains (42). Brain tissue in the inflammatory area had higher levels of proinflammatory cytokines (eg, TNF alpha, IL‐1β, IL‐6, IL‐2) and chemokines (eg, MCP‐1, MCP‐3, SDF‐2). However, histologically normal‐looking areas of cALD brain when compared with control brain also had increased levels of proinflammatory cytokines (TNF‐alpha, IL‐1β, IL‐6, IL‐2) and chemokines (CCL‐2,4,7,11,16,21 and ‐22 and CC‐1,2,4,5), indicating a relationship between metabolic abnormality and inflammatory disease in the absence of vascular infiltrates (42).
As suggested by recent studies, the increased oxidative stress/damage 17, 50 and reduction in plasmalogens 5, 33 participate in the transition of VLCFA‐mediated metabolic disease into neuroinflammatory disease. These conclusions are supported by the following observations: (i) loss of ABCD1 function in cultured astrocytes (56) and in the X‐ALD mouse lead to excessive accumulation of VLCFA and increased oxidative stress/damage 17, 46, 52; (ii) red blood cells and white matter samples of X‐ALD patients contain decreased levels of plasmalogens 5, 33, 39, 64; (iii) and neither ABCD1 null mice nor mice deficient in plasmalogen synthesis develop cerebral demyelination whereas the double mutant do develop microgliosis and moderate myelin abnormalities in CNS (5). These observations indicate that derangements in VLCFA and plasmalogens induce oxidative damage leading to expression of lipolytic enzymes and their products (34) followed by enhanced expression of proinflammatory mediators 33, 56. However, interpretation of studies from mouse mutants lacking plasmalogens deserve some caution as plasmalogen deficiencies have also been described in other neuroinflammatory disorders. Delineation of molecular events required for transition of VLCFA‐induced oxidative stress/damage into neuroinflammatory disease is an active area of investigation. Because over 30% of patients with AMN transition into cerebral ALD, analysis of tissues from these patients may provide clues to events associated with transition of metabolic disease into the neuroinflammatory disease of cALD.
THE THIRD HIT: GENERALIZED LOSS OF PEROXISOMAL FUNCTION AND NEURODEGENERATION IN cALD
The similarities in the neuropathology in the animal model of global peroxisomal dysfunction in oligodendrocytes (the Pex 5 null mice) compared with the double ABCD1/Pex 7 null or to the double ABCD1/ABCD2 null mice suggest that generalized loss of peroxisomal functions may participate in cALD disease pathology 5, 30. Pex 5 and Pex 7 (peroxines) as receptors for peroxisome targeted proteins (PTS1 and PTS2), respectively, play pivotal role in the biogenesis and maintenance of peroxisomes (62).
The inflammatory mediator‐induced downregulation of function of peroxisomes in hepatocytes (3), liver (6), C6 glial cells (31) and CNS of EAE, an animal model of MS (55), indicate a role for decreased peroxisomes in the pathobiology of neuroinflammatory disease of cALD. Second, the cytokine‐mediated compromised peroxisomal integrity, and impaired peroxisomal functions 3, 6, 32 indicate for overall negative consequences for myelin synthesis/repair processes. Further, observed higher levels of VLCFA in demyelinating plaque and demyelinating inflammatory region compared with histologically normal‐looking brain region of cALD brain indicate that peroxisomal functions were altered by the inflammatory mediators (42). Accordingly, cytokine treatment of cultured cells compromised the VLCFA β‐oxidation and other peroxisomal functions 31, 42. Therefore, possibly the decrease in peroxisomes and peroxisomal functions (eg, VLC fatty acids β‐oxidation, plasmalogen and docosahexaenoic acid biosyntheses) in inflammatory regions of X‐ALD brain will further increase the accumulation of VLCFA (31), thus amplifying the inflammatory response (production of cytokines) in brain tissues. As expected, the plaque and inflammatory areas of cALD brain had 2.5‐ and 3.8‐fold greater VLCFA levels compared with