

Abstract
A new route to substituted pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-diones is outlined. The synthesis proceeds via pre-formed hydrazone intermediates, which are then condensed with an activated chlorouracil to build up the entire molecular framework, followed by a reductive ring closure to provide the parent series. The route has been extended to the isomeric pyrimido[5,4-e]-1,2,4-triazine-5,7(6H,8H)-dione class via the use of methylhydrazine as hydrazine surrogate, followed by regiospecific alkylation of the N8-H pyrimidotriazinediones with a range of alkyl and alkaryl substituents. This new methodology permits the generation of a wide range of compounds with variable substitution at the N1, C3, and N8 positions for a heterocyclic scaffold with demonstrated pharmacological activity.
Keywords: pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-diones; pyrimido[5,4-e]-1,2,4-triazine-5,7(6H,8H)-diones; heterocycles; hydrazones; cyclizations
Pyrimido[5,4,e]-1,2,4-triazine-5,7-diones such as the natural products toxoflavin (1) and fervenulin (2) (Figure 1) exhibit antibiotic and other useful pharmacological activity, motivating interest in their exploration since the early 1960s.1 While a variety of analogues have been synthesized and evaluated within both templates, one subclass conspicuously absent is that with aryl substituents at the N1-position, 3, whereas analogues with similar substituents at N8, 4, have been reported.2 We recently have reported on a novel synthesis of compounds related to 1, including hitherto inaccessible N1-phenyl analogues 3.3 In this paper, we report on the further application of this synthesis to a variety of N1-alkyl and N1-aryl compounds, and an efficient process for extending this synthesis to the generation of compounds within the isomeric fervenulin series.
Figure 1.

Structures of natural and synthetic pyrimidotriazinediones
The strategy and range of analogues synthesized within the pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione (toxoflavine) series is shown in Table 1. First, monosubstituted hydrazines 5, either as the free base or hydrochloride salt, were condensed under reflux with aromatic aldehydes 6 in ethanol or tetrahydrofuran to form hydrazones 7, which usually precipitated out of solution upon cooling. The resulting hydrazones 7 were then treated with 6-chloro-3-methyl-5-nitrouracil (8), which had been formed by nitration of commercially available 6-chloro-3-methyluracil according to the published method.4 The addition to 8 required catalysis with aluminum trichloride for those hydrazones formed from arylhydrazines. The resultant 6-(2-arylidene-1-substituted-hydrazinyl)-3-methyl-5-nitrouracils 9 were then cyclized to 3-(4-aryl)-6-methyl-1-substituted-pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-diones 10 upon treatment with zinc dust (4 equiv) and ammonium chloride (2 equiv) while exposed to the air and with vigorous stirring in refluxing 50% aqueous ethanol. The N1-substituted pyrimidotriazinediones were then isolated cleanly, usually in >60% yield, by washing with aqueous 1 M hydrochloric acid. Other solvents and combinations thereof were tried for the zinc-mediated cyclization, but the yields were considerably diminished. Further, additional standard methods were evaluated to reduce the nitro group and promote cyclization. These included standard hydrogenation over palladium on carbon, transfer hydrogenation (Pd/C, ammonium formate), tin in ethyl acetate, tin(II) chloride dihydrate in ethanol, iron in glacial acetic acid, and zinc/hydrochloric acid in ethanol, but none provided clean product.
Table 1.
