

Abstract
Long wavelength voltage-sensitive dyes (VSDs) called Pittsburgh (PGH) dyes were recently synthesized by coupling various heterocyclic groups to a styryl-thiophene intermediate forming extended, partially rigidized chromophores. Unlike most styryl VSDs, dyes with a sulfonic acid anchor directly attached to the chromophore showed no solvatochromic absorption shifts. The limited water solubility of many long wavelength VSDs requires the use of surfactants to transport the dye through aqueous media and effectively label biological membranes. Here, we tested the chemical substitution of the sulfonic acid moiety with polyethyleneglycol (PEG) chains ranging from MW 750 to 5000, to overcome the poor solubility of VSDs while retaining their properties as VSDs. The chemical synthesis of PGH dyes and their PEG derivatives are described. The PEG-derivatives were soluble in aqueous solutions (> 1 mM) and still reported membrane potential changes. In frog and mouse hearts, the voltage sensitivity (ΔF/F per action potential) and spectral properties of PEG dyes were the same as the sulfonated analogs. Thus, the solubility of VSDs can be considerably improved with small polyethyleneglycol chains and can provide an effective approach to improve staining of excitable tissues and optical recordings of membrane potential.
Introduction
Two- and three-dimensional mapping of changes in voltage-sensitive dye (VSD) fluorescence has greatly increased our understanding of the function of nerve networks and the cardiac electrophysiology in normal and pathological states.1-6 Effective VSDs are amphiphilic molecules that contain a water soluble moiety and a lipophilic region. To sense rapid changes in cellular membrane potentials, the sensor portions of these dye molecules associate with the hydrophobic interior of the plasma membrane bilayer where the electric field gradient is the highest. At the same time, the dyes carry charged groups to facilitate transfer of the dyes to the target tissues and to anchor the dyes at cell surfaces, minimizing diffusion of the VSDs to other membranes within the cell.7 Effective imaging deep within tissues requires VSDs with high molar extinction coefficients at red wavelengths and near-IR emission with high quantum yields to avoid intrinsic tissue absorption and fluorescence.6, 8, 9
Styryl dyes are a class of fluorescent VSDs widely used to measure membrane potentials because of their ability to follow voltage changes on a millisecond timescale.3, 6, 10-12 Their structures consist of an electron-rich aminophenyl group linked to a quaternized nitrogen heterocycle by a conjugated polymethine chain. Many styryl dyes, including the series of RH and JPW dyes, have been synthesized with a range of excitation and emission wavelengths and a variety of voltage responses.5, 6, 13, 14 VSDs that have high quantum yields and produce a large change in fractional fluorescence (ΔF/F) are desireable because they generate adequate signals at lower dye concentrations. Recently, Salama et al. reported a series of long-wavelength VSDs designed to report cardiac action potentials (APs) from deeper layers of the heart by incorporating a thienyl group in the polymethine bridge to maintain the rigidity of the styryl chromophore while extending the fluorescence wavelengths.4
Staining of target tissues requires transport of the dye to the plasma membrane and T-tubules in sufficient quantities for voltage measurements. Most styryl VSDs dyes require an additional reagent such as DMSO, Pluronics or cyclodextrins for transport through the aqueous medium to the tissues.9, 15 Labeling conditions are determined for each dye, which can be a time-consuming, trial and error process.4 Attachment of an uncharged hydrophilic anchoring group to the dye structure may eliminate the need for such agents. Poly(ethylene glycols) (PEGs) are commonly used to improve the water-solubility of small molecule pharmaceuticals and exhibit water solubility, high mobility in solution, lack of toxicity, lack of immunogenicity, and are readily cleared from the body.16 Covalent linkage of PEGs to voltage dyes will greatly simplify protocols for probe delivery to excitable tissue.14
In this report, we present the synthesis and spectral characterization of a set of styryl dyes which fluoresce at near infrared wavelengths. The series, PGH-1 through PGH-8, demonstrates the effect of different heterocycles on the spectral properties of the dyes. Analogs of two VSDs were synthesized and coupled to PEG groups of varying chain length (MW 750-5000) to increase water solubility. The enhanced solubility of PEG-derivatives facilitated delivery to target tissues while retaining their spectral properties and voltage sensitivity (ΔF/F per action potential) in cardiac tissues.
Results
Sulfonated Dyes
VSDs were synthesized from heterocycles A or B quaternized with sulfonic acid anchors (Table 1), and a common styrene-thienyl aldehyde intermediate as shown in Scheme 1. The structures of PGH-1 through PGH-10 are listed in Table 2. Briefly, styrene (1) was made from dibutylamino-benzaldehyde via Wittig reaction and then coupled with bromothiophene-carboxaldehyde via a Heck reaction to yield the styryl-thiophene intermediate (2).17 Other syntheses of these intermediates have been reported;18-20 however, the methods used here require minimal purification of the styrene and fewer steps to prepare the aldehyde. Wuskell et. al. recently reported the synthesis of VSDs with similar chromophores utilizing quaternary ammonium anchors.9, 21 The sulfonic acid anchors used here in dyes PGH-1 to PGH-8 produced zwitterionic dyes with the positive charge of the chromophore delocalized between the nitrogen atoms. The benzoxazole dye, PGH-5, was produced; however, the dye rapidly decomposed after chromatographic purification upon drying under mild conditions.
Table 1.
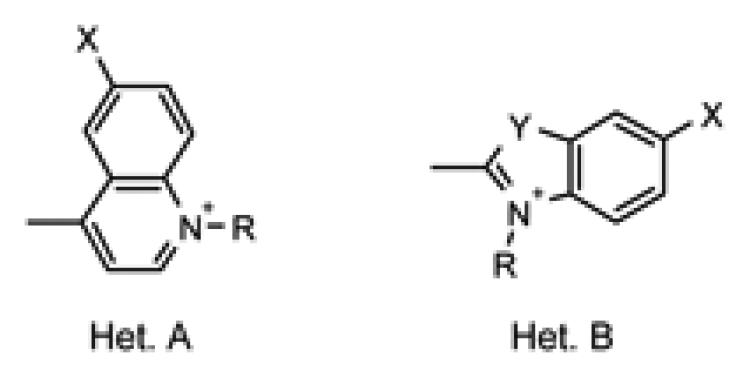
Structures of heterocyclic intermediates used in dye syntheses. Heterocycles (A) are lepidines (3, 10 and 11); or (B) are indolenines (4, 6, 8 and 12); benzothiazole (5); benzoxazole (7); and quinaldine (9). Intermediates 11 and 12 contain carboxylic acid functional groups.
 | ||||
|---|---|---|---|---|
| Intermediate | Het | X | Y | R |
| 3 | A | H | --- | C4H8SO3- |
| 4 | B | H | CMe2 | C4H8SO3- |
| 5 | B | H | S | C4H8SO3- |
| 6 | B | SO3K | CMe2 | C4H8SO3- |
| 7 | B | H | O | C4H8SO3- |
| 8 | B | SO3K | CMe2 | C2H5 |
| 9 | B | OMe | CH=CH | C4H8SO3- |
| 10 | A | OMe | --- | C4H8SO3- |
| 11 | A | H | --- | C5H10CO2H |
| 12 | B | SO3K | CMe2 | C5H10CO2H |
Scheme 1.

