

Abstract
Transplantation of genetically engineered cells into the CNS offers immense potential for the treatment of several neurological disorders. Monitoring expression levels of transgenes and following changes in cell function and distribution over time is critical in assessing therapeutic efficacy of such cells in vivo. We have engineered lentiviral vectors bearing fusions between different combinations of fluorescent and bioluminescent marker proteins and used bioluminescence imaging and intravital-scanning microscopy in real time to study the fate of human neural stem cells (hNSCs) at a cellular resolution in glioma-bearing brains in vivo. Using Renilla luciferase (Rluc)-DsRed2 or GFP-Rluc-expressing malignant human glioma model, transduced hNSCs were shown to migrate extensively toward gliomas, with hNSCs populating gliomas at 10 d after transplantation. Furthermore, transduced hNSCs survived longer in mice with gliomas than in normal brain, but did not modulate glioma progression in vivo. These studies demonstrate the utility of bimodal viral vectors and real-time imaging in evaluating fate of NSCs in diseased models and thus provide a platform for accelerating cell-based therapies for CNS disorders.
Keywords: neural stem cell, bimodal vector, luciferase, fluorescent proteins, glioma, in vivo imaging
Introduction
Neural stem cells (NSCs) are defined by their ability to self-renew and give rise to mature progenitors of neural lineages. The ability of NSCs to migrate to diseased areas of the brain (Snyder and Macklis, 1995; Aboody et al., 2000; Tang et al., 2003; Shah et al., 2005) and their capacity to differentiate into all neural and glial phenotypes (Gage, 2000) provides a powerful tool for targeting the treatment of both diffuse and localized neurologic disorders. Several studies have demonstrated the effectiveness of NSC transplantation in the treatment of neurodegenerative diseases, including spinal cord injury and brain tumors (Snyder and Macklis, 1995; Ehtesham et al., 2002; Lindvall et al., 2004; Hofstetter et al., 2005; Iwanami et al., 2005; Shah et al., 2005). Taking advantage of their homing properties, NSCs have also been modified to deliver selective anti-neoplastic proteins (Ehtesham et al., 2002; Shah et al., 2005), but with mixed results. Although these studies demonstrate the feasibility of NSC-based therapy, cellular delivery of therapeutic proteins via NSC grafts will likely require long-term transgene expression. In vivo assays that permit rapid assessment of the fate of transplanted NSCs, transgene expression, and differentiation are urgently needed to objectively compare therapeutic efficacies of different paradigms.
Several imaging modalities, including magnetic resonance imaging and fluorescence imaging, provide means of tracking transplanted cells in vivo (Lewin et al., 2000; Bulte et al., 2002; Graves et al., 2004), but these techniques are often constrained by limited sensitivity and/or retention of the label (Jendelova et al., 2004; Lee et al., 2004). Previously, we have used dual bioluminescence imaging to demonstrate that mouse NSCs modified to express firefly luciferase (Fluc), implanted into the contralateral hemisphere of murine brain parenchyma with established gliomas, migrate across the brain toward gliomas (Tang et al., 2003; Shah et al., 2005). Although bioluminescence imaging is an efficient method for longitudinal comparison of cell survival and migration, it does not offer the spatial resolution to track single-cell migration in vivo. Laser-scanning microscopic methods (e.g., confocal and multiphoton microscopy) are becoming more widely used to image cellular details in vivo. Confocal and multiphoton microscopy permit in vivo detection of fluorescent reporter proteins within intact tissue and have been used to monitor numerous molecular events, such as recruitment, migration, and survival of single cells (Mempel et al., 2004). In the brain, intravital confocal microscopy (IVM) has been used to visualize vascular microenvironment in gliomas (Vajkoczy et al., 2003; Vajkoczy and Menger, 2004; Farhadi et al., 2005), but has not been applied to monitor stem cell survival or migration in tumor models.
A second vital aspect in successfully imaging the fate of implanted cells is to achieve robust and prolonged reporter gene expression. Lentiviral vectors (LVs) have been used in several studies to genetically modify NSCs ex vivo because these viruses allow stable integration of the transgene into the host genome regardless of their state of division (Steffen and Weinberg, 1978; Naldini et al., 1996). A number of reports have indicated that lentiviral vectors are also resistant to stem cell-specific gene silencing in various types of stem cells compared with MLV-based retrovirus vectors (Lois et al., 2002; Pfeifer et al., 2002). In the current study, we have engineered a variety of lentiviral vectors to express fusions between fluorescent and bioluminescent proteins and show that both fusion partners are fully functional. Using these vectors, we show that human neural stem cells (hNSCs) can be efficiently transduced and tracked by dual bioluminescence imaging and IVM. Using these methods, we study hNSC kinetics and migration to malignant brain tumors. To our knowledge, this is the first report describing the real-time fate of human NSCs in a CNS disease model at a cellular resolution.
Materials and Methods
Generation of fluorescent/bioluminescent lentiviral vectors.
We used a CS-CGW transfer plasmid-based lentiviral vector system (kindly provided by Dr. Esteves, Massachusetts General Hospital, Harvard Medical School) (Miyoshi et al., 1998; Sena-Esteves et al., 2004). To create the LV-GFP-Fluc construct, the cDNA sequences encoding GFP and Fluc were amplified by PCR using the pEGFP-N1 vector (Clontech, Mountain View, CA) and pGL3-Basic (Promega, Madison, WI), respectively, as templates. All the restriction enzyme sequences were incorporated into the primer sequence, and in addition, a 4 aa linker sequence was incorporated into the Fluc forward primer. The resulting GFP fragment was digested with NheI and BamHI, and Fluc fragment was digested with BamHI/XhoI. Both fragments were ligated in-frame into NheI/XhoI-digested CS-CGW plasmid. Similarly, we have created LV-GFP-Renilla luciferase (Rluc), LV-Fluc-DsRed2, and LV-Rluc-DsRed2 lentiviral vectors. To create LV-Lacz vector, the cDNA sequence encoding Lacz was amplified by PCR, and the resulting Lacz fragment was digested with NheI and XhoI and ligated in-frame into NheI/XhoI-digested CS-CGW plasmid. Lentiviral vectors were produced by transient transfection of 293T cells. Cells (15 × 106) were seeded in 150 mm2 tissue culture plates 24 h before transfection in DMEM with 10% FBS, and cells were refed with fresh medium 4 h before transfection. Transfection was performed by calcium phosphate precipitation method using 18 μg of transfer plasmid DNA, transfer vectors constructed above, and the lentiviral helper plasmids pCMVΔ8.91 (18 μg) and glycoprotein expression plasmid pVSVG (12 μg; Clontech). Cells were washed with fresh medium 16–18 h after transfection, and vector supernatants were harvested ∼48 h after transfection. The supernatants were filtered (0.45 μm) and loaded in a Beckman Quick-Seal ultracentrifuge tube (Beckman Coulter, Fullerton, CA) and centrifuged at 28,000 × g for 90 min. Pellets were resuspended in PBS and stored at −80°C. Titers of the GFP- and DsRed2-expressing vectors were determined by counting fluorescent transduced 293T cells. For titering Lacz vectors, cells were stained with X-gal for 6 h, and the number of Lacz-expressing cells was counted.