Substituted Pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-diones 10 from Condensation of 6-Chloro-3-methyl-5-nitrouracil (8) with Hydrazones 7 Followed by Reductive Ring Closure of 9
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar | R | Product | Yielda (%) | Product | Yield (%) from 9 | Overall yield (%) |
| 1 | 4-HOC6H4 | Ph | 9a | 30,b 72c | 10a | 58 | 17,b 41c |
| 2 | 4-MeOC6H4 | Ph | 9b | 41,b 65c | 10b | 78d | 32,b 51c |
| 3 | 4-ClC6H4 | Ph | 9c | 33,b 53c | 10c | 72d | 24,b 38c |
| 4 | 4-HOC6H4 | 4-FC6H4 | 9d | 82c | 10d | 82 | 67 |
| 5 | 4-MeOC6H4 | 4-FC6H4 | 9e | 71c | 10e | 79 | 56 |
| 6 | 5-methyl-2-furyl | 4-FC6H4 | 9f | 38c | 10f | 61 | 23 |
| 7 | 4-MeOC6H4 | 4-BrC6H4 | 9g | 31c | 10g | 76 | 24 |
| 8 | 4-FC6H4 | 4-MeOC6H4 | 9h | 73c | 10h | 40 | 29 |
| 9 | 4-MeC6H4 | (CH2)2OH | 9i | 66e | 10i | 70 | 46 |
| 10 | 3-ClC6H4 | (CH2)2OH | 9j | 68e | 10j | 62 | 42 |
| 11 | 3,4-(MeO)2C6H3 | (CH2)2OH | 9k | 70e | 10k | 56 | 39 |
| 12 | 4-MeOC6H4 | (CH2)2CHMe2 | 9l | 52e | 10l | 62d | 32 |
| 13 | 4-MeOC6H4 | i-Pr | 9m | 80e | 10m | 51 | 41 |
| 14 | various | Me | –e | ||||
Overall yield for first two steps.
Hydrazone isolated in separate step prior to reaction with 8 with AlCl3 catalysis (Method A).
Hydrazone formation in situ and subsequent reaction with 8 with AlCl3 catalysis (Method B).
Ref. 3.
See Table 2. Hydrazone formation in situ and subsequent reaction with 8 without AlCl3 catalysis (Method C).
For Ar and R = aryl, the method was amenable to a variety of substituents on either ring (Table 1, entries 1–8). Both electron-donating and electron-withdrawing substituents were tolerated on either component and there was variation in overall yield depending on the combination of moieties examined. However, the hydrazone formed from 2-pyridylhydrazine was unreactive, perhaps due to catalyst deactivation via complexation of the adjacent pyridyl nitrogen with aluminum trichloride. An attempt to conduct this reaction in the absence of catalyst and at higher temperatures led only to recovered hydrazone.
In optimizing the reaction sequence, it was found that hydrazones 7, where R = aryl, did not need to be isolated in a separate step (Table 1, Method A) and that yields of 9 were often improved by forming the hydrazone in situ. Hence, this became the preferred method. In the improved protocol (Table 1, Method B), 6-chloro-3-methyl-5-nitrouracil (8) and aluminum trichloride were added after 1–2 hours of reaction time between the arylhydrazine 5 and arylaldehyde 6 in an appropriate solvent. After a solvent screen that included standard chlorinated solvents (no reaction) and acetonitrile (modest reaction), tetrahydrofuran was found to provide superior yields. In addition to arylhydrazines, the methodology was investigated for a variety of alkylhydrazines 5 (R = alkyl), and here the hydrazones 7 formed in situ were found to be sufficiently reactive with chlorouracil 8 that activation with aluminum trichloride was unnecessary. Table 1 (Method C; entries 9–13) summarizes reactions for successful alkylhydrazines. In addition to those shown, the hydrazones of methylhydrazine also reacted quickly and efficiently with 8; however, in the subsequent cyclization of 9 to 10 with zinc and ammonium chloride, demethylation at N1 was a significant side reaction. Varying the reaction time and temperature could minimize this, but it was never completely suppressed. Hence, for R = methyl, the preferred method of synthesis would be one of the standard methods commonly used to make N1-methylpyrimidotriazinediones,5 but for most other R substituents, our new methodology is advantageous with regard to both synthesis time and efficiency. Moreover, for Methods B and C, the sequence of in situ formation of hydrazone 7 followed by condensation with chloronitrouracil 8 and subsequent cyclization to 10 can be carried out with yields comparable to or better than when 7 is isolated separately.
There are some shortcomings to this methodology, however. The reaction between hydrazones 7 and 6-chloro-3-methyl-5-nitrouracil (8) was the more general of the last two steps. An example where the desired Michael-type addition to 8 did not occur included the hydrazone of tert-butylhydrazine. For the cyclization of 9 to 10, reaction conditions were robust for aryl and some alkyl substituents at the N1-position. However, problems were encountered for both benzyl and methyl (as described above) substituents. While N1-methylpyrimidotriazinediones could be isolated to some extent, despite the competing demethylation, no desired N1-benzylpyrimidotriazinediones 10 could be detected as debenzylation appeared to rapidly follow cyclization. A similar outcome was observed for R = (CH2)2NEt2 with N1–C bond cleavage likely occurring via an aziridinium intermediate.