Structures of voltage sensitive dyes described in this report.
Table 2.
| Dye | Het. | X | Y | R |
|---|---|---|---|---|
| PGH-1 | A | H | --- | C4H8SO3- |
| PGH-2 | B | H | CMe2 | C4H8SO3- |
| PGH-3 | B | H | S | C4H8SO3- |
| PGH-4 | B | SO3K | CMe2 | C4H8SO3- |
| PGH-5 a | B | H | O | C4H8SO3- |
| PGH-6 | B | SO3K | CMe2 | C2H5 |
| PGH-7 | B | OMe | CH=CH | C4H8SO3- |
| PGH-8 | A | OMe | --- | C4H8SO3- |
| PGH-9 | A | H | --- | C5H10CO2H |
| PGH-10 | B | SO3K | CMe2 | C5H10CO2H |
| PGH-9-PEG750 | A | H | --- | C5H10CONH[C2H4O]~17Me |
| PGH-9-PEG2000 | A | H | --- | C5H10CONH[C2H4O]~45Me |
| PGH-9-PEG5000 | A | H | --- | C5H10CONH[C2H4O]~113Me |
| Bis-PGH-9-PEG3400 a | A | H | --- | {C5H10CONH[C2H4O]~37CH2}2 |
| PGH-10-PEG5000 | B | SO3K | CMe2 | C5H10CONH[C2H4O]~113Me |
Decomposed after purification.
Contains two PGH-9 dyes with both R substitutions being either end of the same bis-functional PEG3400 linker.
The spectral properites of these new dyes, PGH-1 to PGH-8, were characterized in alcohols of different polarity to verify the solvatochromic sensitivity of the various heterocycles. The absorption maxima and extinction coefficients are shown in Table 3, while the emission maxima and relative fluorescence intensities are given in Table 4. The observed increased Stokes shift with increasing solvent polarity is typical for styryl VSDs.22 For two of the dyes, PGH-4 and PGH-6, the increased Stokes shift was assymetrical; the absorption maxima did not change with solvent polarity. These anomalous dyes have a sulfonic acid anchor directly attached to the chromophore. This difference in absoprtion behavior is illustrated in Figure 1. PGH-1 shows a wavelength shift with similar extinction values, while PGH-6 gives an absorbance difference with similar wavelength maxima.
Table 3.
Absorption Properties of VSDs in Various Solvents.
| Methanol | Ethanol | 2-Propanol | n-Butanol | n-Octanol | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dye | Amax | ε | Amax | ε | Amax | ε | Amax | ε | Amax | ε |
| (nm) | (M−1cm−1) | (nm) | (M−1cm−1) | (nm) | (M−1cm−1) | (nm) | (M−1cm−1) | (nm) | (M−1cm−1) | |
| 1 | 601 | 24,900 | 608 | 24,800 | 612 | 24,300 | 619 | 24,800 | 620 | 24,700 |
| 2 | 664 | 32,200 | 673 | 31,500 | 670 | 33,800 | 678 | 35,200 | 678 | 33,300 |
| 3 | 614 | 47,400 | 620 | 46,800 | 626 | 45,750 | 630 | 47,100 | 636 | 43,000 |
| 4 | 686 | 53,600 | 690 | 55,200 | 680 | 53,900 | 688 | 60,600 | 686 | 55,800 |
| 6 | 690 | 62,500 | 690 | 67,600 | 690 | 65,800 | 696 | 52,900 | 692 | 45,500 |
| 7 | 587 | 42,500 | 594 | 42,100 | 594 | 43,100 | 604 | 42,000 | 606 | 43,400 |
| 8 | 588 | 28,400 | 598 | 34,100 | 596 | 30,300 | 602 | 32,000 | 602 | 32,000 |
Table 4.
Emission Properties of VSDs in Various Solvents. λEX = 610 nm.
| Methanol | Ethanol | 2-Propanol | n-Butanol | n-Octanol | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dye | λ max | Fl. | λ max | Fl. | λ max | Fl. | λ max | Fl. | λ max | Fl. |
| (nm) | rel. int. | (nm) | rel. int. | (nm) | rel. int. | (nm) | rel. int. | (nm) | rel. int. | |
| 1 | 1025 | 107 | 966 | 300 | 934 | 714 | 900 | 795 | 868 | 2859 |
| 2 | 935 | 487 | 914 | 968 | 887 | 1855 | 890 | 1882 | 855 | 4392 |
| 3 | 928 | 707 | 901 | 1489 | 890 | 2621 | 888 | 2534 | 848 | 6626 |
| 4 | 940 | 341 | 923 | 832 | 888 | 1947 | 888 | 2046 | 849 | 2174 |
| 6 | 940 | 341 | 927 | 707 | 892 | 1397 | 893 | 486 | 861 | 1607 |
| 7 | 952 | 187 | 888 | 496 | 870 | 1121 | 866 | 1370 | 835 | 4062 |
| 8 | 959 | 108 | 960 | 351 | 940 | 686 | 898 | 930 | 866 | 2283 |
Fig. 1.

Absorption spectra of PGH-1 (Panel A) and PGH-6 (Panel B) in methanol ( black lines) and octanol (
black lines) and octanol ( gray lines).
gray lines).