Cell lines and cell culture.
Mouse C17.2 neural stem cells (mNSCs) were cultured as described previously (Shah et al., 2005). hNS1 is a human fetal neural stem cell line (hNSC) derived from the human diencephalic and telencephalic regions of 10–10.5 weeks gestational age from an aborted human Caucasian embryo. hNSCs were immortalized using retrovirally transduced v-myc (Villa et al., 2000). Their in vitro and in vivo properties (including the absence of transformation, clonality, multipotency, stability, and survival) have been described in detail previously (Rubio et al., 2000; Villa et al., 2004; Navarro-Galve et al., 2005). Cells were cultured in 4:1 culturing medium [DMEM/F-12 (Invitrogen, Carlsbad, CA), 0.6% d-glucose (Sigma-Aldrich, St. Louis, MO), 0.5% albumax (Invitrogen), 0.5% glutamine (Invitrogen), recombinant human FGF (40 ng/ml; R & D Systems, Minneapolis, MN), recombinant human EGF (40 ng/ml; R & D Systems), N2 supplements (Invitrogen), and 1% nonessential amino acids (Cellgro; Mediatech, Manassas, VA)] and growth medium [DMEM with 5% fetal bovine serum (Sigma-Aldrich), 1 mm sodium pyruvate (Cellgro; Mediatech), and 26 mm sodium bicarbonate]. For hNSC differentiation, cells were grown in differentiation medium (DMEM/F-12, 0.6% d-glucose, 0.5% albumax, 0.5% glutamine, N2 supplements, and 1% nonessential amino acids) for 2 weeks, and immunohistochemistry was performed as described below. 293T/17 cells (from Dr. David Baltimore, Massachusetts Institute of Technology, Cambridge, MA), and Gli36 human glioma cells (from Dr. Anthony Campagnoni, University of California, Los Angeles, Los Angeles, CA) were grown in DMEM with 10% fetal bovine serum. mNSCs were grown as described previously (Shah et al., 2005). All cells were cultured at 37°C in a humidified atmosphere with 5% CO2 and 1% penicillin/streptomycin (Invitrogen). hNSCs, mNSCs, and Gli36 glioma cells were transduced or cotransduced with lentiviral vectors in a growth medium containing 12 μg/ml polybrene (Thermo Fisher Scientific, Waltham, MA), and 24 h later cells were visualized for GFP or DsRed2 expression by fluorescence microscopy and assayed for luciferase activity by bioluminescence imaging as discussed below.
In vitro bioluminescence.
To determine the correlation between the number of transduced cells and the bioluminescence signal, cells were seeded in different concentrations and substrates for luciferases (1 μg/ml coelenterazine for Rluc and 1.5 μg/ml d-luciferin for Fluc) were added to the medium. Luciferase activity was measured using a cryogenically cooled high-efficiency CCD camera system (Roper Scientific). Each experiment was performed in triplicate.
Immunocytochemistry.
To assess transgene expression and differentiation of hNSCs into terminal cells, GFP-Fluc-expressing hNSCs were fixed, permeabilized, and incubated with anti-nestin monoclonal antibody (clone 10C2 1:200; Millipore Bioscience Research Reagents, Temecula, CA) directed specifically against human nestin, for 1 h at 37°C. Cells were then washed and incubated with goat anti-mouse Alexa dye 496 nm-conjugated secondary antibody (Invitrogen) for 1 h, then washed, mounted, and examined microscopically. Nontransduced hNSCs and hNSCs transduced with LV-Lacz were seeded on poly-l-lysine (50 μg/ml; Sigma-Aldrich)-coated glass coverslips at a density of 2 × 104 cells/cm2. After 24 h of proliferation, the growth factors were withdrawn and cells were allowed to differentiate for 7 d. Cultures were fixed, permeabilized, and incubated overnight at 4°C with mouse monoclonal antibodies against βIII-tubulin (1:2000; Sigma-Aldrich) and rabbit antiserum against glial fibrillary acidic protein (GFAP) (1:200; Dako, Carpinteria, CA). On the next day, the cultures were incubated with CY3-conjugated antibody (goat anti mouse; 1:500; Jackson ImmunoResearch Laboratories, West Grove, PA) or CY5-conjugated antibody (donkey anti-rabbit; 1:300; Jackson ImmunoResearch Laboratories). Cell nuclei were counterstained with Hoescht 33258 (Invitrogen) at 0.2 μg/ml in PBS and detected as described above.
Cell transplantation.
Athymic nude mice (nu/nu; 6–7 weeks old) and SCID mice (6–8 weeks old; Charles River Laboratories, Wilmington, MA) were stereotactically implanted with LV-transduced Gli36 glioblastoma cells and mNSCs or hNSCs. All animal protocols were approved by an institutional review board. Five sets of experiments were performed: (1) For correlation studies, GFP-Fluc or Rluc-DsRed2 hNSCs (n = 4 in each case) were implanted stereotaxically in the right frontal lobe [from bregma, anteroposterior (AP), −2 mm; mediolateral (ML), 2 mm; ventral (V; from dura), 2 mm] in varying concentrations (1 × 103 to 1 × 105), and 2 d later mice were imaged for Fluc or Rluc activity as described below. (2) To compare hNSC and mNSC survival in nude and SCID mice, either GFP-Fluc-expressing hNSCs or mNSCs (5 × 105 in 4 μl of PBS; n = 5 in each case) were implanted stereotaxically in the right frontal lobe [from bregma, AP, −2 mm; ML, 2 mm; V (from dura), 2 mm], and mice were imaged on days 3, 7, and 10 for Fluc activity as described below. (3) To study migration of hNSCs toward gliomas by bioluminescence imaging and hNSC fate, Gli36 or Gli36-Rluc-DsRed2 cells (5 × 104 in 4 μl of PBS) were implanted stereotaxically into the right frontal lobe [from bregma, AP, −2 mm; ML, 2 mm; V (from dura), 1.0 mm] of SCID mice (n = 10), and 2 d later, GFP-Fluc hNSCs (n = 8; 5 × 105 in 4 μl of PBS) or saline (n = 4) were injected into the right frontal lobe [from bregma, AP, −2 mm; ML, 1 mm; V (from dura), 1.0 mm] of tumor-bearing mice. GFP-Fluc hNSCs were also implanted into the same right frontal lobe of non-tumor-bearing mice (n = 4). (4) To study the influence of glioma on hNSC survival in vivo, Gli36-Rluc-DsRed2 cells (5 × 104) were mixed with GFP-Fluc hNSCs (5 × 105) in 4 μl of PBS and implanted stereotactically into the right frontal lobe of nude mice (n = 20), and mice were imaged for hNSC survival and glioma growth as described below. (5) Finally to image cells in vivo, Gli36-GFP-Rluc glioma cells were implanted as described above and 7 d later, a small circular portion of the skull (∼7 mm diameter) was removed, and the dura was gently peeled back from the cortical surface. hNSCs expressing Fluc-DsRed2 (5 × 105 in 4 μl of PBS) were implanted in non-tumor-bearing mice and also in glioma-bearing mice in a close proximity (1 mm lateral) to the gliomas.