histologically normal‐looking areas in cALD brain 31, 42. Indeed, studies from our laboratory using cultured C6 cells demonstrated that cytokine (TNF‐alpha, IL‐1β, INF‐γ) treatment of these cells negatively affect the metabolism of VLCFA such as inhibiting their oxidation and thereby accumulation of these fatty acids (31). By using inhibitors of nitric oxide synthase, NO was identified as one of the intermediates in this inhibition (9). These observations indicate that in addition to downregulation of peroxisomes/proteins, the inflammatory mediators (cytokines) induce inactivation of peroxisomal activities via NO‐mediated mechanisms. Further, the downregulation of peroxisomal proteins/biogenesis under inflammatory disease conditions is supported by the observed decrease in peroxisomes/protein in the liver of rats treated with sublethal doses of endotoxin, a Peroxisome proliferator activated receptor alpha (PPARa)‐dependent mechanism 6, 43. A sublethal injection of bacterial endotoxin produced drastic changes in peroxisomal structure and function. Structural changes were characterized by alternation in membrane lipid composition and hence the fluidity properties 6, 32. Changes in function were associated with a decrease in β‐oxidation of VLCFA. Overall, there was a marked decrease in the population of peroxisomes of normal density (6). These observations indicate a generalized loss of peroxisomes and thereby their functions in the inflammatory regions of CNS suggesting that a generalized loss of peroxisomal function participates in X‐ALD neuropathology. Accordingly, mice in which most peroxisome functions are selectively disrupted by conditional knockout of Pex 5 in oligodendrocytes exhibited not only an accumulation of VLCFA and deficiency of plasmalogens but also modeled the entire neuropathological phenotype of human X‐ALD including axonal degeneration, cerebral demyelination and neuroinflammation (30). Recent report showing deficiency of peroxisomes/peroxisomal dysfunction in neuroinflammatory disease models of EAE, Krabbe disease and cerebral palsy indicate that in addition to cALD and other peroxisomal disorders, inflammatory‐compromised peroxisomal functions may also participate in the pathobiologies in these disease conditions (56).
CONCLUDING REMARKS
In light of recently generated evidences, we propose a three‐hit hypothesis as a framework to study the disease pathogenesis of X‐ALD. The metabolic derangements characterized by excess of VLCFA and lower plasmalogens levels and oxidative stress (first hit), which in turn, generates inflammatory disease (second hit) with the participation of environmental, stochastic, genetic or epigenetic factors. Subsequently, the cytokine and chemokine mediators (inflammatory response) further cause a generalized loss of peroxisomes/peroxisomal functions (third hit), creating a vicious circle resulting in cell loss and progressive inflammatory demyelinating disease. Therefore, therapy for X‐ALD inflammatory disease should also be considered while developing treatment strategies for the metabolic and oxidative stress disease aspects of X‐ALD. Pathophysiological events of ABCD1 dysfunction may vary in different cell types (eg, astrocytes/microglia vs. oligodendrocytes vs. neurons) of CNS and are likely to be dependent on the relative distribution of peroxisomal ABC transporters and cellular metabolic needs for VLCFA by different cell types. Elucidation of cell autonomous vs. noncell autonomous mechanisms are needed to direct future cell therapeutic approaches.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health [NS‐22576 and NS‐37766 to I.S.], European Commission [FP7‐241622], the European Leukodystrophy Association [ELA 2006‐043I4], the Spanish Institute for Health Carlos III [FIS PI080991] and the Autonomous Government of Catalonia [2009SGR85] to A.P. The work was developed under the COST action BM0604 [to A.P.]. The CIBER de Enfermedades Raras is an initiative of the ISCIII.