A dye that exhibits a large ΔF/F response also must be soluble in solvents or buffers that are compatible with physiological conditions for excitable tissues. Great care must be taken to avoid the precipitation of dye or the formation of dye particles in the capillaries which would block flow and render the organ ischemic. The solubilities of selected dyes were tested by diluting a known concentration of dye in DMSO into various solvents. At micromolar concentrations, the dyes remained soluble in alcohol solvents as well as aqueous buffers, even buffered saline solutions. However, effective labeling of tissues and intact organs with a VSD is typically achieved by perfusing the tissue with a bolus injection of dye at 1-2 mM resulting in a homogeneous and intense VSD labeling on the membranes of the tissue. Figure 2 shows the excitation and emission spectra of PGH-1 in cardiac tissue. PGH dyes have desirable spectral properties in tissue but in order to get sufficient loading, DMSO and Pluronic L-64 are needed to keep the dyes from coming out of solution as they course through the vascular bed.4 The search for alternative methods to solubilize the dyes without extraneous surfactants led to the development of PEG derivatives.
Fig. 2.

Excitation-Emission Curve Pair of PGH-1 in Frog Cardiac Tissue. Parameters: λEM = 875 nm for excitation scan, λEX = 600 nm for emission scan.
PEG Derivatives
VSDs with functional groups (PGH-9 and PGH-10) were synthesized as in Scheme 1. The highly water-soluble PEG derivatives were prepared from different molecular weight amino-methoxy-PEGs (mPEG-NH2) using standard methods and are listed in Table 2.16, 23 The combination of the hydrophilic polymer and the hydrophobic styryl dye made purification of these PEG dyes less straight forward than the sulfonated dyes. On acidic alumina the blue chromophore irreversibly disappeared for PGH-9 derivatives of mPEG molecular weights 2000 to 5000. These larger PEG-derivatives were successfully purified by C2-reversed phase chromatography with water-methanol gradients containing 1% acetic acid. After purification, the PGH-9 derivatives had an increased intensity of the short wavelength band (~400 nm) relative to long wavelength band (~600 nm). Table 5 shows a comparison of Ashort/Along of the PEG-dye derivatives, their corresponding parent dyes (PGH-9 and PGH-10) and the sulfonate analogs (PGH-1 and PGH-6). When compared with PGH-1 (containing no PEG group), the ratio of Ashort/Along increased as molecular weight of PEG increased, ranging from 1.4 to 2.7 for PEG750 and PEG5000 derivatives respectively. This increased absorbance ratio could not be reversed. Syntheses of two PGH-9 PEG-derivatives were repeated with closer stoichiometric control. Product isolation by precipitation from ether yielded sufficiently pure material (single component by TLC), eliminating the need for chromatographic purification. The Ashort/Along increase seen previously was not observed with these resynthesized preparations and the ratio (0.6) was the same as underivatized PGH-9. Mass spectral analyses confirmed that the PGH-9 PEG-derivatives were intact. This enhanced short wavelength absorption was not observed with PGH-10-PEG5000 following chromatographic purification.
Table 5.
Absorbance ratios (short/long) were calculated for PGH-1, PGH-6 and PEG-derivatives of PGH-9 and PGH-10 in MeOH.
| Lepidine Dyes | Short λ Max. |
Long λ Max. |
Ashort/Along |
|---|---|---|---|
| PGH-1 | 392 | 601 | 0.71 |
| PGH-9 | 398 | 606 | 0.56 |
| PGH-9-PEG750 (LC) | 396 | 596 | 1.39 |
| PGH-9-PEG2000 (LC) | 390 | 590 | 1.99 |
| PGH-9-PEG3400 (LC) | 390 | 588 | 1.44 |
| PGH-9-PEG5000 (LC) | 390 | 588 | 2.71 |
| PGH-9-PEG750 (No LC) | 398 | 610 | 0.58 |
| PGH-9-PEG5000 (No LC) | 398 | 608 | 0.60 |
|
| |||
| Indolenine Dyes |
Short λ Max. |
Long λ Max. |
Ashort/Along |
|
| |||
| PGH-6 | 406 | 690 | 0.21 |
| PGH-10 | 410 | 692 | 0.22 |
| PGH-10-PEG5000 | 405 | 691 | 0.23 |
The mass profile of the PEG derivatives showed polydispersity as evidenced by a distribution of mass spectral peaks spaced 44 m/z units apart. The products showed polydispersity similar to the starting mPEG-amine reagents, but shifted by approximately the mass of the dye.16 A marked change in the TLC migration of the products from the starting materials, with no change in spectral properties, gave further evidence of dye-PEG conjugation.
The PEG dye derivatives had much higher water-solubility than the sulfonated dyes. The aqueous solubilities of PGH-1, PGH-6, PGH-9-PEG5000 and PGH-10-PEG5000 are shown in Table 6. With the aid of DMSO, PGH-1 showed limited solubility in water and saline; however, PGH-6 was essentially insoluble. Both PEG dye derivatives showed high water solubility without the need for DMSO. The fluorescence properties of PGH-10-PEG5000 and the four PGH-9 PEG-derivatives were similar to PGH-6 and PGH-1, respectively.
Table 6.
Percentage Aqueous Solubility of Voltage Dyes and PEG-Derivatives.
| Solubility at Stock Solution | ||
|---|---|---|
| Dye | Water | PBS |
| PGH 1 | 40.7% | 7.3% |
| PGH 9-PEG5000 | 77.5% | 83.2% |
| PGH 6 | 0%a | 0%a |
| PGH 10-PEG5000 | 71.4% | 78.9% |
Solution was colorless.
PEG-derivatives of PGH dyes had similar spectral, voltage-sensitive responses and retention in cardiac tissues indicating that these dyes provide similar voltage sensing and tissue retention as their non-PEGylated analogs with the added advantage of being highly water soluble. Figure 3 illustrates action potentials recorded from mouse hearts stained with PGH-1 and its PEG-analogs. Action potentials recorded from PGH-1 delivered with a pH 6 aqueous stock solution (Panel A) were similar to those recorded with PGH-1 delivered from a DMSO plus Pluronic stock solution (Panel B). Similar results were obtained with PGH-9-PEG750 derivative delivered from a Tyrode's stock solution in the absence Pluronic (Panel C). PGH-9-PEG750 (Panel D) shows the same inversion of the action potential signal as observed with PGH-1 when the excitation wavelength was shifted from 540 to 690 nm.4 PGH-9-PEG2000 (Panel E) had similar spectral response characteristics and exhibited action potential signals with similar signal to noise (S/N) ratio compared to the PGH-9-PEG750 derivative. In contrast, the PEG-5000 derivative yielded considerably lower S/N ratio compared to the other dyes, most likely due to reduced insertion in the membrane (Panel F).