In vivo bioluminescence imaging.
Mice were imaged for Rluc activity by injecting coelenterazine (100 μg/animal in 150 μl of saline) intravenously via the tail vein and 5 min later, photon counts were recorded over 5 min using a cryogenically cooled high-efficiency CCD camera system (Roper Scientific, Trenton, NJ). To image Fluc, mice were given intraperitoneal injection of d-luciferin (4.5 mg/animal in 150 μl of saline), and photon counts were recorded 5 min after d-luciferin administration over 5 min. Images were processed and visualized as described previously (Shah et al., 2003). Mice were imaged every 2–5 d for Fluc or Rluc activity.
Intravital fluorescence microscopy.
A prototype multichannel upright laser-scanning fluorescent microscope (IV100; Olympus, Tokyo, Japan) with a custom-designed stage and scanning unit for intravital observations was used (Alencar et al., 2005). The stage was equipped with a heating plate regulated by a thermostat (37°C). Images were acquired with Fluoview imaging software (Olympus). Lasers used for excitation included a 488 nm argon laser, a 561 nm solid-state yellow laser, and a 633 nm HeNe-R laser. Emission signal was filtered using 505–525 nm, 586–615 nm, and 660–730 nm bandpass filters, respectively. Cell migration quantitation was performed using Image J analysis software.
Tissue processing.
Immediately after the last imaging session, mice were killed and brains were immersed in Tissue-Tek (Sakura Finetek, Torrance, CA) on dry ice, and 7 μm coronal brain sections were cut. For TO-PRO-3 (Invitrogen) nucleic acid staining, brain sections on slides were incubated with 1 μm TO-PRO-3 for 10 min, and slides were washed in PBS and mounted for microscopy to be visualized for TO-PRO-3 staining, GFP and DsRed2 fluorescence on a confocal microscope (Bio-Rad, Hercules, CA). For nestin, GFAP, MAP-2, and Ki67 staining, sections were incubated for 1 h in a blocking solution (0.3% BSA, 8% goat serum, and 0.3% Triton X-100) at room temperature, followed by incubation at 4°C overnight with the following primary antibodies diluted in blocking solution: (1) anti-human nestin (clone 10C2; Millipore Bioscience Research Reagents), (2) anti-human GFAP (Millipore Bioscience Research Reagents), and (3) anti-Ki67 (clone MIB-1; Dako) and (4) anti-MAP-2 (Millipore Bioscience Research Reagents). Sections were washed three times with PBS, incubated in the appropriate secondary antibody, and visualized using confocal microscope (LSM Pascal; Zeiss, Thornwood, NY). For quantitative analysis, the number of cells immunoreactive for a specific antigen was counted in at least three nonoverlapping fields in each sample and expressed as a percentage of GFP-positive hNSCs.
Statistical analysis.
Data were analyzed by Student's t test when comparing two groups and by ANOVA, followed by Dunnett's posttest when comparing more than two groups. Data were expressed as mean ± SEM, and differences were considered significant at p < 0.05.
Results
A number of different lentiviral vector constructs encoding bioluminescent and fluorescent fusion proteins were generated (Fig. 1A). To test the efficiency of these vectors, human Gli36 glioma cells and hNSCs were transduced with LVs at a multiplicity of infection (MOI) of 1. All viral vectors exhibited >90% infectivity for both human Gli36 glioma cells and hNSCs (Fig. 1B). Engineered fusion proteins (GFP-Fluc, GFP-Rluc, Fluc-DsRed2, and Rluc-DsRed2) were expressed at high intensity and were retained through several passages of cells. hNSCs transduced with LV-GFP-Fluc retained the characteristics of hNSCs as revealed by their nestin expression and proliferation rate for >2 weeks in culture (percentage of transduced cells: day 3, 98 ± 5%; day 7, 93 ± 4%; day 15, 97 ± 5%) (Fig. 2A,B). Furthermore, transduced hNSCs retained their potential to differentiate into neuronal cells and astrocytes as revealed by βIII-tubulin (percentage of positive cells: transduced, 12 ± 1%; nontransduced, 10 ± 3%) and GFAP staining (percentage of positive cells: transduced, 37 ± 2%; nontransduced, 32 ± 2%) (Fig. 2C–E). These experiments demonstrate the retention of the NSC multipotency in lentiviral-transduced cells.
Figure 1.

Expression of bimodal imaging transgenes using lentiviral vectors. A, A self-inactivating lentiviral system based on HIV-1 (CS-CGW) was used to construct the following vectors: fusion between GFP and Fluc, GFP and Rluc, Fluc and DsRed2, and Rluc and DsRed2 under the CMV promoter. B, hNSCs and glioma cells were transduced in culture with these lentiviral vectors at MOI = 1 and visualized for GFP or DsRed2 fluorescence. Magnification, 20×.
Figure 2.