The demethylation that occurred when R = Me in the formation of 10 could be used to advantage, however, when the goal was an N8-H pyrimidotriazinedione. While hydrazine itself could be used in the initial condensation with aldehydes 6 to form hydrazones 7 (R = H), subsequent reaction of these with chlorouracil 8 followed by reductive ring closure with zinc and ammonium chloride never provided clean N1(N8)-H pyrimidotriazinediones. Hence, methylhydrazine could be used as a surrogate to attain the same objective. Table 2 lists several examples where 3-aryl-substituted pyrimidotriazinediones were made in this two-pot three-step process. Here, slightly lower yields were obtained in the ring closure/demethylation step when there was an electron-donating substituent at the 4-position of the aryl substituent (Table 2, entries 6 and 7), whereas the highest yields were obtained with the strongly electron-withdrawing fluoro group at the same position (Table 2, entries 3 and 8). Both of these observations are consistent with the ease with which demethylation of N1-methylpyrimidotriazinediones occurred. This involves a likely mechanism of SN2 nucleophilic attack of the N1-methyl group by solvent followed by C–N bond cleavage with the core pyrimidotriazinedione heterocycle serving as a leaving group.
Table 2.
N 8-H Pyrimido[5,4-e]-1,2,4-triazine-5,7(6H,8H)-diones 10 from Methylhydrazine as a Hydrazine Surrogate
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ar | Producta | Yield (%) | Product | Yield (%) from 9 | Overall yield (%) |
| 1 | Ph | 9n | 75 | 10n | 50 | 38 |
| 2 | 3-FC6H4 | 9o | 82 | 10o | 61 | 50 |
| 3 | 4-FC6H4 | 9p | 87 | 10p | 79 | 68 |
| 4 | 4-ClC6H4 | 9q | 78 | 10q | 63 | 49 |
| 5 | 3-MeOC6H4 | 9r | 72 | 10r | 65 | 46 |
| 6 | 4-MeOC6H4 | 9s | 90 | 10s | 43 | 38 |
| 7 | 3-F-4-MeOC6H3 | 9t | 85 | 10t | 47 | 40 |
| 8 | 4-F-3-MeOC6H3 | 9u | 84 | 10u | 86 | 72 |
Hydrazone formation in situ and subsequent reaction with 8 without AlCl3 catalysis (Method C).
The demethylated pyrimidotriazinediones could then be alkylated to produce a variety of novel N8-substituted pyrimidotriazinedione analogues, as shown in Table 3. This reaction, which has been previously documented in the literature,6 can be subjected to conditions that provide either N1- or N8-alkyl products or mixtures thereof. Our conditions, which involve the use of cesium carbonate as base, provide N8 products exclusively for a wide range of alkyl and alkaryl substituents. While yields are variable, these were not optimized except for 11i and 11j.
Table 3.
N8-Substituted Pyrimido[5,4-e]-1,2,4-triazine-5,7(6H,8H)-diones 11 from Regiospecific Alkylation of 10
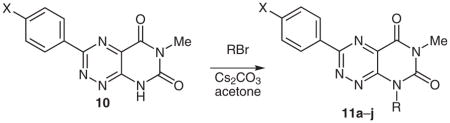 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | X | R | Product | Yield (%) |
| 1 | 10q | Cl | Pr | 11a | 47 |
| 2 | 10s | OMe | (CH2)2NEt2a | 11b | 22 |
| 3 | 10n | H | (CH2)2OH | 11c | 43 |
| 4 | 10n | H | (CH2)2Ph | 11d | 39 |
| 5 | 10p | F | 4-t-BuC6H4CH2 | 11e | 71 |
| 6 | 10q | Cl | 2-FC6H4CH2 | 11f | 57 |
| 7 | 10n | H | 3-FC6H4CH2 | 11g | 66 |
| 8 | 10s | OMe | 4-FC6H4CH2 | 11h | 81 |
| 9 | 10p | F | 3,4-F2C6H3CH2 | 11i | 91 |
| 10 | 10s | OMe | 3,4-F2C6H3CH2 | 11j | 87 |
Chloride reacted instead of bromide.