REFERENCES
- 1. Aubourg P, Dubois‐Dalcq M (2000) X‐linked adrenoleukodystrophy enigma: how does the ALD peroxisomal transporter mutation affect CNS glia? Glia 29:186–190. [DOI] [PubMed] [Google Scholar]
- 2. Basso AS, Frenkel D, Quintana FJ, Costa‐Pinto FA, Petrovic‐Stojkovic S, Puckett L et al (2008) Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest 118:1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beier K, Volkl A, Fahimi HD (1992) Suppression of peroxisomal lipid beta‐oxidation enzymes of TNF‐alpha. FEBS Lett 310:273–276. [DOI] [PubMed] [Google Scholar]
- 4. Berger J, Pujol A, Aubourg P, Forss‐Petter S (2010) Current and future pharmacological treatment strategies in X‐linked adrenoleukodystrophy. Brain Pathol 20:845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brites P, Mooyer PA, El Mrabet L, Waterham HR, Wanders RJ (2009) Plasmalogens participate in very‐long‐chain fatty acid‐induced pathology. Brain 132:482–492. [DOI] [PubMed] [Google Scholar]
- 6. Contreras MA, Khan M, Smith BT, Cimini AM, Gilg AG, Orak J et al (2000) Endotoxin induces structure‐function alterations of rat liver peroxisomes: Kupffer cells released factors as possible modulators. Hepatology 31:446–455. [DOI] [PubMed] [Google Scholar]
- 7. Deon M, Wajner M, Sirtori LR, Fitarelli D, Coelho DM, Sitta A et al (2006) The effect of Lorenzo's oil on oxidative stress in X‐linked adrenoleukodystrophy. J Neurol Sci 247:157–164. [DOI] [PubMed] [Google Scholar]
- 8. Di Benedetto R, Denti MA, Salvati S, Attorri L, Di Biase A (2009) PMP70 knock‐down generates oxidative stress and pro‐inflammatory cytokine production in C6 glial cells. Neurochem Int 54:37–42. [DOI] [PubMed] [Google Scholar]
- 9. Dobashi K, Pahan K, Chahal A, Singh I (1997) Modulation of endogenous antioxidant enzymes by nitric oxide in rat C6 glial cells. J Neurochem 68:1896–1903. [DOI] [PubMed] [Google Scholar]
- 10. Dubois‐Dalcq M, Feigenbaum V, Aubourg P (1999) The neurobiology of X‐linked adrenoleukodystrophy, a demyelinating peroxisomal disorder. Trends Neurosci 22:4–12. [DOI] [PubMed] [Google Scholar]
- 11. Dumser M, Bauer J, Lassmann H, Berger J, Forss‐Petter S (2007) Lack of adrenoleukodystrophy protein enhances oligodendrocyte disturbance and microglia activation in mice with combined ABCD1/Mag deficiency. Acta Neuropathol 114:573–586. [DOI] [PubMed] [Google Scholar]
- 12. Eichler FS, Ren JQ, Cossoy M, Rietsch AM, Nagpal S, Moser AB et al (2008) Is microglial apoptosis an early pathogenic change in cerebral X‐linked adrenoleukodystrophy? Ann Neurol 63:729–742. [DOI] [PubMed] [Google Scholar]
- 13. Ferrer I, Kapfhammer JP, Hindelang C, Kemp S, Troffer‐Charlier N, Broccoli V et al (2005) Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late‐onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum Mol Genet 14:3565–3577. [DOI] [PubMed] [Google Scholar]
- 14. Ferrer I, Aubourg P, Pujol A (2010) General aspects and neuropathology of X‐linked adrenoleukodystrophy. Brain Pathol 20:817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fischer LR, Glass JD (2010) Oxidative stress induced by loss of Cu,Zn‐superoxide dismutase (SOD1) or superoxide‐generating herbicides causes axonal degeneration in mouse DRG cultures. Acta Neuropathol 119:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Forss‐Petter S, Werner H, Berger J, Lassmann H, Molzer B, Schwab MH et al (1997) Targeted inactivation of the X‐linked adrenoleukodystrophy gene in mice. J Neurosci Res 50:829–843. [DOI] [PubMed] [Google Scholar]