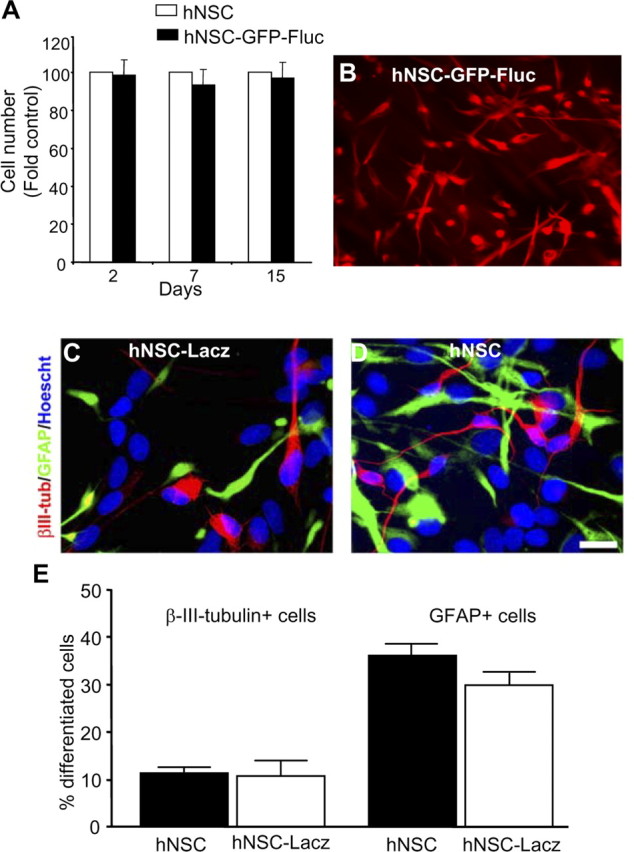
Characterization of transduced hNSCs. A, B, hNSCs were transduced with LV-GFP-Fluc, and growth of transduced and nontransduced cells in culture was compared over 15 d (A); immunohistochemistry was performed with anti-nestin antibody and detected with Alexa-540-conjugated secondary antibody (B). C, D, hNSCs or LV-Lacz transduced hNSCs were differentiated for 7 d, and immunocytochemistry was performed with βIII-tubulin and GFAP antibodies and detected with CY3- or CY5-conjugated antibody. Cell nuclei were counterstained with Hoescht 33258. E, Percentage of βIII-tubulin+ cells and GFAP+ cells. Data represent means ± SEM (n = 4). Magnification: B, 20×; C, D, 40×.
To characterize the activity of luciferase fusion proteins, we performed correlation studies between the cell number and the bioluminescent signal both in culture and in vivo in living mice. Different concentrations of GFP-Fluc and Rluc-DsRed2 hNSCs (ranging from 1 × 104 to 5 × 105) were either plated or implanted into the frontal lobe of nude mice, and bioluminescence imaging was performed. The bioluminescence signal generated from both GFP-Fluc and Rluc-DsRed2 hNSCs in culture (Fig. 3A,B) and in vivo (Fig. 3C,D) correlated linearly with cell number within the ranges tested. This demonstrates that we can quantify the cell number of hNSCs implanted in the mouse brain based on the bioluminescence imaging signal.
Figure 3.

Bioluminescence characteristics of hNSCs. Different concentrations of either NSCs transduced with LV-GFP-Fluc or LV-Rluc-DsRed2 were either plated or implanted intraparenchymally in the mouse brain. A, B, Transduced hNSCs were plated in concentrations ranging from 2 × 104 to 1.5 × 105, and 12 h later, cells were incubated with 150 μg/ml d-luciferin or 1 μg/ml coelenterazine and imaged under the CCD with a scan time of 1 min. C, D, Mice bearing transduced hNSCs were injected either with 4.5 mg/mouse of d-luciferin intraperitoneally or 100 μg/mouse of coelenterazine and imaged under the CCD with a scan time of 5 min. The photon intensities from GFP-Rluc and Rluc-DsRed2-expressing hNSCs both in vitro and in vivo are plotted.
To compare human and mouse NSCs, transduced cells were implanted into the frontal lobe of mice and followed in real time. In vitro bioluminescence imaging revealed similar Fluc expression levels of both mNSCs and hNSCs (Fig. 4A). At all time points studied (3, 7, and 10 d) in grafted nude mice, the bioluminescence signal was higher in mNSCs (day 3,100%; day 7, 92 ± 6%; day 10, 75 ± 5%) than in hNSCs (day 3, 83 ± 4%; day 7, 35 ± 2%; day 10, 17 ± 1%) (Fig. 4B). When hNSCs were implanted in SCID mice, the detected Fluc signal substantially increased (day 3, 88 ± 6%; day 7, 55 ± 3%; day 10, 31 ± 2%) compared with nude mice, but it did not reach the levels obtained for mNSCs in nude mice. Because SCID mice are further immune compromised than nude mice, these results may implicate immune rejection as a factor in hNSC survival in the brain.
Figure 4.

Bioluminescence imaging of NSCs in mice brains. A, hNSCs or mNSCs transduced with LV-GFP-Fluc were plated and 24 h later imaged for bioluminescence. The photon intensities from mNSCs and hNSCs expressing GFP-Fluc are plotted as percentage relative to hNSCs. B, hNSCs or mNSCs expressing GFP-Fluc were implanted intraparenchymally in nude and SCID mice and followed by Fluc bioluminescence imaging on day 3, 7, and 10 d of implantation. One representative image of each mice group on day 10 is shown. The photon intensities from mNSCs and hNSCs expressing GFP-Fluc are plotted as percentage survival relative to mNSCs in nude mice at 3 d (*p < 0.05 vs mNSCs nude).
Next, to determine the effect of gliomas on hNSC survival, we implanted either GFP-Fluc hNSCs or a mix of GFP-Fluc hNSCs and Rluc-DsRed2 glioma cells in the brain parenchyma and followed both the hNSC and glioma burden in real time by dual bioluminescence imaging (Fig. 5A). As shown in the summary graph and representative images (Fig. 5B), there was a considerable increase in hNSC cell numbers in the presence of glioma compared with the hNSCs implanted without glioma cells (day 10 hNSCs: without gliomas, 15 ± 10%; with gliomas, 120 ± 20%; *p < 0.05 vs hNSCs). Although the presence of gliomas influenced hNSC net yield, hNSCs did not influence the proliferation of glioma or changes in the glioma burden over time as revealed by Rluc bioluminescence imaging (Fig. 5C). The presence of a reduced number of GFP-expressing hNSCs in day 7 and 10 compared with day 3 brain sections from mice without gliomas confirmed their reduced survival (Fig. 5Da,Dc,De). Furthermore, the presence of a very small number of GFP-expressing hNSCs in day 3 and 7 brain sections and their absence in day 10 brain sections from deeper tissues confirmed that hNSCs did not migrate deeper into the parenchyma (Fig. 5Db,Dd,Df). These results indicate that glioma cells or host response modulates hNSC survival either through secretion of growth factors or by inhibition of molecules involved in foreign cell rejection.
Figure 5.