In summary, we have discovered a facile route to pyrimido[5,4-e]-1,2,4-triazine-5,7-diones with variable substitution at the N1, C3, and N8 positions, including previously undisclosed N1-aryl variants. We also have exemplified the use of methylhydrazine as a surrogate for hydrazine to provide an entry to a variety of N1(N8)-unsubstituted con-geners, which can be mildly and regioselectively functionalized at the N8-position with alkyl and alkaryl halides. We expect this chemistry to find wide application toward the discovery of novel pharmacological agents.
All starting materials were obtained from commercial suppliers and were used without further purification. THF was distilled prior to use over Na/benzophenone. Reactions were run under a blanket of N2 unless specified otherwise. Glassware was oven-dried before use for reactions run under anhydrous conditions. Melting points were determined in open capillary tubes on a Laboratory Devices Mel-Temp apparatus and are uncorrected. The NMR spectra were recorded on a Bruker instrument at 500 MHz for 1H, 125 MHz for 13C, and 470 MHz for 19F spectra. Mass spectra were recorded on a Micromass TofSpec-2E Matrix-Assisted, Laser-Desorption, Time-of-Flight mass spectrometer.
All known compounds had spectroscopic (NMR and MS) and physical properties identical to those given in the literature. There are preparative procedures in the literature for the synthesis of hydrazones 7a,7 7b8 and 7c9 of Table 1, (hydrazinyl)nitropyrimidinediones 9n,10 9q10 and 9s10 of Table 2, pyrimidinotriazines 10n,10 10q10 and 10s10 of Table 2, and alkylated pyrimidinotriazines 11a11 and 11c12 of Table 3.
2-Substituted 1-(4-Arylidene)hydrazines 7; General Procedure for Method A
A mixture of aryl- or alkylhydrazine 5, arylaldehyde 6 (1.1 equiv), and abs EtOH (0.5–0.7 M) was heated at reflux for 90 min. The soln was left standing at r.t. for several hours and the formed precipitate was collected by filtration, rinsed thoroughly with EtOH, and dried. It was used directly in the next step.
1-(4-Chlorobenzylidene)-2-phenylhydrazine (7c)9
Pale peach powder; yield: 290 mg (52%); mp 124–126 °C.
1H NMR (DMSO-d6): δ = 6.76 (t, J = 7.3 Hz, 1 H), 7.07 (d, J = 8.2 Hz, 1 H), 7.22 (dd, J = 7.5 Hz, J′ = 8.1 Hz, 2 H), 7.44 (d, J = 8.2 Hz, 2 H), 7.66 (d, J = 8.5 Hz, 2 H), 7.85 (s, 1 H), 10.45 (s, 1 H).
13C NMR (DMSO-d6): δ = 112.52, 119.42, 127.60, 129.13, 129.59, 132.52, 135.29, 135.43, 145.54.
MS: m/z = 231.1 (M + H).
6-(2-Arylidene-1-substituted-hydrazinyl)-3-methyl-5-nitropy-rimidine-2,4(1H,3H)-diones 9; General Procedure for Method A
Hydrazone 7 was suspended in anhyd THF (0.3M), and treated with AlCl3 (1.0 equiv) and chlorouracil 8 (0.9 equiv). The mixture was heated at reflux for 4 h, during which time it became homogeneous and usually a darker color in appearance. The mixture was cooled on ice for several hours and the thus formed precipitate was collected by filtration, rinsed successively with THF and EtOH, and dried.
6-[2-(4-Chlorobenzylidene)-1-phenylhydrazinyl]-3-methyl-5-nitropyrimidine-2,4(1H,3H)-dione (9c)
Yellow powder; yield: 95 mg (63%); mp 210–212 °C (dec).