- 17. Fourcade S, Lopez‐Erauskin J, Galino J, Duval C, Naudi A, Jove M et al (2008) Early oxidative damage underlying neurodegeneration in X‐adrenoleukodystrophy. Hum Mol Genet 17:1762–1773. [DOI] [PubMed] [Google Scholar]
- 18. Fourcade S, Ruiz M, Camps C, Schluter A, Houten SM, Mooyer PA et al (2009) A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am J Physiol Endocrinol Metab 296:E211–E221. [DOI] [PubMed] [Google Scholar]
- 19. Fourcade S, Ruiz M, Guilera C, Hahnen E, Brichta L, Naudi A et al (2010) Valproic acid induces antioxidant effects in X‐linked adrenoleukodystrophy. Hum Mol Genet [Epub ahead of print; doi:10.1093/hmg/ddq082. [DOI] [PubMed] [Google Scholar]
- 20. Garashi M, Belchis D, Suzuki K (1976) Brain gangliosides in adrenoleukodystrophy. J Neurochem 27:327–328. [DOI] [PubMed] [Google Scholar]
- 21. Gilg AG, Singh AK, Singh I (2000) Inducible nitric oxide synthase in the central nervous system of patients with X‐adrenoleukodystrophy. J Neuropathol Exp Neurol 59:1063–1069. [DOI] [PubMed] [Google Scholar]
- 22. Griffin DE, Moser HW, Mendoza Q, Moench TR, O'Toole S, Moser AB (1985) Identification of the inflammatory cells in the central nervous system of patients with adrenoleukodystrophy. Ann Neurol 18:660–664. [DOI] [PubMed] [Google Scholar]
- 23. Hayashi H, Oohashi M (1995) Incorporation of acetyl‐CoA generated from peroxisomal beta‐oxidation into ethanolamine plasmalogen of rat liver. Biochim Biophys Acta 1254:319–325. [DOI] [PubMed] [Google Scholar]
- 24. Hayashi H, Sato A (1997) Fatty alcohol synthesis accompanied with chain elongation in liver peroxisomes. Biochim Biophys Acta 1346:38–44. [DOI] [PubMed] [Google Scholar]
- 25. Hein S, Schonfeld P, Kahlert S, Reiser G (2008) Toxic effects of X‐linked adrenoleukodystrophy‐associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum Mol Genet 17:1750–1761. [DOI] [PubMed] [Google Scholar]
- 26. Hettema EH, Van Roermund CW, Distel B, Van Den Berg M, Vilela C, Rodrigues‐Pousada C et al (1996) The ABC transporter proteins Pat1 and Pat2 are required for import of long‐chain fatty acids into peroxisomes of Saccharomyces cerevisiae . EMBO J 15:3813–3822. [PMC free article] [PubMed] [Google Scholar]
- 27. Ho JK, Moser H, Kishimoto Y, Hamilton JA (1995) Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest 96:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hubbard WC, Moser AB, Tortorelli S, Liu A, Jones D, Moser H (2006) Combined liquid chromatography‐tandem mass spectrometry as an analytical method for high throughput screening for X‐linked adrenoleukodystrophy and other peroxisomal disorders: preliminary findings. Mol Genet Metab 89:185–187. [DOI] [PubMed] [Google Scholar]
- 29. Hubbard WC, Moser AB, Liu AC, Jones RO, Steinberg SJ, Lorey F et al (2009) Newborn screening for X‐linked adrenoleukodystrophy (X‐ALD): validation of a combined liquid chromatography‐tandem mass spectrometric (LC‐MS/MS) method. Mol Genet Metab 97:212–220. [DOI] [PubMed] [Google Scholar]
- 30. Kassmann CM, Lappe‐Siefke C, Baes M, Brugger B, Mildner A, Werner HB et al (2007) Axonal loss and neuroinflammation caused by peroxisome‐deficient oligodendrocytes. Nat Genet 39:969–976. [DOI] [PubMed] [Google Scholar]
- 31. Khan M, Pahan K, Singh AK, Singh I (1998) Cytokine‐induced accumulation of very long‐chain fatty acids in rat C6 glial cells: implication for X‐adrenoleukodystrophy. J Neurochem 71:78–87. [DOI] [PubMed] [Google Scholar]