hNSC survival in mice bearing gliomas. hNSCs expressing GFP-Fluc alone or mixed with Gli36 glioma cells expressing Rluc-DsRed2 were implanted intraparenchymally in SCID mice, and the presence of hNSCs and glioma cells was imaged by dual bioluminescence imaging. A, Fluc imaging showing the presence of hNSCs and Rluc imaging showing the presence of glioma cells. B, Fluc bioluminescence intensities of hNSCs expressing GFP-Fluc over time. One representative image of mice with hNSCs and with (+) or without glioma cells is shown. The photon intensities are plotted as percentage hNSC survival (*p < 0.05 vs hNSCs). C, Rluc bioluminescence intensities of glioma cells expressing Rluc-DsRed2 implanted with and without hNSCs and plotted as percentage glioma volumes over time. D, Mice brains implanted with hNSCs expressing GFP-Fluc alone were sectioned, and confocal microscopy was performed. Photomicrographs of brain sections at the implantation depth (i.e., 2 mm) (a, c, e) and 3 mm (b, d, f) are shown. Magnification: D, 10×.
To investigate the migratory capabilities of hNSCs toward gliomas when implanted at distinct sites, we used dual bioluminescence imaging and intravital microscopy. GFP-Fluc-expressing hNSCs were implanted at a 1 mm distance from established Gli36-Rluc-DsRed2 gliomas in SCID mice. Bioluminescence imaging (Fig. 6A–C) was indicative of the migration of hNSCs toward gliomas (Fig. 6D). Intravital microscopy on Fluc-DsRed2 hNSCs implanted at a 1 mm distance from established GFP-Rluc gliomas (Fig. 6E) confirmed the robust migration of Fluc-DsRed2 hNSCs toward gliomas at day 7 after hNSC implantation (Fig. 6F; supplemental video, available at www.jneurosci.org as supplemental material). At day 10, hNSCs had completely homed to the glioma site and were seen to penetrate gliomas (Fig. 6G). Quantification of hNSC-DsRed2 fluorescence intensity at the hNSC implantation site and glioma site further confirmed the robust migration of hNSCs within 10 d of implantation (Fig. 6H). Intravital microscopy on non-tumor-bearing mice implanted with Fluc-DsRed2 hNSCs confirmed the reduction of hNSCs in SCID mice (Fig. 6I–K). Fluorescence confocal microscopy on brain sections from mice 10 d after implantation revealed the presence of DsRed2 hNSCs specifically in the GFP-positive gliomas and not in the surrounding normal brain tissue (Fig. 7A–G). For immunohistochemical analysis, brain sections from mice bearing gliomas and implanted with hNSCs expressing GFP-Fluc 10 d after implantation revealed a robust expression of neural stem cell marker nestin in hNSCs (98 ± 0.4%) (Fig. 7H,L,P) and not in the normal mouse brain. Furthermore, only a very small percentage of GFP-expressing hNSCs stained for the proliferation marker Ki-67 (2 ± 0.2%) (Fig. 7I,M,Q). hNSCs did not stain for astrocytic marker GFAP (Fig. 7J,N,R) or neuronal marker MAP-2 (Fig. 7K,O,S). In contrast, robust Ki67 staining was seen in glioma cells (Fig. 7I,M,Q), and GFAP (Fig. 7J,N,R) and MAP-2 (Fig. 7K,O,S) expression was seen in normal brain. These results show that hNSCs implanted in the mice with established gliomas migrate specifically into the gliomas; a majority of them do not proliferate and remain in an undifferentiated state.
Figure 6.
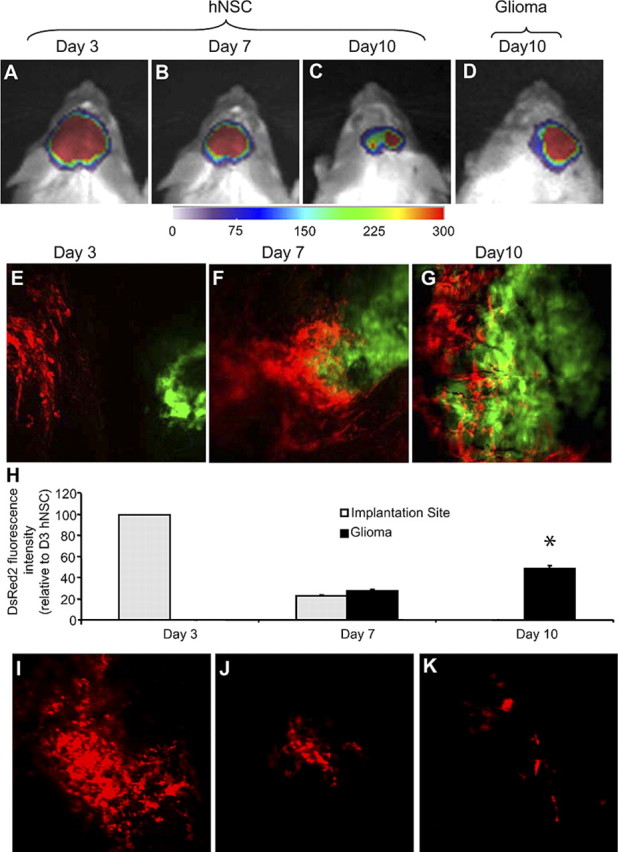
hNSCs migrate into gliomas in vivo. A–D, Bioluminescence imaging of mice implanted with GFP-Fluc-expressing hNSCs in mice with established Rluc-DsRed2 gliomas. Fluc images of mice on day 3 (A), day 7 (B), and day 10 (C) and Rluc image on day 10 (D) are shown. E–G, Intravital microscopy of Fluc-DsRed2 hNSCs implanted in mice with established GFP-Rluc gliomas on days 3 (E), 7 (F), and 10 (G) after hNSC implantation. Magnification, 30×. H, Summary data of hNSC cell density at the site of hNSC implantation and colocalized with established glioma at 3, 7, and 10 d after implantation (*p < 0.05 vs hNSCs at implantation site). I–K, Intravital microscopy of Fluc-DsRed2 hNSCs implanted in non-tumor-bearing mice on days 3 (I), 7 (J), and 10 (K) after hNSC implantation.
Figure 7.