1H NMR (DMSO-d6): δ = 3.14 (s, 3 H), 7.42 (d, J = 7.7 Hz, 2 H), 7.50 (m, 3 H), 7.58 (m, 3 H), 7.70 (d, J = 7.7 Hz, 2 H), 11.90 (br s, 1 H).
13C NMR (DMSO-d6): δ = 27.72, 112.52, 122.27, 127.83, 128.78, 129.11, 130.22, 131.17, 132.73, 134.97, 135.29, 142.61, 145.53, 157.75.
MS: m/z = 422.1 (M + Na).
6-(2-Arylidene-1-substituted-hydrazinyl)-3-methyl-5-nitropy-rimidine-2,4(1H,3H)-diones 9 via In Situ Generation of Hydrazone 7; General Procedure for Methods B and C
Method B: A mixture of aryl- or alkylhydrazine 5 and arylaldehyde 6 (1.1 equiv) was dissolved in anhyd THF (0.3 M) and the soln was heated at reflux for 1.5 h. The soln was cooled to r.t, and chlorouracil 8 (0.9 equiv) was added, followed by AlCl3 (1.0 equiv). The mixture was returned to reflux for 4 h and then cooled on ice for several hours. The formed precipitate was collected by filtration, rinsed with THF and EtOH, and dried.
Method C: The same initial steps for Method B were followed, except that the catalyst was omitted. A precipitate began to form within 5–10 min after addition of 8. The mixture was heated for an additional 20–45 min. Further processing as per Method B provided product 9.
6-[2-(4-Fluorobenzylidene)-1-(4-methoxyphenyl)hydrazinyl]-3-methyl-5-nitropyrimidine-2,4(1H,3H)-dione (9h)
Fluorescent yellow powder; yield: 556 mg (73%); mp 228–229 °C.
1H NMR (DMSO-d6): δ = 3.15 (s, 3 H), 3.86 (s, 3 H), 7.14 (d, J = 8.85 Hz, 2 H), 7.23 (dd, J = 8.85 Hz, J′ = 8.85 Hz, 2 H), 7.33 (d, J = 8.8 Hz, 2 H), 7.46 (s, 1 H), 7.75 (dd, J = 8.3 Hz, J′ = 5.7 Hz, 2 H).
13C NMR (DMSO-d6): δ = 27.66, 55.99, 115.83, 124.39, 126.33, 129.68, 130.12, 131.64, 143.71, 147.29, 149.11, 158.83, 160.83, 162.18, 165.48.
19F NMR (DMSO-d6): δ = −107.2.
MS: m/z = 414.1 (M + H).
6-[1-Isopropyl-2-(4-methoxybenzylidene)hydrazinyl]-3-methyl-5-nitropyrimidine-2,4(1H,3H)-dione (9m)
Pastel yellow powder; yield: 74 mg (80%); mp 280–285 °C (dec).
1H NMR (DMSO-d6): δ = 1.40 (d, J = 6.1 Hz, 6 H), 3.20 (s, 3 H), 3.79 (s, 3 H), 4.76 (m, 1 H), 6.97 (d, J = 8.3 Hz, 2 H), 8.15 (d, J = 8.2 Hz, 2 H), 12.25 (s, 1 H).
13C NMR (DMSO-d6): δ = 22.04, 27.57, 49.37, 55.59, 95.54, 113.88, 124.76, 129.86, 143.21, 147.78, 151.18, 158.64, 160.04.
MS: m/z = 362.1 (M + H), 384.0 (M + Na).
6-[2-(4-Fluoro-3-methoxybenzylidene)-1-methylhydrazinyl]-3-methyl-5-nitropyrimidine-2,4(1H,3H)-dione (9u)
Pastel yellow powder; yield: 295 mg (84%); mp 218–222 °C.
1H NMR (DMSO-d6): δ = 3.15 (s, 3 H), 3.41 (s, 3 H), 3.95 (s, 3 H), 7.35 (m, 1 H), 7.55 (m, 1 H), 7.90 (d, J = 8.5 Hz, 1 H), 8.71 (s, 1 H), 11.30 (br s, 1 H).
13C NMR (DMSO-d6): δ = 27.63, 34.98, 56.34, 111.01, 116.49, 122.42, 131.20, 131.65, 142.36, 149.47, 157.62.