Fig. 3.

Comparison of Mouse ventricular action potentials recorded with PGH-1 and PGH-9 PEG-derivatives. Excitation at 540 ± 25 nm and emission monitored > 650nm (except Panel D). Panel A: PGH-1 in Ringer's solution at pH 6. Panel B: PGH 1 in a stock solution of 18% Pluronic L64 and DMSO. Panel C: PGH-9-PEG750 in Ringer's at pH 7.4. Panel D: PGH-9-PEG750 stained as Panel C, excited at 690 ± 25 nm and the emission >750 nm. Panel E PGH-9-PEG2000 in Ringer's stock solution at pH 7.0. Panel F PGH-9-PEG5000 in Ringer's stock solution at pH 7.0.
Discussion
For a dye to function satisfactorily as a potentiometric probe, it must exhibit the following essential properties: the dye must get to target tissue and cells; must interact in the proper orientation to sense membrane potentials only in the plasma membrane; should exhibit large and specific optical changes that vary only with membrane potential changes; should induce minimal chemical- and photo-toxicity to the cells and should be optically and spatially stable for the duration of the experiment.5, 7, 10 The voltage sensor component of the dye should lie in the membrane where the electric field and charge movements are most significant during electrical activity. Molecular design features include (1) extended conjugation for long wavelength absorption while maintaining photostability; (2) various nitrogen-heterocycles contributing to the long wavelength absorption and providing spectral sensitivity to voltage changes (3) hydrophobic groups for stable partitioning into the lipid bilayer; and (4) a hydrophilic group making the dye membrane impermeant and facilitating tissue staining.6, 24 Through the structural modifications of the dyes, we have reached near IR excitation wavelengths with increased brightness and improved the delivery of the dyes to the tissues.
Increasing the absorption wavelength is typically done by extending the conjugated system. Incorporation of thienyl unit in the conjugated system extended the wavelength and was effective in expanding conjugation of donor-acceptor molecules providing increased photostability and greater oscillator strength.20, 25 Extension of fluorescence to the near to infra-red range improves penetration of the excitation and transmission of emitted light through thick muscle tissue. Intrinsic chromophores in tissue are avoided and Rayleigh scatter is reduced at the longer wavelengths. Far-red absorption was achieved with PGH-2, PGH-4 and PGH-6.
The length of the alkyl chains on the styryl portion of the dye can determine the depth of penetration into the lipid bilayer and the rate of dye washout.15 Because of their effectiveness in other dyes, butyl groups were selected to maximize tissue retention without an excessive compromise on water solubility.14 The depth of chromophore penetration into the lipid bilayer also increases with the number of carbons between it and the covalently linked anchor.15 The type of hydrophillic group is significant. Loew & Wuskell developed a family of styryl dyes which excite at 600-650 nm and emit >750 nm, but have restricted application for cardiac tissue study.9, 26 While these dyes have improved spectral ranges, they exhibit limited photostability and low S/N ratio signals. The hydrophilic anchor in these dyes is the quaternary ammonium group, which can be drawn through the plasma membranes of cells with large negative membrane potential and thus is not effective for anchoring the dyes at the cell surface.4 We have used sulfonic acid counterions to anchor our dyes to the membrane surface.
The choice of the heterocycle can impact the quantum yield and voltage sensitivity of the dye within the membrane.6 In this study, styryl dyes, PGH-1 through PGH-8, were synthesized from lepidine, quinoline, indolenine, benzothiazole and benzoxazole heterocycles. While most of the heterocycles give absorption maxima near 600 nm, the indolenine dyes (PGH-2, PGH-4 and PGH-6) showed maxima near 690 nm. Unlike most styryl VSDs, the indolenine dyes with the sulfonic acid group attached directly to the chromophore (PGH-4 and PGH-6) did not show absorbance solvatochromism. The close proximity of the anchor to the aromatic system may maintain a polar environment for the chromophore. The sulfonate group will effectively orient the hydroxyl groups of the solvent molecules near the chromophore even in octanol. While PGH-1 yielded larger ΔF/F signals in heart, PGH-6 also was effective as a VSD; even though it shows minimal absorption wavelength solvatochromism.4
The surfactant-like properties of PGH-9 PEG-derivatives presented additional challenges for purification. Column purified PGH-9 PEG-derivatives showed an increased Ashort/Along ratio while alternate synthesis of these derivatives, and the PGH-10 derivative, showed the same ratios as the parent dyes. As the “greasy” voltage dye core and the highly water soluble PEG group became more dissimilar (as with increasing molecular weight of PEG group), the Ashort/Along ratio increased. Interactions between the PEG groups, the hydrophobic dye moiety and the chromatographic stationary phase may have induced these spectral changes. A shift in absorption maximum from 600 nm to 400 nm, while maintaining an intact dye structure, can be caused by a disruption of the pi electron system. Encapsulation by the lengthy PEG group could stabilize a folded conformation of the dye. Johnsson et al incubated other types of PEG derivatives at 60-70 C for 3 hrs to obtain clear solutions.27 Heating had no effect on the absorption spectra of our voltage dye-PEGs. PEG-lipids with molecular weight >1000 form spherical micelles because of the steric hindrance of the larger molecular weight PEG groups.27 Because of the duality inherent in their structure, these new VSD PEG-derivatives may behave like PEG-lipids. Practical considerations such as concentration may be necessary in applications where critical micellar concentration and formation of spherical or lamellar aggregates are a factor.27
Conjugation of PEG groups to VSDs significantly improved water-solubility and simplified delivery to excitable tissues. Derivatives with a longer PEG group (MW 5000) allowed for easier isolation from reaction mixture by precipitation with ether. While derivatives with larger PEG groups are easier to synthesize, the 2 mM stock concentrations of these materials approach 1% solute concentration. At these concentrations, the viscosity of the solution is high and can be problematic in applications where this is a concern. Derivatives with a shorter PEG group (MW 750) could not be isolated by precipitation, thus making product recovery more arduous. However, the shorter PEG-dyes labelled the tissues better in mouse hearts than longer PEG-dyes and did not cause ischemia. The attachment of PEG groups to styryl VSDs, regardless of their lengths, have made it possible to deliver these dyes to tissues without the need for DMSO or pluronics. The simplified delivery method greatly improves the productivity of excitable membrane experimentation.