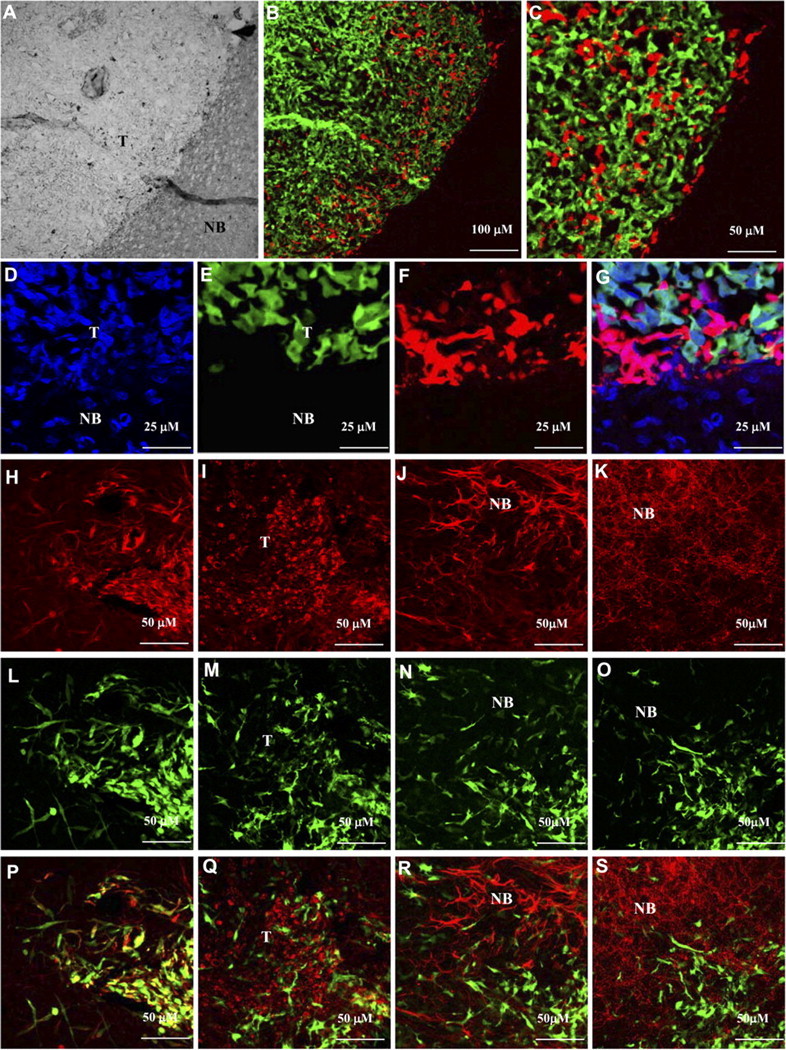
hNSCs are present specifically in the gliomas in an undifferentiated state. Mice implanted with Gli36-GFP-Rluc glioma cells stereotactically into the right frontal lobe were implanted with Fluc-DsRed2 hNSCs 2 d later. Mice were imaged by intravital microscopy (see Fig. 6) and killed on day 10 after hNSC implantation. Mice brains were sectioned, and confocal microscopy was performed. A, Light image showing the normal brain (NB) and the tumor (T). B, Fluorescent image of A showing hNSCs (red) infiltrating the tumor (green); 10× magnification. C, Higher magnification (40×) image of B. D, Brain sections were stained with 1 μm TO-PRO-3 (nucleic acid staining) and confocal microscopy was performed. D, TO-PRO staining (blue). E, GFP-positive glioma cells. F, DsRed2 hNSCs. G, Merge of D–F. H–S, Immunohistochemistry on brain sections from Gli36-glioma-bearing mice implanted with hNSCs expressing GFP-Fluc, 10 d after implantation. Representative images of brain sections immunostained for nestin (H, L, P), Ki67 (I, M, Q), GFAP (J, N, R), and MAP-2 (K, O, S) are shown. Green, GFP expression; red, Ki67, GFAP, or MAP-2 expression; yellow, coexpression of GFP and nestin.
Discussion
We have created a number of lentiviral vectors bearing novel fusions for in vivo multimodality imaging of NSCs. We show that transduced human NSCs have enhanced surviving ability in immune-compromised mice bearing gliomas, migrate efficiently toward established gliomas, and do not effect glioma growth in vivo.
One of the major obstacles in the in vivo evaluation of stem cell therapies has been the challenge to visualize cell populations in the same animal over time. To engineer efficient stem cell therapies, the specifications of the maximal local cell delivery, the integration of delivered cells into the host organ structure, and the long-term survival and potential growth of the delivered cells are of prime importance. An efficient and robust way to follow cells both in culture and in vivo is to transduce them with lentiviral vectors expressing fusions of bioluminescent and fluorescent marker genes. These vectors have the ability to integrate transgenes into the genome of dividing and nondividing cells (Naldini et al., 1996) and provide means of efficient long-term expression in cells and their progeny without using any antibiotic selection marker. The fluorescent marker then serves to determine the efficiency of transduction, and in conjunction with the bioluminescent marker, serve as an in vivo cell tracking protein. Other studies have used lentiviral vectors expressing either the bioluminescent or the fluorescent proteins to transduce neural stem cells in culture and in vivo (Consiglio et al., 2004; Vroemen et al., 2005) and shown that lentivirus transduction of human neural stem cells has no detrimental effect on stem cell differentiation (Okada et al., 2005). In the current studies, we have shown that NSCs and glioma cells transduced with dual imaging lentiviral vectors showed sustained and robust expression of these markers while maintaining the characteristic features of NSCs. A very high expression of both the bioluminescent and fluorescent proteins is attributable to the fact that both the proteins are expressed as a single transcript under the robust CMV promoter. This is in contrast to the majority of other viral vectors, in which bioluminescence imaging reporters are expressed under the CMV promoter and fluorescent reporters are translated via an IRES, which typically leads to bioluminescent proteins being expressed at higher levels than the fluorescent proteins. In the present study, bioluminescence imaging allowed real-time imaging of the fate of NSCs in individual mice over successive time intervals. In contrast to the other studies that have imaged the fate of gliomas and NSCs simultaneously, our current studies give an insight into the survival and influence of hNSCs and glioma cells when implanted in the vicinity of each other. This ability to image both the fate of NSCs and gliomas in vivo is critical in assessing the efficacy of gene delivery and in quantitating therapeutic effects. However, bioluminescent imaging is limited by a lower spatial resolution compared with other modalities such as intravital microscopy. We explored the possibility of using intravital microscopy that offers higher spatial resolution and have shown that the migration of neural stem cells to glioblastomas can be imaged at a single-cell resolution, thus proving highly useful to study migratory speed and to gain a more complete insight into tumoral homing of NSCs. Furthermore, we show that fusions between fluorescent and bioluminescent proteins offer possible means to circumvent limitations of bioluminescence imaging, because combined use of bioluminescence imaging and intravital microscopy can be complementary and can be used to address different questions simultaneously.
