

Abstract
Motivation
Progress in systems biology depends on developing scalable informatics tools to predictively model, visualize, and flexibly store information about complex biological systems. Scalability of these tools, as well as their ability to integrate within larger frameworks of evolving tools, is critical to address the multi-scale and size complexity of biological systems.
Results
Using current software technology, such as self-generation of database and object code from UML schemas, facilitates rapid updating of a scalable expert assistance system for modeling biological pathways. Distribution of key components along with connectivity to external data sources and analysis tools is achieved via a web service interface.
Keywords: Bioinformatics, Biosynthetic, Database, Metabolic, Modeling, Signal transduction, Simulation, Systems biology
58.1 Introduction
Here, we describe Sigmoid, a generative, scalable software infrastructure for systems biology designed to facilitate global modeling of biological systems. If deciphered as an acronym, SIGMOID would translate to SIGnal MOdeling Interface and Database. Here, the term Signal, in a biological sense, would be broadly interpreted. Sigmoid supports the process of cycling between model building, hypothesis generation, biological experimentation, and data gathering, by integrating the hypothesis and discovery phases of the research process.
In Sigmoid, we address the problem of creating a scalable expert assistance system for modeling biological pathways, using current software technology to decrease the difficulty and cost of building the system. The reason for building such a system is to provide computational support to biologists and computational scientists who need to create and explore predictive dynamical models of complex biological systems such as metabolic, gene regulation, or signal transduction pathways in living cells [1].
58.1.1 Overview of the Software Infrastructure
The Sigmoid modeling system core consists of distributed modules implementing: (1) pathway/cell model generation and simulation (Cellerator; [2]), (2) a pathway modeling database (Sigmoid Proper), (3) a Web service-oriented middleware, (4) a World Wide Web model browser, and (5) a graphical user interface (Sigmoid Model Explorer (SME)) friendly to a biologist user. From there, other components have been integrated into the system such as a parameter optimization module and functional connections to compatible external data sources. These modules are organized in a classical three-tier architecture (Fig. 58.1). The back end currently consists of the database, the simulator, and other model manipulators. The GUI front end does not access the back-end modules directly but rather via a Web service middleware module. The extra development overhead introduced by the middle layer is more than compensated by the advantages in terms of distributed computing, performance, flexibility, and scalability. With the exception of rapid model retrieval, the middleware layer brokers all communications between the GUI and the back-end components and also among the back-end components themselves. We have found that storing binary instances of models in a database cache can provide significant improvements in model retrieval times in comparison to full model reconstruction and retrieval through the middleware layer. In the event that the rapid model retrieval interface is not accessible, the system will shift access to the database through the middleware. This infrastructure was created by a close collaboration between bioinformaticians and biologists by having the design of many of the essential software objects and their relationships be visible as implementation proceeded.
Fig 58.1.

Sigmoid three-tier architecture. Separation of modules into a communicating distributed system increases scalability of the architecture. Our simulator is the xCellerator model generator/simulator; the database is Sigmoid (autogenerated from a UML schema); user interface is the Sigmoid Model Explorer (SME)
We have coordinated the development of various software modules in Sigmoid by using the Universal Modeling Language (UML) to diagram the most important biological objects – notably reactions and molecular reactants. This UML diagram is used as a template to automatically generate several parts of Sigmoid, in particular a realization of the Sigmoid pathway modeling database (in SQL) and the corresponding Java object hierarchy along with support files for facilitating the object-relational mapping and end-user documentation. Also the Graphical User Interface (GUI) relies heavily on the Java reflection utility to automatically discover much of what it needs to know about the Sigmoid schema. Thus, there is a guarantee that the software actually implements something very close to the UML construction of biological objects. In addition, coding time for different modules of the system is reduced.
To keep the infrastructure flexible and manageable as it grows, we have resorted to a “generative” approach that seeks to partially automate the generation of both executable code and mathematical models. We have applied this approach to as many of the modules in Fig. 58.1 as possible, starting from high-level inputs such as UML diagrams and reaction notations understandable to noncomputer scientists.