CONCLUSION
Use of voltage sensitive dyes has been valuable for basic research and medical physiology of excitable tissues like heart and nerve networks. Improvements in the sensitivity of the probes significantly enhance the data quality and range of possible experiments. Furthermore, advantages can be obtained through improved dye delivery and imaging of excitable cells from deeper layers in heart and brain tissues. In developing new methods, we have uncovered a better probe and enhanced membrane staining using water-soluble VSDs, but these developments seem to provide only incremental improvements. It would be interesting to venture into new paradigms that can produce major increments in dye performance rather than small quantum jumps in performance.
Experimental
Chemical Synthesis
All reactions were carried out under an argon atmosphere in oven dried glassware. Unless specified, all reagents were purchased from either Aldrich Chemicals or ACROS Chemicals. Chromatographic material sources: silica gel TLC cards (Fluka 60778), RP-TLC plates (Analtech 52521), bulk silica gel (Aldrich 28,859-4), bulk reversed phase C-18 silica gel (Separation Method Technologies BOD-35-150) and bulk C-2 silica gel (SMT BMEB2-35-150). mPEG-amine derivatizing reagents were obtained from Fluka (Sigma-Aldrich): O-(2-Aminoethyl)-O′-methylpolyethylene glycol-750 (mPEG750-NH2, Fluka 07964); O-(2-Aminoethyl)-O′-methylpolyethylene glycol-2,000 (mPEG2000-NH2, Fluka 06676); Polyoxyethylene bisamine (3,350 MW) (di-NH2-PEG3400, Sigma P9906-5G); and O-(2-Aminoethyl)-O′-methylpolyethylene glycol – 5,000 (mPEG5000-NH2, Fluka 06679).
Synthetic procedures for some of the intermediates used in the syntheses of the dyes reported here, namely 4-(2,3,3-trimethyl-5-sulfonato-indolinium-1-yl)butane-1-sulfonate (6), 1-ethyl-(2,3,3-trimethyl-5-sulfonato-indolinium) bromide (8), and 6-(2,3,3-trimethyl-3H-indolium-1-yl)hexanoic acid (12), were previously reported by Mujumdar et. al.28
Dyes were characterized by NMR (300 or 500 MHz). Some dyes required the use of mixed solvent systems due to limited solubility in single solvents. Mass Spectra were obtained using a PerSeptive Voyager STR MS matrix assisted laser desorption ionization (MALDI) time of flight (TOF) mass spectrometer. Samples were prepared using equal volumes of analyte and matrix (10mg/mL alpha-cyano-4-hydroxycinnamic acid in 50:50 H2O:MeOH + 0.1% AcOH v/v). Calibration was done using a mixture of peptides (FMRF, Bradykinin, Angiotensin, Fibrinopeptide, Renin Substrate, ACTH) each at approx. 10uM in 50:50 H2O:MeOH + 0.1% AcOH(v/v). Absorption spectra and extinction coefficients were determined using a Hewlett Packard Spectrophotometer (8452A Diode Array) or Perkin Elmer Lambda 45 Spectrophotometer. Corrected excitation and emission spectra were obtained using a PTI QuantaMaster fluorescence system configured with double grating excitation and emission monochrometers and a Hamamatsu R5108 photomultiplier tube cooled with dry ice and run at 1100V in photon-counting mode. Data acquisition and analysis was done with Felix32 software. Fluorescence samples were prepared by 1:10 dilution of dye solution used for absorption spectrum in to appropriate solvent. Excitation wavelength was set 10-20 nm below the absorption maximum of the dye (for lepidine-type dyes at 600 nm; for indolenine-type dyes, at 675 nm). Real-time correction software was used.
Spectral Analysis of Dyes
Extinction coefficients presented in the synthetic methods were calcuated from samples typically prepared by dissolving 1.00 mg of dye in 100.0 ml of methanol or ethanol with the aid of sonication. Dye samples in DMSO (1.00 mg/ml) were diluted into methanol or ethanol to produce an optical density of approximately 1.0 AU. The same volume of DMSO stock solution was diluted into solvents for measurement of absorption spectra. Further dilutions of these solutions were made into corresponding solvents yielding optical densities of approximately 0.1 AU for measurement of fluorescence spectra. Fluorescence samples were analysed as described previously in the text.
Solubility of Dyes and PEG Derivatives
Dye solids were dissolved to make 2 mM solutions in water and PBS. While PEG derivatives dissolved directly, PGH 1 and PGH 6 required initial dissolution in 10 μl of DMSO followed by dilution into aqueous solvents (1000 μl). After brief vortex mixing and 1 hour of sonication, samples were filtered through 0.22 μm filters then diluted into methanol for concentration determination by absorption spectroscopy.
Tissue Labeling Experiments
A frog heart was prepared and then perfused with concentrated dye solution until it was noticeably stained.4, 7 The heart was placed in a 10 mm × 10 mm cuvette and right-angle fluorescence measurements were taken from re-emergent light through the tissue. Emission spectrum parameters: excitation 600 nm, emission 620-1000 nm; Excitation spectrum parameters: emission 875 nm, excitation 500-865 nm.
Mouse Ventricular Action Potential Experiments
Mouse hearts were isolated and perfused in a Langendorff apparatus, with a Tyrode's solution containing (in mM): NaCl 136, KCl 5.4, MgCl2 1, NaH2PO4 0.33, CaCl2 2.5 HEPES 10, glucose 10, pH 7.4. Hearts were placed in a chamber designed to reduce contraction artifacts, with temperature controlled to 37 ± 1 °C via a feedback control, as previously described.29 Hearts were stained with a VSD by adding a bolus of 30 μl dye from a stock solution of 1 mM dye dissolved in different solvents. The amount of VSD delivered to each heart was thus kept constant to compare the different dyes through the S/N ratio of their optical action potentials (APs) when the dye was excited at 540 ± 25 nm and emission with a long pass filter at 650 nm. All animal experiments complied with the University of Pittsburgh's Animal Care and Usage Committee and the National Institute of Health.