We have previously shown that mouse and human NSCs are attractive candidates for delivering therapeutic proteins and can be used to target both the primary tumor mass and invasive tumor foci (Martinez-Serrano and Bjorklund, 1997; Aboody et al., 2000; Martinez-Serrano et al., 2001; Shah et al., 2005). In this study, we demonstrate that hNSCs implanted into mice with an established glioma migrate preferentially toward the glioma (1 mm away), and colocalize with the tumor by 10 d after implantation. Several studies have demonstrated the migratory properties of hNSCs in mice. Aboody et al. (2000) showed that hNSCs implanted in the contralateral hemisphere of mice with a glioma had sufficient migratory properties to cross the corpus callosum and infiltrate the glioma by 1 week after implantation. Rubio et al. (2000), using rats, also demonstrated that hNSCs can colonize the neostriatum from a single, centrally placed deposit in as short time as 1 month. Although the specific factors responsible for mediating hNSC homing to gliomas are still unclear, studies have suggested that factors such as VEGF and SDF-1 are upregulated by gliomas and function as chemoattractants (Hong et al., 2006). Additionally, evidence from Ziu et al. (2006) suggests that the extracellular matrix of gliomas may play a role in NSC migration and survival by creating a permissive environment for NSCs. Although further research will be required to fully understand the factors that regulate NSC migration, noninvasive serial imaging of hNSCs using bioluminescent imaging or IVM may serve as a powerful tool for investigating the migratory signals responsible for the homing property of hNSCs to gliomas.
Our studies reveal the persistence of hNSCs in the brains of tumor-bearing mice compared with normal mice. A number of studies have shown that the tumor microenvironment exerts profound immunosuppressive activity on APCs (antigen-presenting cells) and T-effector cells by secretion of bioactive growth factors and cytokines, such as VEGF, transforming growth factor-h, or IL-10 (interleukin-10) (Maeurer et al., 1995); (Vieweg et al., 2007). Moreover, tumors are infiltrated by Tregs (regulatory T-cells) and MSCs (myeloid suppressor cells) that actively inhibit T-cell responses at the tumor site through direct cell–cell contact (Zou, 2006), secretion of nitric oxide (NO), or reactive oxygen species (Kusmartsev et al., 2004; Vieweg et al., 2007). All these factors favor conditions that allow tumors and might also allow tumor-associated hNSCs in our model to escape immune recognition and foster their proliferation and survival.
In conclusion, we have used engineered LVs and novel imaging methods to dissect the homing properties of neural stem cells and the influence of gliomas on their survival in mouse brains. Using this study as a template, advances can be made in the way neural stem cells can be engineered with therapeutic agents, and both the target (e.g., glioma) and the delivery vehicles (neural stem cells) can be followed at cellular resolution in mouse models of CNS disease.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant P50 CA86355 (R.W., K.S., X.B.), an American Brain Tumor Association grant (K.S.), the Goldhirsh Foundation (K.S.), and NIH Grant P01 CA69246 (R.W.). Work by A.M.-S.'s group was supported by Ministerio de Educación y Ciencia Grant SAF 2004-03405, Foundation La Caixa Grant BM05-22-0, Instituto de Salud Carlos III Grant RD06/0010/0009, and the Foundation Ramon Areces Institutional Grant to the Centro de Biología Molecular Serevo Ochoa. We thank Dr. Pradeep Bhide and Dr. Stephen Yip for their helpful comments.
References
- Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM, Breakefield XO, Snyder EY. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alencar H, Mahmood U, Kawano Y, Hirata T, Weissleder R. Novel multiwavelength microscopic scanner for mouse imaging. Neoplasia. 2005;7:977–983. doi: 10.1593/neo.05376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulte JW, Duncan ID, Frank JA. In vivo magnetic resonance tracking of magnetically labeled cells after transplantation. J Cereb Blood Flow Metab. 2002;22:899–907. doi: 10.1097/00004647-200208000-00001. [DOI] [PubMed] [Google Scholar]
- Consiglio A, Gritti A, Dolcetta D, Follenzi A, Bordignon C, Gage FH, Vescovi AL, Naldini L. Robust in vivo gene transfer into adult mammalian neural stem cells by lentiviral vectors. Proc Natl Acad Sci USA. 2004;101:14835–14840. doi: 10.1073/pnas.0404180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehtesham M, Kabos P, Gutierrez MA, Chung NH, Griffith TS, Black KL, Yu JS. Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2002;62:7170–7174. [PubMed] [Google Scholar]
- Farhadi MR, Capelle HH, Erber R, Ullrich A, Vajkoczy P. Combined inhibition of vascular endothelial growth factor and platelet-derived growth factor signaling: effects on the angiogenesis, microcirculation, and growth of orthotopic malignant gliomas. J Neurosurg. 2005;102:363–370. doi: 10.3171/jns.2005.102.2.0363. [DOI] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Graves EE, Weissleder R, Ntziachristos V. Fluorescence molecular imaging of small animal tumor models. Curr Mol Med. 2004;4:419–430. doi: 10.2174/1566524043360555. [DOI] [PubMed] [Google Scholar]
- Hofstetter CP, Holmstrom NA, Lilja JA, Schweinhardt P, Hao J, Spenger C, Wiesenfeld-Hallin Z, Kurpad SN, Frisen J, Olson L. Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nat Neurosci. 2005;8:346–353. doi: 10.1038/nn1405. [DOI] [PubMed] [Google Scholar]
- Hong X, Jiang F, Kalkanis SN, Zhang ZG, Zhang XP, DeCarvalho AC, Katakowski M, Bobbitt K, Mikkelsen T, Chopp M. SDF-1 and CXCR4 are up-regulated by VEGF and contribute to glioma cell invasion. Cancer Lett. 2006;236:39–45. doi: 10.1016/j.canlet.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Iwanami A, Kaneko S, Nakamura M, Kanemura Y, Mori H, Kobayashi S, Yamasaki M, Momoshima S, Ishii H, Ando K, Tanioka Y, Tamaoki N, Nomura T, Toyama Y, Okano H. Transplantation of human neural stem cells for spinal cord injury in primates. J Neurosci Res. 2005;80:182–190. doi: 10.1002/jnr.20436. [DOI] [PubMed] [Google Scholar]