58.2 Methods
58.2.1 Model Generation and Simulation: xCellerator
In order to facilitate the modeling of biochemical reactions, a library of reusable reaction models that can be expressed in a simple higher-level language that specifies the molecular species and the type of reaction is required. Cellerator [2] code is implemented as a Mathematica notebook and designed to facilitate biological modeling via automated equation generation. Sigmoid now supports xCellerator [3], the most recent version of Cellerator.
Many models of molecular interactions have been implemented in xCellerator using different formalisms, such as differential equations or stochastic molecular simulation formalism ranging from the law of mass action and simple Michaelis-Menten models to more complex models of enzyme reactions (e.g., the Monod–Wyman–Changeaux or MWC model for allosteric enzymes [4]) and gene regulation [5]. The list of reaction models continues to expand along with the library of actual pathway models comprising sets of coordinated reactions with parameters derived from the literature whenever possible. In addition, an extended set of enzyme mechanism models for single and multi-substrate, positively and negatively regulated, and allosteric enzymes, called kMech, has been written for xCellerator and continues to develop [6]. Sigmoid currently supports all the available xCellerator and kMech reaction models.
To illustrate xCellerator utility, consider the example of a three-stage catalytic model. This reaction is a composite representation of three reversible reactions; substrate–enzyme complex formation, the conversion of the substrate to product within the complex, and subsequent disassociation of the enzyme–product complex into free enzyme and product. When presented with the correct input notation, xCellerator will translate the symbolic reaction to differential equations. The resulting differential equations and variable definitions are passed to Mathematica where they are solved by the numeric solver function (NDSolve) and time plots generated. See example in Fig. 58.2. The parameters for this enzyme mechanism are stored in the Sigmoid Pathways Database. In short, xCellerator converts symbolic reactions to mathematical equations and solves the corresponding equations.
Fig 58.2.
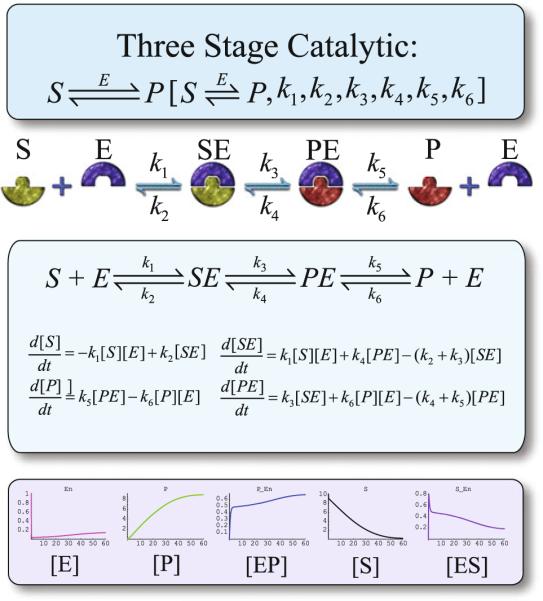
Sigmoid Three Stage Catalytic model. From Top to bottom. xCellerator input notation, reaction cartoon, resulting differential equations and an example of numerical output
58.2.2 Sigmoid Pathway Database
The pathway model database is defined by a UML schema. Comprehensive UML class diagrams of the Sigmoid Schema can be found at http://www.sigmoid.org. The schema is organized into four main diagrams. The first diagram consists of the various top-level container classes such as the Model Class and the Gene Ontology source class. The first diagram also contains the parameter set hierarchy, classes for graphical layout in SME, and various classes to handle units and measures. The three remaining diagrams consist, respectively, of three major class hierarchies: Reactions, Reactants, and Knowledge Sources. Reactions utilize Reactants for their products, substrates, and enzymes; Models are composed of parameterized Reactions, and these three class hierarchies utilize Knowledge Sources to reference external information about themselves.