19F NMR (DMSO-d6): δ = –129.7.
MS: m/z = 350.1 (M – H).
Anal. Calcd for C14H14FN5O5: C, 47.87; H, 4.02; N, 19.94. Found: C, 47.82; H, 3.89; N, 19.87.
Pyrimidotriazinediones 10; General Procedure
Table 1: To a suspension of 9 in 50% aq EtOH (0.2 M) was added Zn dust (4.0 equiv) and NH4Cl (2.0 equiv). The vigorously stirred suspension was heated at reflux overnight with exposure to the air. The mixture was cooled to r.t. and diluted with aq 1 M HCl (1–3 mL per mmol of Zn used). After stirring at r.t. for 1 h, the formed precipitate was collected by filtration, washed successively with aq 1 M HCl and EtOH, and dried to provide 10.
Table 2: The same procedure as described above was followed except that reaction was carried out in 50% aq DMF (0.08–0.1 M).
1-(4-Fluorophenyl)-3-(4-methoxyphenyl)-6-methylpyrimi-do[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione (10e)
Bright red-orange solid; yield: 188 mg (79%); mp 310–315 °C (dec).
1H NMR (DMSO-d6): δ = 3.29 (s, 3 H), 3.86 (s, 3 H), 7.14 (d, J = 8.8 Hz, 2 H), 7.51 (t, J = 8.7 Hz, 2 H), 7.82 (dd, J1 = 8.8 Hz, J2 = 4.9 Hz, 2 H), 8.14 (d, J = 8.8 Hz, 2 H).
13C NMR (DMSO-d6): δ = 26.03, 52.04, 112.45, 115.67, 117.07, 119.19, 124.06, 124.19, 128.12, 129.45, 144.84, 154.96, 162.24.
19F NMR (DMSO-d6): δ = −105.6.
MS: m/z = 380.1 (M + H), 402.1 (M + Na).
Anal. Calcd for C19H14FN5O3: C, 60.16; H, 3.72; N, 18.46. Found: C, 59.88; H, 3.50; N, 18.19.
1-(2-Hydroxyethyl)-6-methyl-3-(4-methylphenyl)pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione (10i)
Bright reddish-orange powder; yield: 57 mg (70%); mp 199–201 °C (dec).
1H NMR (DMSO-d6): δ = 2.41 (s, 3 H), 3.34 (s, 3 H), 3.93 (t, J = 5.2 Hz, 2 H), 4.53 (t, J = 5.3 Hz, 2 H), 4.98 (br s, 1 H), 7.39 (d, J = 7.3 Hz, 2 H), 8.21 (d, J = 7.3 Hz, 2 H).
13C NMR (DMSO-d6): δ = 21.50, 28.75, 57.15, 58.09, 127.07, 130.28, 132.64, 141.67, 146.92, 149.95, 151.75, 154.59, 159.49.
MS: m/z = 314.1 (M + H).
3-(4-Methoxyphenyl)-6-methylpyrimido[5,4-e]-1,2,4-triazine-5,7(6H,8H)-dione (10s)10
Mustard yellow powder; yield: 897 mg (43%); mp >325 °C.
1H NMR (TFA-d): δ = 3.55 (s, 3 H), 3.90 (s, 3 H), 7.09 (d, J = 7.2 Hz, 2 H), 8.31 (d, J = 7.2 Hz, 2 H).
13C NMR (TFA-d): δ = 19.49, 45.95, 116.54, 121.91, 132.06, 138.08, 147.51, 150.95, 161.72, 166.36.
MS: m/z = 284.1 (M – H).
8-Alkylpyrimido[5,4-e]-1,2,4-triazine-5,7(6H,8H)-diones 11; General Procedure
A mixture of 3-arylpyrimidotriazinedione 10, Cs2CO3 (1.5 equiv), alkyl halide (1.2 equiv), and acetone (0.1 M) was stirred at r.t. overnight (bromides) or heated to 50 °C in a sealed vessel overnight (chlorides). The mixture was diluted with H2O, and refrigerated for 3–4 h. The resulting precipitate was collected, washed with H2O, and dried to provide 11.