- 32. Khan M, Contreras M, Singh I (2000) Endotoxin‐induced alterations of lipid and fatty acid compositions in rat liver peroxisomes. J Endotoxin Res 6:41–50. [DOI] [PubMed] [Google Scholar]
- 33. Khan M, Singh J, Singh I (2008) Plasmalogen deficiency in cerebral adrenoleukodystrophy and its modulation by lovastatin. J Neurochem 106:1766–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khan M, Singh J, Gilg AG, Uto T, Singh I (2010) Very long chain fatty acid accumulation causes lipotoxic response via 5‐lipoxygenase in cerebral adrenoleukodystrophy. J Lipid Res [Epub ahead of print; doi:10.1194/jlr.M002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Knazek RA, Rizzo WB, Schulman JD, Dave JR (1983) Membrane microviscosity is increased in the erythrocytes of patients with adrenoleukodystrophy and adrenomyeloneuropathy. J Clin Invest 72:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu J, Sabeva NS, Bhatnagar S, Li XA, Pujol A, Graf GA (2010) ABCD2 is abundant in adipose tissue and opposes the accumulation of dietary erucic acid (C22:1) in fat. J Lipid Res 51:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu JF, Lawler AM, Watkins PA, Powers JM, Moser AB, Moser HW, Smith KD (1997) A mouse model for X‐linked adrenoleukodystrophy. Proc Natl Acad Sci U S A 94:9366–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGuinness MC, Griffin DE, Raymond GV, Washington CA, Moser HW, Smith KD (1995) Tumor necrosis factor‐alpha and X‐linked adrenoleukodystrophy. J Neuroimmunol 61:161–169. [DOI] [PubMed] [Google Scholar]
- 39. Moser AB, Jones DS, Raymond GV, Moser HW (1999) Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochem Res 24:187–197. [DOI] [PubMed] [Google Scholar]
- 40. Moser H, Smith KD, Watkins PA, Powers J, Moser AB (2001) X‐linked adrenoleukodystrophy. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C (ed.), pp. 3257–3301. McGraw‐Hill: New York. [Google Scholar]
- 41. Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H et al (1993) Putative X‐linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361:726–730. [DOI] [PubMed] [Google Scholar]
- 42. Paintlia AS, Gilg AG, Khan M, Singh AK, Barbosa E, Singh I (2003) Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X‐ALD: implications for potential therapies. Neurobiol Dis 14:425–439. [DOI] [PubMed] [Google Scholar]
- 43. Paintlia MK, Paintlia AS, Contreras MA, Singh I, Singh AK (2008) Lipopolysaccharide‐induced peroxisomal dysfunction exacerbates cerebral white matter injury: attenuation by N‐acetyl cysteine. Exp Neurol 210:560–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pestronk A (1991) Invited review: motor neuropathies, motor neuron disorders, and antiglycolipid antibodies. Muscle Nerve 14:927–936. [DOI] [PubMed] [Google Scholar]
- 45. Powers JM (1995) The pathology of peroxisomal disorders with pathogenetic considerations. J Neuropathol Exp Neurol 54:710–719. [PubMed] [Google Scholar]
- 46. Powers JM (2005) Adreno‐leukodystrophy: a personal historical note. Acta Neuropathol 109:124–127. [DOI] [PubMed] [Google Scholar]
- 47. Powers JM, Moser HW (1998) Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol 8:101–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Powers JM, Liu Y, Moser AB, Moser HW (1992) The inflammatory myelinopathy of adreno‐leukodystrophy: cells, effector molecules, and pathogenetic implications. J Neuropathol Exp Neurol 51:630–643. [DOI] [PubMed] [Google Scholar]
- 49. Powers JM, DeCiero DP, Cox C, Richfield EK, Ito M, Moser AB, Moser HW (2001) The dorsal root ganglia in adrenomyeloneuropathy: neuronal atrophy and abnormal mitochondria. J Neuropathol Exp Neurol 60:493–501. [DOI] [PubMed] [Google Scholar]