While initial versions of the Sigmoid database were implemented by hand, we wished to automatically transform the class descriptions contained in the high-level UML diagram of this hierarchy into a set of instantiable objects upon which applications may be built. Our current approach to the process of auto-generating software components from a master UML diagram relies on the capabilities of several existing open-source projects [1]. These pre-existing projects remove much of the core software development responsibilities and allow us to focus on tying them together to produce the specific software products needed for our own use. Object-relational database code autogeneration from UML is itself a contribution of potentially general interest in database software engineering. The current version of Sigmoid is implemented using PostgreSQL, the main Open Source database software.
An essential function of Sigmoid is to assist in the translation of biological knowledge into mathematical form. The representation of Reactions in Sigmoid is aimed at this goal. Sigmoid Reactions represent biochemical processes that transform molecular or other biological objects that are represented as Sigmoid Reactants. A major design feature of Sigmoid is to support translation of biology into mathematics. Reactions are defined in two subhierarchies: Biological Reactions and Mathematical Reactions. The Biological Reaction hierarchy is intended to provide biologically oriented users with symbolic representations of a biochemical reaction or process. Attributes that represent the basic reactants with primary roles are included. The kinetics of the reaction are abstracted out and delegated to Mathematical Reactions. Mathematical Reactions constitute a type hierarchy of mathematical models of reactions or other processes in the Sigmoid schema. Such representations include particular rate laws, as well as the translation of compound reactions into a subnetwork of more elementary reactions, each of which has a more elementary mathematical model. Most Mathematical Reactions currently have direct xCellerator/kMech implementation functions associated with them. Numerical parameters associated with each reaction are contained by reference, which enables key reaction parameters to be shared within a Mathematical Reaction or across a full reaction network.
This way, the Sigmoid architecture can offer explicit support for the translation of biological processes into mathematical process models. Each type of biological reaction may, in principle, be translated into several alternative mathematical reaction models, and each mathematical reaction model can serve as the translation of several different biological reactions. An example of the importance of many-to-many reaction translations is shown in Fig. 58.3.
Fig 58.3.

Simplified version of the Sigmoid Schema Reaction hierarchy. (a) There may exist one to many relations between a particular biological reaction and potential functions (Mathematical Reactions) that may be assigned to model the kinetics of the interaction. For instance, numerous mathematical functions can be assigned to model a catalytic process. (b) In reverse, the functional application of a particular set of differential equations may be conserved over a variety of biological phenomena, so there also may be one to many associations between a particular mathematical function (Reaction) and the biological scenarios it may be applied to. For instance, a hill equation may provide useful in modeling a catalytic reaction, transcriptional regulation, or even a transport process
58.2.3 Sigmoid Web Middleware for Distributed Computing and Web Services
A new distributed Web middleware layer was built which accesses the Sigmoid database and translates reaction sets into the input language of the xCellerator cell model generator. It then calls xCellerator with requests for model generation and simulation and receives output plots in response. All these functions are exposed as Web services available to Java application programs and/or other clients. In addition to load balance and security management, the middleware provides a gateway between the front end and the back end of the architecture, allowing each one to evolve independently as long as the interface to the middleware is properly maintained. Furthermore, the middleware allows scalability in terms of the number of users who can be served simultaneously simply by increasing the computational and database server resources [1].
58.2.4 The Graphical User Interface: Sigmoid Model Explorer User Interface
The last component of the system to be initiated, and the most recent to achieve functional maturity, is the SME Web-compatible Graphical User Interface. The GUI allows the user to visualize, design, edit, and store pathway models, parameters, and initial conditions and their properties, to simulate the models by calling the simulator through the middleware, and to view and compare the properties of simulated models by viewing the temporal evolution of the concentration of chemical species under different conditions. The GUI runs from any Web browser as a Webstart or as a local client program.
Recent enhancements to SME are: (1) For model creation: there exists a new mechanism to create biological models completely from within SME and save them locally or commit them to the database. To facilitate the construction of more complex biological processes, one to many mathematical reactions can be assigned to each biological reaction. Also, there are utilities to facilitate the use of webpages as source of information for data input and perform queries to the Gene Ontology database from within SME. Gene Ontology entities can be either used to tag Sigmoid objects or instantiated directly as Sigmoid objects, i.e., Reactants or Biological reactions. (2) Numerous enhanced display features. (3) Model translation: SME can preform local translation of Sigmoid models to xCellerator code and perform translation of SBML 1.0 to Mathematica code. (4) Model simulation: SME supports simulation through a local Mathematica license using the JLink library as well as through the remote server, and there is an option to retrieve and display the output graphs for intermediate complexes generated by xCellerator/kMech reaction types. (5) Connectivity: SME now supports the Web Services Description Language (WSDL), which is an XML grammar for describing network services. Supporting WSDL expedites adoption of supplementary datasets and functionalities from other systems that support this standard.