SYNTHESIS OF INTERMEDIATES
N,N-dibutyl-4-ethenylaniline (1)
Butylithium (2.5 M in hexane, 13.6 ml; 34.0 mmol) was added slowly to a solution of methyltriphenylphosphonium bromide (14.0 g; 39.2 mmol) in anhydrous THF (100 ml). Dibutylaminobenzaldehyde (5.0g; 21.6 mmol) was added and the reaction stirred under argon at room temperature. After 24 hours the reaction was poured into hexane (1000 ml) and stirred for 15 minutes. The yellow precipitate that formed was removed by filtration through Celite and the filtrate was concentrated to approximately 20 ml by rotary evaporation and passed through a 0.2 μm ultrafilter. Solvent removal by rotary evaporation produced a light yellow oil, which solidified on standing; yield 4.52g. (19.5 mmol, 90%). Silica gel TLC Rf = 0.8 (ethyl acetate-Hexane,1:9). 1H NMR (CDCl3, 300 mHz): 0.897 (6 H, t, J = 7.1, -CH2CH2CH2CH3); 1.299 (4 H, m, -CH2CH2CH2CH3); 1.516 (4 H, m, -CH2CH2CH2CH3); 3.216 (4 H, t, J = 7.6, -CH2CH2CH2CH3); 4.915 (1 H, d, J = 11.1, -CH=CHH); 5.437 (1 H, d, J = 18.0, -CH=CHH); 6.548 (3 H, m, overlapping, -CH=CH2, Phe); 7.221 (2 H, d, J= 6.4, Phe).
5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}thiophene-2-carboxaldehyde (2)
Freshly distilled 5-bromo-2-thiophene carboxaldehyde (1.91 mL,160.7 mmol) was mixed with dibutylaminovinylbenzene (1) (3.11g, 13.4 mmol), palladium (II) acetate (30.9 mg), tris-(o-tolyl)phosphine (84.7 mg) and triethylamine (6.15 ml) under argon in a thick-walled glass reaction vessel. The mixture was stirred magnetically and heated at 115-120 °C for 24 hours. The resulting deep brown mixture was cooled, stirred at room temperature overnight, then partitioned between methylene chloride (50 ml) and water (50 ml). The aqueous layer was washed with methylene chloride (2 × 40 ml). The organic layers were combined, dried over MgSO4, and concentrated by rotary evaporation. Product residue was dissolved in 2:8 EtOAc: Hexane (20 ml) and chromatographed over silica (80 g) eluted with a gradient of ethyl acetate in hexane. The product was collected as a single component by silica gel TLC with Rf=0.23 (ethyl acetate-hexane, 1:9); isolated yield of 2.75g (8.05 mmol, 60%). 1H NMR (CDCl3, 300 mHz): 0.994 (6 H, t, J = 7.3, -CH2CH2CH2CH3), 1.394 (4 H, m, -CH2CH2CH2CH3), 1.589 (4H, m, -CH2CH2CH2CH3), 3.257 (4H, t, J = 7.6, -CH2CH2CH2CH3), 6.580 (2 H, d, J = 8.9, -Phe), 6.996 (1 H, d, J = 16.3, =CH-); 7.061 (1 H, d, J = 3.9, -thiophene); 7.109 (1 H, d, J = 16.1, =CH-); 7.382 (2 H, d, J = 8.9, -Phe ); 7.563 (1 H, d, J = 3.9, -thiophene ); 9.755 (1 H, s, -CHO).
4-(4-methylquinolinium-1-yl)butane-1-sulfonate (3)
4-Methyl-quinoline (3 ml, 22.7 mmol) and 1,4-butanesultone (6.3 ml, 61.6 mmol) were heated at 100°C for 16 hours. The resulting solid was triturated with methanol-ethyl acetate (1:3) and recrystallized from methanol/ether yielding a light purple solid. Isolated yield of 5.23g (18.7 mmol, 82%). RP-TLC Rf = 0.10 (methanol-water 1:9); 1H NMR (DMSO-d6 300 MHz) 1.958 (2 H, m, -CH2CH2CH2CH2SO3); 2.274 (2 H, m, -CH2CH2CH2CH2SO3); 2.923 (2 H, t, J = 7.2, -CH2CH2CH2CH2SO3); 3.082 (3 H, s, -Me); 5.094 (2 H, t, J = 7.6, -CH2CH2CH2CH2SO3); 7.971 (1 H, d, J = 5.6, Pyr); 8.067 (1 H, t, J = 7.6, -Phe); 8.283 (1 H, t, J = 8.1, -Phe); 8.580 (1 H, overlapping d, J = 8.2 -Phe); 8.619 (1 H, overlapping d, J = 8.8, -Phe ); 9.268 (1 H, d, J = 6.5, Pyr).
4-(2,3,3-trimethyl-3H-indolium-1-yl)butane-1-sulfonate (4)
This intermediate was synthesized as described previously by Ernst et. al.30 Briefly, 2,3,3-trimethyl-3H-indole (4.8 g) and 1,4-butanesultone (4.2 ml) were heated for 18 hours at 110°C. The resulting solid was triturated with ethyl acetate (50 ml). The product was collected by vacuum filtration and washed with ether (2 × 50 ml) yielding 9.5 g pink solid. RP-TLC Rf = 0.73 (acetonitrile-water, 6:4); 1H NMR (D2O, 300 MHz) – 1.519 (6 H, s, Ind-Me2); 1.849 (2 H, m, -CH2CH2CH2CH2SO3); 2.079 (2 H, m, -CH2CH2CH2CH2SO3); 2.924 (2 H, t, J = 7.6, -CH2CH2CH2CH2SO3); 4.476 (2 H, t, J = 7.8, -CH2CH2CH2CH2SO3); 7.586 (2 H, overlapping, m, indolenine); 7.687 (1 H, m, indolenine); 7.755 (1 H, m, indolenine).
4-(2-methyl-1,3-benzothiazol-3-ium-3-yl)butane-1-sulfonate (5)
Similarly, 2-methylbenzothiazole (25 ml, 197 mmol), 1,4-butanesultone (18.1 ml, 177 mmol) and 1,2-dichlorobenzene (100 ml, anhydrous) were heated overnight at 110°C. After cooling the mixture was diluted with ether (350 ml). The product was collected by vacuum filtration, washed with ether (2 × 100 ml), then dried in vacuo, yielding a crystalline solid, 6.87 g (24.1 mmol, 14%) showing a single component by RP-TLC (water) Rf=0.28. E 210 nm = 10,921 M−1cm−1; 1H NMR (D2O, 300 MHz) – 1.889 (2 H, m, -CH2CH2CH2CH2SO3); 2.083 (2 H, m, -CH2CH2CH2CH2SO3); 2.938 (2 H, t, J = 7.6, -CH2CH2CH2CH2SO3); 3.141 (3 H, s, -Me); 4.715 (obscured by D2O, -CH2CH2CH2CH2SO3); 7.728 (1 H, t, J = 7.4, Phe); 7.833 (1 H, t, J = 7.4, Phe); 8.115 (2 H, overlapping doublets, J = 11.0, J = 10.5, Phe).