8-(3-Fluorobenzyl)-6-methyl-3-phenylpyrimido[5,4-e]-1,2,4-triazine-5,7(6H,8H)-dione (11g)
Yield: 47 mg (66%); mp 229–232 °C.
1H NMR (DMSO-d6): δ = 3.40 (s, 3 H), 5.60 (s, 2 H), 7.11 (t, J = 7.2 Hz, 1 H), 7.31 (m, 2 H), 7.39 (m, 1 H), 7.63 (d, J = 2.6 Hz, 3 H), 8.43 (m, 2 H).
13C NMR (DMSO-d6): δ = 29.31, 45.42, 114.42, 123.70, 127.67, 129.77, 130.80, 131.99, 134.42, 139.46, 149.93, 150.19, 159.99, 160.20, 160.51.
MS: m/z = 364.1 (M + H).
Acknowledgments
A.J.T. gratefully acknowledges financial support of the American Chemical Society Division of Medicinal Chemistry, Sheila B. Cresswell, and Fred and Dee Lyons Graduate Fellowships. This publication was also made possible by grant number GM007767 from NIGMS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS.
References
- 1.(a) Lacrampe JFA, Connors RW, Ho CY, Richardson A, Freyne EJE, Buijnsters PJJ, Bakker AC. 2004,007,498. WO. 2004; (b) Nagamatsu T, Yamasaki H, Hirota T, Yamato M, Kido Y, Shibata M, Yoneda F. Chem Pharm Bull. 1993;41:362. doi: 10.1248/cpb.41.362. [DOI] [PubMed] [Google Scholar]; (c) Liao T, Baiocchi F, Cheng CC. J Org Chem. 1966;31:900. doi: 10.1021/jo01341a061. [DOI] [PubMed] [Google Scholar]; (d) Levenberg B, Linton SN. J Biol Chem. 1966;241:846. [PubMed] [Google Scholar]
- 2.Nagamatsu T, Yamasaki H, Yoneda F. Heterocycles. 1994;37:1147. [Google Scholar]
- 3.Turbiak AJ, Showalter HDH. Tetrahedron Lett. 2009;50:1996. doi: 10.1016/j.tetlet.2009.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black HT. J Heterocycl Chem. 1987;24:1373. [Google Scholar]
- 5.(a) Nagamatsu T, Yamasaki H, Hirota T, Yamato M, Kido Y, Shibata M, Yoneda F. Chem Pharm Bull. 1993;41:362. doi: 10.1248/cpb.41.362. [DOI] [PubMed] [Google Scholar]; (b) Yoneda F, Nagamatsu T. Chem Pharm Bull. 1975;23:2001. doi: 10.1248/cpb.23.2001. [DOI] [PubMed] [Google Scholar]; (c) Yoneda F, Nagamatsu T, Shinomura K. J Chem Soc, Perkin Trans. 1976;1:713. [PubMed] [Google Scholar]
- 6.(a) Nagamatsu T, Yamasaki H. J Chem Soc, Perkin Trans. 2001;1:130. [Google Scholar]; (b) Yoneda F, Nagamatsu T. J Heterocycl Chem. 1974;11:271. [Google Scholar]; (c) Yoneda F, Nagamatsu T. Tetrahedron Lett. 1973;14:1577. [Google Scholar]
- 7.Dabideen DR, Cheng KF, Aljabari B, Miller EJ, Pavlov VA, Al-Abed Y. J Med Chem. 2007;50:1993. doi: 10.1021/jm061477+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao HC, Resnick P. J Org Chem. 1965;30:2832. [Google Scholar]
- 9.Deng X, Mani NS. J Org Chem. 2008;73:2412. doi: 10.1021/jo7026195. [DOI] [PubMed] [Google Scholar]
- 10.Yoneda F, Nagamatsu T. Synthesis. 1975:177. [Google Scholar]
- 11.Yoneda F, Higuchi M, Nitta Y. J Heterocycl Chem. 1980;17:869. [Google Scholar]
- 12.Yoneda F, Nagamatsu T. J Heterocycl Chem. 1974;11:271. [Google Scholar]