- 50. Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, Moser HW et al (2005) Adreno‐leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol 64:1067–1079. [DOI] [PubMed] [Google Scholar]
- 51. Press C, Milbrandt J (2008) Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci 28:4861–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL (2002) Late onset neurological phenotype of the X‐ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet 11:499–505. [DOI] [PubMed] [Google Scholar]
- 53. Pujol A, Ferrer I, Camps C, Metzger E, Hindelang C, Callizot N et al (2004) Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X‐adrenoleukodystrophy. Hum Mol Genet 13:2997–3006. [DOI] [PubMed] [Google Scholar]
- 54. Shani N, Watkins PA, Valle D (1995) PXA1, a possible Saccharomyces cerevisiae ortholog of the human adrenoleukodystrophy gene. Proc Natl Acad Sci U S A 92:6012–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Singh I, Paintlia AS, Khan M, Stanislaus R, Paintlia MK, Haq E et al (2004) Impaired peroxisomal function in the central nervous system with inflammatory disease of experimental autoimmune encephalomyelitis animals and protection by lovastatin treatment. Brain Res 1022:1–11. [DOI] [PubMed] [Google Scholar]
- 56. Singh J, Khan M, Singh I (2009) Silencing of ABCD1 and ABCD2 genes sensitizes astrocytes for inflammation: implication for X‐adrenoleukodystrophy. J Lipid Res 50:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Theda C, Moser AB, Powers JM, Moser HW (1992) Phospholipids in X‐linked adrenoleukodystrophy white matter: fatty acid abnormalities before the onset of demyelination. J Neurol Sci 110:195–204. [DOI] [PubMed] [Google Scholar]
- 58. Uto T, Contreras MA, Gilg AG, Singh I (2008) Oxidative imbalance in nonstimulated X‐adrenoleukodystrophy‐derived lymphoblasts. Dev Neurosci 30:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Valianpour F, Selhorst JJ, Van Lint LE, Van Gennip AH, Wanders RJ, Kemp S (2003) Analysis of very long‐chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab 79:189–196. [DOI] [PubMed] [Google Scholar]
- 60. Van Roermund CW, Visser WF, Ijlst L, Van Cruchten A, Boek M, Kulik W et al (2008) The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl‐CoA esters. FASEB J 22:4201–4208. [DOI] [PubMed] [Google Scholar]
- 61. Vargas CR, Wajner M, Sirtori LR, Goulart L, Chiochetta M, Coelho D et al (2004) Evidence that oxidative stress is increased in patients with X‐linked adrenoleukodystrophy. Biochim Biophys Acta 1688:26–32. [DOI] [PubMed] [Google Scholar]
- 62. Wanders RJ, Waterham HR (2006) Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem 75:295–332. [DOI] [PubMed] [Google Scholar]
- 63. Whitcomb RW, Linehan WM, Knazek RA (1988) Effects of long‐chain, saturated fatty acids on membrane microviscosity and adrenocorticotropin responsiveness of human adrenocortical cells in vitro . J Clin Invest 81:185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wilson R, Sargent JR (1993) Lipid and fatty acid composition of brain tissue from adrenoleukodystrophy patients. J Neurochem 61:290–297. [DOI] [PubMed] [Google Scholar]
- 65. Yanagisawa N, Shimada K, Miyazaki T, Kume A, Kitamura Y, Sumiyoshi K et al (2008) Enhanced production of nitric oxide, reactive oxygen species, and pro‐inflammatory cytokines in very long chain saturated fatty acid‐accumulated macrophages. Lipids Health Dis 7:48. [DOI] [PMC free article] [PubMed] [Google Scholar]