4-(6-methoxy-2-methylquinolinium-1-yl)butane-1-sulfonate (9)
1,4-Butanesultone (1.08ml, 10.5mmol) was added to a solution of 6-methoxyquinaldine (2.0g, 11.55 mmol) in tetramethylene sulfone (5.0 ml). After heating at 110 °C for 3 days, the mixture was cooled and diluted with ethyl acetate (20 ml). The resulting solid was collected on a vacuum funnel and washed with diethyl ether (2×10 ml), ethyl acetate (10 ml) and dried under vacuum. C18-RP-TLC (MeOH-H2O, 1:1) gave Rf = 0.50. Yield: 1.86g (6.01 mmol, 52%). 1H NMR (DMSO-d6, 300 mHz): 1.897 (2 H, m, -CH2CH2CH2CH2SO3); 2.034 (2 H, m, -CH2CH2CH2CH2SO3); 2.663 (2 H, t, J = 7.0, -CH2CH2CH2CH2SO3); 3.055 (3 H, s, -Me); 3.98 (3 H, s, -OMe); 4.886 (2 H, t, J = 8.2, -CH2CH2CH2CH2SO3); 7.736-7.838 (2 H, m, overlapping, Phe, Phe); 7.983 (1 H, d, J = 8.3, Phe); 8.527 (1 H, d, J = 9.5, Phe); 8.883 (1 H, d, J = 9.0, Pyr).
4-(6-methoxy-4-methylquinolinium-1-yl)butane-1-sulfonate (10)
6-Methoxy-4-methylquinoline, (1.0g, 5.77 mmol) dissolved in tetramethylene sulfone (2.5 ml) was mixed with 1,4-butanesultone (0.54ml, 5.27mmol) and heated at 110 °C for 3 days. After cooling, the reaction was diluted with ethyl acetate (20 ml). The resulting solid was collected, washed with ether (2×10 ml) and ethyl acetate (10 ml), and dried in vacuo. RP-TLC (MeOH-H2O, 1:1) Rf = 0.50 Yield 1.50g (4.85 mmol, 84.0%). 1H NMR (D2O, 300 mHz): 1.805 (2 H, m, -CH2CH2CH2CH2SO3); 2.154 (2 H, m, -CH2CH2CH2CH2SO3); 2.876-2.183 (5 H, overlapping (s+t), J = 7.4, -Me, -CH2CH2CH2CH2SO3); 3.987 (3 H, s, -OMe); 4.910 (2 H, t, J = 7.3, -CH2CH2CH2CH2SO3); 7.542 (1 H, d, J = 3.1, Phe between -Me and -OMe); 7.732 (1 H, overlapping, s, Phe); 7.774 (1 H, overlapping, d, J = 6.1, Pyr); 8.268 (1 H, d, J = 10.1, Phe); 8.883 (1 H, d, J = 6.0, Pyr).
1-(5-carboxypentyl)-4-methylquinolinium bromide (11A) (Isolated as COOH)
Lepidine (4.80 ml, 0.0363 mol) and 6-bromohexanoic acid (35.4g, 0.182 mol) were heated 110°C overnight. After cooling, the mixture was diluted with water (200 ml) and extracted with ethyl acetate (200 ml). The aqueous layer was washed with dichloromethane (2 × 50 ml) and then concentrated by rotary evaporation. The resulting oily residue was dissolved in 15 ml of methanol and crystallized upon addition of ethyl acetate, yielding 6.1g (0.0180 mol, 50.0%). C18-RP-TLC (MeOH-H2O, 1:1) Rf = 0.30 1H NMR (D2O, 300 mHz): 1.383 (2 H, m, -CH2CH2CH2CH2CH2COOH), 1.589 (2 H, m, -CH2CH2CH2CH2CH2COOH), 2.004 (2 H, m, -CH2CH2CH2CH2CH2COOH), 2.301 (2 H, t, J = 7.1, -CH2CH2CH2CH2CH2COOH), 2.955 (3 H, s, -Me), 4.921 (2 H, t, J = 7.6, -CH2CH2CH2CH2CH2COOH), 7.813 (1 H, d, J = 6.3, Pyr), 7.946 (1 H, t, J = 7.4, Phe), 8.147 (1 H, t, J = 7.3, Phe), 8.332 (1 H, d, J =8.7, Phe), 8.422 (1 H, d, J = 8.3, Phe), 8.982 (1 H, d, J = 5.7, Pyr).
(11B) (Isolated as DIPEA salt)
Lepidine (15 ml, 113 mmol), 6-bromohexanoic acid (22.07 g, 113 mmol), and diisopropylethyl amine (14.6 g, 113 mmol) were mixed and heated at 120 °C for 18 hours. Reaction progress was monitored by C18-RPTLC (MeOH-H2O, 1:1, Rf 0.35). After cooling, the mixture was diluted with water (200 ml) and extracted with dichloromethane (200 ml). The aqueous layer was made alkaline (pH 8-9) using 20 ml of saturated sodium bicarbonate solution followed by extraction with 250 ml of dichloromethane. The organic layer was separated and washed with water (100 ml). Aqueous layers were combined and dried by rotary evaporation. Product was purified by flash chromatography (C18-RP) eluting with a methanol in water gradient (0 to 20%). Fractions containing pure DIPEA salt were combined and dried yielding 23.37 g of a hygroscopic solid (44%). 1H NMR (D2O, 300 mHz): 1.302 (15 H, m, DIPEA –CH3); 1.375 (2 H, m, -CH2CH2CH2CH2CH2COOH), 1.595 (2 H, m, -CH2CH2CH2CH2CH2COOH), 2.029 (2 H, m, -CH2CH2CH2CH2CH2COOH), 2.200 (2 H, t, J = 7.3, -CH2CH2CH2CH2CH2COOH), 3.178 (2 H, m, DIPEA –H{CH3}2); 3.694 (2 H, m, DIPEA –CH2CH3); 4.912 (2 H, t, J = 7.3, -CH2CH2CH2CH2CH2COOH), 7.829 (1 H, d, J = 6.5, Pyr), 7.931 (1 H, t, J = 7.6, Phe), 8.147 (1 H, t, J = 7.9, Phe), 8.328 (1 H, d, J =9.0, Phe), 8.397 (1 H, d, J = 8.9, Phe), 8.995 (1 H, d, J = 6.3, Pyr).