58.3 Results
58.3.1 Sigmoid Database Population
The generative version of Sigmoid has been successfully populated with over 20 published models that range from simple molecular interactions to complex cell-fate decision networks. A majority of the models in the database focus on virtual representation of intracellular pathways that include examples in signaling, metabolism, the cell cycle, and gene regulation. Large-scale models of the signaling pathways include the mammalian Epidermal Growth Factor Receptor (EGFR) pathway [7] and the yeast pheromone response pathway [8], while other models represent common aspects of metabolism that include the anabolic Calvin cycle in plants [9], two models of branched chain amino acid biosynthesis in bacteria [10, 11], and catabolic glycolysis [12]. Furthermore, a simple model of the circadian clock [13] and two models of intracellular calcium flux [14] demonstrate oscillating outputs. Separate models of the NFkB [15], calcineurin [16], and the p53 [17] regulatory networks demonstrate how transcription factors and their ability to activate or inhibit gene expression are regulated. Lastly, some models in the database represent diverse processes, including the mechanism of degradation of enzymes during industrial food processing [18] and the cell-fate decisions of protists in the presence of far-red light under starvation conditions [19].
Finally, computational models of the mitogen-activated protein kinase (MAPK) cascade are also present in the Sigmoid database. Several models derived from [20] examine the same MAPK cascade with two separate mechanisms, mass action and Michaelis-Menten, for each of the phosphorylation and dephosphorylation events. For each of these mechanisms, the models increase in complexity as the site and order of phosphorylation are taken into account in the set of reactions. In contrast to these models, Huang_1996_MAPK and its xCellerator notebook “MAPK cascade: Huang and Ferrell 1996,” present the celebrated (1996) model that demonstrates the connection between a nonprocessive, two-collision dual-phosphorylation mechanism of kinase activation and an ultrasensitive, switch-like response. The model Bardwell_ 2007_MAPK_Variable Feedback and the corresponding notebook “MAPK Cascade with Variable Feedback” extend this model to include a simple feedback phosphorylation of an upstream kinase by the MAPK (Fig. 58.4). The effects of the feedback loop on the system depend upon the nature of the feedback: if feedback phosphorylation increases the activity of the upstream kinase (positive feedback), a bistable, all-or-none response may result [21]. In contrast, if feedback phosphorylation decreases the activity of the upstream kinase (negative feedback), then the result may be damped or sustained oscillation of the activity of the kinases in the cascade [22]. The notebook contains examples of parameter values that will generate either of these outcomes, illustrating how complex, diverse, and biologically useful behaviors can emerge from the combination of an ultrasensitive cascade architecture and a simple feedback loop.
Fig 58.4.

Sigmoid Model Explorer showing portion of MAPK pathway. (a) Global Network View; (b) TreeView of compositional hierarchy; (c) network layout visualization; (d) parameter-editing panel; (e) output plot preview panel. Along the top are various action buttons for saving and running the model, and for switching the main panel to view output plots. User can select reaction icons.
Since the flexible but comprehensive schema of the Sigmoid database allows us to easily leverage other databases, we are developing “populator” programs which capture community input from diverse sources and make it available to a biologist end-user in an integrated manner. For example, without much effort we were able to populate Sigmoid with the yeast GOnet database [23], which contains information about yeast ORFs and their annotations, gene ontology (GO), and protein–protein interactions. The GOnet database itself is periodically updated and integrates information from three different sources: (1) ORFs (description, mutant phenotype, gene product, etc.) from the Saccharomyces Genome DataBase (SGD); (2) GO term annotation from the Gene Ontology Consortium arranged in the three categories of Molecular Function, Biological Process, and Cellular Component; and (3) genetic and physical interactions information from the General Repository for Interaction Datasets (GRID).