- Jendelova P, Herynek V, Urdzikova L, Glogarova K, Kroupova J, Andersson B, Bryja V, Burian M, Hajek M, Sykova E. Magnetic resonance tracking of transplanted bone marrow and embryonic stem cells labeled by iron oxide nanoparticles in rat brain and spinal cord. J Neurosci Res. 2004;76:232–243. doi: 10.1002/jnr.20041. [DOI] [PubMed] [Google Scholar]
- Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- Lee IH, Bulte JW, Schweinhardt P, Douglas T, Trifunovski A, Hofstetter C, Olson L, Spenger C. In vivo magnetic resonance tracking of olfactory ensheathing glia grafted into the rat spinal cord. Exp Neurol. 2004;187:509–516. doi: 10.1016/j.expneurol.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Kokaia Z, Martinez-Serrano A. Stem cell therapy for human neurodegenerative disorders-how to make it work. Nat Med. 2004;10(Suppl):S42–S50. doi: 10.1038/nm1064. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Maeurer MJ, Martin DM, Castelli C, Elder E, Leder G, Storkus WJ, Lotze MT. Host immune response in renal cell cancer: interleukin-4 (IL-4) and IL-10 mRNA are frequently detected in freshly collected tumor-infiltrating lymphocytes. Cancer Immunol Immunother. 1995;41:111–121. doi: 10.1007/BF01527407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Serrano A, Bjorklund A. Immortalized neural progenitor cells for CNS gene transfer and repair. Trends Neurosci. 1997;20:530–538. doi: 10.1016/s0166-2236(97)01119-3. [DOI] [PubMed] [Google Scholar]
- Martinez-Serrano A, Rubio FJ, Navarro B, Bueno C, Villa A. Human neural stem and progenitor cells: in vitro and in vivo properties, and potential for gene therapy and cell replacement in the CNS. Curr Gene Ther. 2001;1:279–299. doi: 10.2174/1566523013348562. [DOI] [PubMed] [Google Scholar]
- Mempel TR, Scimone ML, Mora JR, von Andrian UH. In vivo imaging of leukocyte trafficking in blood vessels and tissues. Curr Opin Immunol. 2004;16:406–417. doi: 10.1016/j.coi.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Navarro-Galve B, Villa A, Bueno C, Thompson L, Johansen J, Martinez-Serrano A. Gene marking of human neural stem/precursor cells using green fluorescent proteins. J Gene Med. 2005;7:18–29. doi: 10.1002/jgm.639. [DOI] [PubMed] [Google Scholar]
- Okada S, Ishii K, Yamane J, Iwanami A, Ikegami T, Katoh H, Iwamoto Y, Nakamura M, Miyoshi H, Okano HJ, Contag CH, Toyama Y, Okano H. In vivo imaging of engrafted neural stem cells: its application in evaluating the optimal timing of transplantation for spinal cord injury. FASEB J. 2005;19:1839–1841. doi: 10.1096/fj.05-4082fje. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Ikawa M, Dayn Y, Verma IM. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc Natl Acad Sci USA. 2002;99:2140–2145. doi: 10.1073/pnas.251682798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio FJ, Bueno C, Villa A, Navarro B, Martinez-Serrano A. Genetically perpetuated human neural stem cells engraft and differentiate into the adult mammalian brain. Mol Cell Neurosci. 2000;16:1–13. doi: 10.1006/mcne.2000.0854. [DOI] [PubMed] [Google Scholar]
- Sena-Esteves M, Tebbets JC, Steffens S, Crombleholme T, Flake AW. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods. 2004;122:131–139. doi: 10.1016/j.jviromet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Shah K, Tang Y, Breakefield X, Weissleder R. Real-time imaging of TRAIL-induced apoptosis of glioma tumors in vivo. Oncogene. 2003;22:6865–6872. doi: 10.1038/sj.onc.1206748. [DOI] [PubMed] [Google Scholar]
- Shah K, Bureau E, Kim DE, Yang K, Tang Y, Weissleder R, Breakefield XO. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann Neurol. 2005;57:34–41. doi: 10.1002/ana.20306. [DOI] [PubMed] [Google Scholar]
- Snyder EY, Macklis JD. Multipotent neural progenitor or stem-like cells may be uniquely suited for therapy for some neurodegenerative conditions. Clin Neurosci. 1995;3:310–316. [PubMed] [Google Scholar]
- Steffen D, Weinberg RA. The integrated genome of murine leukemia virus. Cell. 1978;15:1003–1010. doi: 10.1016/0092-8674(78)90284-2. [DOI] [PubMed] [Google Scholar]
- Tang Y, Shah K, Messerli SM, Snyder E, Breakefield X, Weissleder R. In vivo tracking of neural progenitor cell migration to glioblastomas. Hum Gene Ther. 2003;14:1247–1254. doi: 10.1089/104303403767740786. [DOI] [PubMed] [Google Scholar]
- Vajkoczy P, Menger MD. Vascular microenvironment in gliomas. Cancer Treat Res. 2004;117:249–262. doi: 10.1007/978-1-4419-8871-3_15. [DOI] [PubMed] [Google Scholar]
- Vajkoczy P, Blum S, Lamparter M, Mailhammer R, Erber R, Engelhardt B, Vestweber D, Hatzopoulos AK. Multistep nature of microvascular recruitment of ex vivo-expanded embryonic endothelial progenitor cells during tumor angiogenesis. J Exp Med. 2003;197:1755–1765. doi: 10.1084/jem.20021659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieweg J, Su Z, Dahm P, Kusmartsev S. Reversal of tumor-mediated immunosuppression. Clin Cancer Res. 2007;13:727s–732s. doi: 10.1158/1078-0432.CCR-06-1924. [DOI] [PubMed] [Google Scholar]
- Villa A, Snyder EY, Vescovi A, Martinez-Serrano A. Establishment and properties of a growth factor-dependent, perpetual neural stem cell line from the human CNS. Exp Neurol. 2000;161:67–84. doi: 10.1006/exnr.1999.7237. [DOI] [PubMed] [Google Scholar]
- Villa A, Navarro-Galve B, Bueno C, Franco S, Blasco MA, Martinez-Serrano A. Long-term molecular and cellular stability of human neural stem cell lines. Exp Cell Res. 2004;294:559–570. doi: 10.1016/j.yexcr.2003.11.025. [DOI] [PubMed] [Google Scholar]
- Vroemen M, Weidner N, Blesch A. Loss of gene expression in lentivirus- and retrovirus-transduced neural progenitor cells is correlated to migration and differentiation in the adult spinal cord. Exp Neurol. 2005;195:127–139. doi: 10.1016/j.expneurol.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Ziu M, Schmidt NO, Cargioli TG, Aboody KS, Black PM, Carroll RS. Glioma-produced extracellular matrix influences brain tumor tropism of human neural stem cells. J Neurooncol. 2006;79:125–133. doi: 10.1007/s11060-006-9121-5. [DOI] [PubMed] [Google Scholar]
- Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]