58.3.2 Parameter Optimization
A Simulated Annealing Optimizer [24] has been integrated into Sigmoid through the web services interface. It uses a global optimization technique and Lam–Delosme schedule to make the optimization process faster and more efficient when compared with other general schedules available [25]. It aims to reverse engineer model parameters (e.g., kinetic rate constants) given both the model structure (represented as ordinary differential equations) and empirical system dynamics as expressed by time series experimental data.
58.3.3 Parameter Analysis
The Parameter Analysis routine in Sigmoid allows one to quickly sample the parameter space of a particular model and quantify the diversity of model outputs resulting from variation of the parameters in specified ranges. First, free parameters are defined within the model that will be part of the analysis. Then, a simulation function is defined that accepts a particular parameter variation and returns the model's output. Users have options to select Sigmoid output functions, such as the temporal sequence of a particular state variable. The output variation is measured using preset or user-defined metrics aimed at focusing on particular aspects of output behavior. For example, one can measure the difference between the obtained output and some reference time state or determine the time points at which the output might have peaks or troughs in an oscillatory response. The value of the metric might reflect on how sensitive a certain model is to simultaneous variation of any number of parameters, from one to all. This information can then be used in investigation of robustness of the model and the corresponding biological process. The values of the varied parameters, model output, and resulting metrics are stored in a database table using Mathematica's Database Link package. Using a database provides a convenient method for storing the vast amounts of tabular data and allows for rapid remote access. Since model evaluations are independent, the procedure is easily parallelized. The same notebook can run on multiple computers simultaneously, as long as all can connect to the same database. Lastly, Mathematica's powerful visualization and analysis features can be used to observe correlations between parameter values and associated metrics. (See Fig. 58.5.)
Fig 58.5.
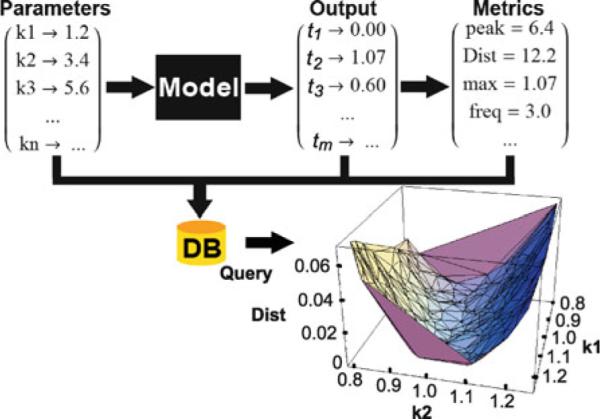
Sensitivity of model output to parameter variations is handled by a set of operations integrated into the Sigmoid environment. These functions or their user-defined variants can allow fast and efficient generation of a set of solutions corresponding to variation of any parameter number from one to all and storage of these solutions in a database that can be queried to form various metrics of model performance. The results can be used to analyze the robustness of various models of a specific biochemical system of interest
58.4 Conclusions
We have described the Sigmoid intelligent software infrastructure for systems biology. A version of each of the main components is available today, and there are clear signs that the infrastructure can already be used to yield biologically relevant results. Since Sigmoid is based upon a computer algebra representation tool, it stands poised to serve as a formidable engine in model analysis. For instance, the Escherichia coli metabolic pathway model correctly predicts the effect of certain mutations, and the MAP Kinase cascade model shows that, depending on the parameter sets and initial conditions chosen, it can generate a switch-like or graded input–output relationship, or even produce oscillatory behavior.
Development and expansion of Sigmoid continues at all levels. As the mediator of the user experience with Sigmoid, the GUI and web interface are bound to attract the largest number of feature requests from users. Because the overall architecture is now functional, many of these requests can be met at reasonable levels of effort and cost. Depending on their accessibility to software agents, an opportunity exists to import relevant data from other sources such as KEGG, Systems Biology Workbench, SiBML/GeneNet, SabioRK, Biomodels etc. There is also a need for new reaction types in xCellerator to deal with various kinds of (nontranscriptional) feedback. Other reaction types already in xCellerator and kMech (such as various enzymatic models, GMWC, GRN, etc.) will need to be exposed for further pathway modeling. An essential aspect of the scale-up of Sigmoid will be expert curation of the allowed and suggested mappings from biological reaction mechanisms to mathematical reaction models.
Sigmoid capitalizes on the robust mathematical software tools and the problem-solving environment that Mathematica offers (along with the xCellerator/kMech packages designed to facilitate biological modeling via automated equation generation). Sigmoid implements the web services framework [1] to create a truly distributed system. This flexible, scalable architecture offers powerful modularity that, in conjunction with the generative nature of the Sigmoid coding cycle, allows for manageable, cost-effective adoption of new system components (new simulation engines, analysis tools, and data structures) while opening the ability to play within yet larger bioinformatics frameworks.
Acknowledgment
This work has been supported by NSF grant EIA-0321390 and NIH grant T15 LM007443 to PB, a Laurel Wilkening faculty innovation award to PB, a UC Systemwide Biotechnology Research and Education Program 2002–2006 award to PB, NIH grant GM069013 to EM, and NCI Director's Challenge support to Children's Hospital Los Angeles for EM. B.C. was supported by NIH grant T15LM07443 from the National Library of Medicine; A.L. was supported by NIH grants: GM69013 and GM072024, KS and TW were supported by NIH P50 grant GM76516, NASA Intelligent Systems Program support of EM, and by the Institute for Genomics and Bioinformatics at UCI. We would like to thank students, programmers, and colleagues who have provided us with valuable feedback or have helped implement particular components of the infrastructure. They include Ben Bornstein, G. Wesley Hatfield, Peter Hebden, Elliot Meyerowitz, Kirill Petrov, Lucas Scharenbroich, Tarek Najdi, Li Zhang, Bruce Shapiro, Diane Trout, and Chin-ran Yang.
Footnotes
Availability: All sigmoid modeling software components and supplementary information are available through: http://www.igb.uci.edu/servers/sb.html.
References
- 1.Cheng J, Scharenbroich L, Baldi P, Mjolsness E. Sigmoid: Towards a generative, scalable software infrastructure for pathway bioinformatics and systems biology. IEEE Intelligent Systems. 2005;20(3):68–75. [Google Scholar]
- 2.Shapiro BE, Levchenko A, Meyerowitz EM, Wold BJ, Mjolsness ED. Cellerator: Extending a computer algebra system to include biochemical arrows for signal transduction simulations. Bioinformatics. 2003;19(5):677–678. doi: 10.1093/bioinformatics/btg042. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro BE, Lu J, Hucka M, Mjolsness E. Mathematica platforms for modeling in systems biology: Recent developments in MathSBML and Cellerator. Poster Abstract G24, Eighth International Conference on Systems Biology (ICSB), Long Beach, 2007; [last accessed on June 29, 2010]. URL http://icsb-2007.org/ [Google Scholar]
- 4.Najdi TS, Yang CR, Shapiro BE, Wesley Hatfield G, Mjolsness ED. The generalized Monod, Wyman, Changeux model for mathematical modeling of metabolic enzymes with allosteric regulation. IEEE Computational Systems Bioinformatics Conference; Stanford, CA: Stanford University; 2005. [DOI] [PubMed] [Google Scholar]
- 5.Segel IH. Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. Wiley; New York, NY: 1992. Enzyme Kinetics. [Google Scholar]
- 6.Yang CR, Shapiro BE, Mjolsness ED, Hatfield GW. An enzyme mechanism language for the mathematical modeling of metabolic pathways. Bioinformatics. 2005;21:774–780. doi: 10.1093/bioinformatics/bti068. [DOI] [PubMed] [Google Scholar]
- 7.Kholodenko BN, Demin OV, Moehren G, Hoek JB. Quantification of short term signaling by the epidermal growth factor receptor. J Biol Chem. 1999;274(42):30169–30181. doi: 10.1074/jbc.274.42.30169. [DOI] [PubMed] [Google Scholar]
- 8.Kofahl B, Klipp E. Modelling the dynamics of the yeast pheromone pathway. Yeast. 2004;21(10):831–850. doi: 10.1002/yea.1122. [DOI] [PubMed] [Google Scholar]
- 9.Poolman MG, Assmus HE, Fell DA. Applications of metabolic modelling to plant metabolism. J Exp Bot. 2004;55(400):1177–1186. doi: 10.1093/jxb/erh090. [DOI] [PubMed] [Google Scholar]
- 10.Najdi TS, Yang CR, Shapiro BE, Hatfield GW, Mjolsness ED. Application of a generalized MWC model for the mathematical simulation of metabolic pathways regulated by allosteric enzymes. J Bioinform Comput Biol. 2006;4(2):335–355. doi: 10.1142/s0219720006001862. [DOI] [PubMed] [Google Scholar]
- 11.Yang CR, Shapiro BE, Hung SP, Mjolsness ED, Hatfield GW. A mathematical model for the branched chain amino acid biosynthetic pathways of Escherichia coli k12. J Biol Chem. 2005;280(12):11224–11232. doi: 10.1074/jbc.M411471200. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen K, Sarensen PG, Hynne F, Busse HG. Sustained oscillations in glycolysis: An experimental and theoretical study of chaotic and complex periodic behavior and of quenching of simple oscillations. Biophys Chem. 1998;72(1–2):49–62. doi: 10.1016/s0301-4622(98)00122-7. [DOI] [PubMed] [Google Scholar]
- 13.Tyson JJ, Hong CI, Thron CD, Novak B. A simple model of circadian rhythms based on dimerization and proteolysis of PER and TIM. Biophys J. 1999;77(5):2411–2417. doi: 10.1016/S0006-3495(99)77078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borghans JM, Dupont G, Goldbeter A. Complex intracellular calcium oscillations. A theoretical exploration of possible mechanisms. Biophys Chem. 1997;66(1):25–41. doi: 10.1016/s0301-4622(97)00010-0. [DOI] [PubMed] [Google Scholar]
- 15.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298(5596):1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 16.Hilioti Z, Gallagher DA, Low-Nam ST, Ramaswamy P, Gajer P, Kingsbury TJ, Birchwood CJ, Levchenko A, Cunningham KW. GSK-3 kinases enhance calcineurin signaling by phosphorylation of RCNS. Genes Dev. 2004;18(1):35–47. doi: 10.1101/gad.1159204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1(1):68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- 18.Brands CM, van Boekel MA. Kinetic modeling of reactions in heated monosaccharidecasein systems. J Agric Food Chem. 2002;50(23):6725–6739. doi: 10.1021/jf011164h. [DOI] [PubMed] [Google Scholar]
- 19.Marwan W. Theory of time-resolved somatic complementation and its use to explore the sporulation control network in Physarum polycephalum. Genetics. 2003;164(1):105–115. doi: 10.1093/genetics/164.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Markevich NI, Hoek JB, Kholodenko BN. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J Cell Biol. 2004;164(3):353–359. doi: 10.1083/jcb.200308060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrell JE, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280:895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 22.Kholodenko BN. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur J Biochem. 2000;267:1583–1588. doi: 10.1046/j.1432-1327.2000.01197.x. [DOI] [PubMed] [Google Scholar]
- 23.Irwin B, Aye M, Baldi P, Beliakova-Bethell N, Cheng H, Dou Y, Liou W, Sandmeyer S. Retroviruses and yeast retrotransposons use overlapping sets of host genes. Genome Res. 2005;15:641–654. doi: 10.1101/gr.3739005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L. PhD thesis. Information and Computer Science: University of California; Irvine: 2008. Dynamic Biological Signaling Pathway Modeling and Parameter Estimation Through Optimization. LD 791.9 I5 2008 Z43, OCLC:276454918. [Google Scholar]
- 25.Lam J, Delosme J. Performance of a New Annealing Schedule. 25th ACM/IEEE DAC.1988. pp. 306–311. [Google Scholar]