SYNTHESIS OF DYES & DERIVATIVES
Dyes (PGH-1 through PGH-10) were synthesized by the following general method. Equi-molar quantities of anilino-thiophene-carboxaldehyde (2) in ethanol (~0.25M) and quanternized heterocyclic intermediate (1 through 12) dissolved in methanol (~0.25M) were stirred at room temperature. Quinoline and Lepidine intermediates (9, 3, 10 and 11) required the addition of 1% (v/v) piperidine to facilitate the reaction. At various times a sample of the mixture was diluted in ethanol and measured by UV/Vis spectroscopy. Reaction progress was monitored by appearance of the product dye (Table 1a) and decrease of the aldehyde (A444). If significant starting aldehyde remained unreacted, additional heterocycle was added and the reaction continued. When the Adye / A444 ratio remained constant, the reaction was concentrated to dryness by rotary evaporation. Dyes were purified by either normal or reversed phase column chromatography.
4-{4-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]quinolinium-1-yl}butane-1-sulfonate (PGH-1)
Intermediates (2) and (3) (7.17 mmol) condensed with the aid of piperidine to form the product dye which was purified by column chromatography over silica gel (75g), and eluted with a methanol-ethyl acetate gradient. Fractions containing pure PGH 1 dye [silica TLC (MeOH-EtOAc, 1:1); Rf = 0.20] were combined and dried by rotary evaporation. Yield 1.3g (2.16 mmol, 30%). λmax(MeOH)/nm 602 (ε/dm3 mol−1 cm−1 24 895). 1H NMR (DMSO-d6, 300 mHz): 0.926 (6 H, t, J = 7.4, -CH2CH2CH2CH3); 1.271-1.395 (4 H, m, -CH2CH2CH2CH3); 1.469-1.544 (4 H, m, -CH2CH2CH2CH3); 1.684-1.734 (2 H, m, -CH2CH2CH2CH2SO3); 2.035-2.082 (2 H, m -CH2CH2CH2CH2SO3); 2.541 (obscured by DMSO-d6 signal, -CH2CH2CH2CH2SO3); 3.338 (obscured by H2O signal, -CH2CH2CH2CH3); 4.950 (2 H, t, J = 7.01, -CH2CH2CH2CH2SO3); 6.656 (2 H, d, J = 8.5, Phe); 7.016 (1 H, d, J = 16.2, -CH=); 7.180-7.231 (2 H, overlapping signals s+d, J = 4.3 and 15.3, thiophene, -CH=); 7.422 (2 H, d, J = 8.9, Phe); 7.644 (1 H, d, J = 4.2, thiophene); 7.830 (1 H, d, J = 15.6, -CH=); 7.992 (1 H, t, J = 7.5, Phe); 8.211 (1 H, t, J = 7.5, Phe); 8.358-8.405 (2 H, t, J = 7.5 and 6.5, Phe, Pyr); 8.565 (1 H, d, J = 8.5 Phe); 8.909 (1 H, d, J = 8.5 Phe); 9.282 (1 H, d, J = 6.5, Pyr). MALDI-TOF m/z 603 (M+, 100%), 625 (84).
4-{2-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-3,3-dimethyl-3H-indolium-1-yl}butane-1-sulfonate (PGH-2)
The dye made from intermediates (2) and (4) (1.18 mmol) was separated over C18-reversed phase silica gel eluting with a step gradient of methanol in water (10% steps, 100 ml/step). Fractions containing pure PGH-2 dye [C18-RP TLC (MeOH-water, 6:4); Rf = 0.20] were combined and dried, yielding 0.22 g (0.36 mmol, 30%). λmax(iPrOH)/nm 670 (ε/dm3 mol−1 cm−1 33 760). 1H NMR (DMSO-d6, 300 mHz): 0.931 (6 H, t, J = 7.4, -CH2CH2CH2CH3); 1.303-1.400 (4 H, m, -CH2CH2CH2CH3); 1.478-1.576 (4 H, m, -CH2CH2CH2CH3); 1.765 (6 H, s, Ind-Me2); 1.802-1.849 (2 H, m, -CH2CH2CH2CH2SO3); 1.849-1.972 (2 H, m, -CH2CH2CH2CH2SO3); 2.559 (2 H, t, J = 7.1, -CH2CH2CH2CH2SO3); 3.333 (obscured by D2O signal, -CH2CH2CH2CH3); 4.561 (2 H, t, J = 7.2, - CH2CH2CH2CH2SO3); 6.681 (2 H, d, J = 8.7, Phe); 7.140 (1 H, d, J = 15.7, -CH=); 7.279 (2 H, d, J = 2.0, -HC=CH-); 7.387 (1 H, d, J = 3.9, thiophene); 7.494 (2 H, d, J = 8.7, Phe); 7.534-7.584 (2 H, dd, J = 6.8 and 1.4, Ind); 7.805 (1 H, d, J = 7.3, Ind), 7.873 (1 H, d, J = 7.7, Ind); 8.140 (1 H, d, J = 4.0, thiophene); and 8.604 (1 H, d, J = 15.6, -CH=).
4-{2-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-1,3-benzothiazol-3-ium-3-yl}-butane-1-sulfonate (PGH-3)
Intermediates (2) and (5) (2.34 mmol) combined to form a blue product. The residue was dissolved in a mixture of methanol (8 ml), trifluoroacetic acid (0.36 ml), and dichloromethane (70 ml), and separated over a silica gel column (30 g) eluting with a methanol in dichloromethane gradient. Fractions containing pure dye (silica TLC developed with methanol-ethyl acetate, 1:1, Rf = 0.56) were combined and dried. The resulting film was dissolved in methanol-dichloromethane (1:3) and precipitated with hexane (100 ml). Product was collected by filtraton and dried in vacuo yielding 0.54 g of blue powder (0.89 mmol, 38%). λmax(MeOH)/nm 614 (ε/dm3 mol−1 cm−1 47 384). 1H NMR (CDCl3 with 10% MeOH-d4, 300 mHz): 0.917 (6 H, t, J = 7.3, -CH2CH2CH2CH3); 1.261-1.384 (4 H, m, -CH2CH2CH2CH3); 1.481 -1.582 (4 H, m, -CH2CH2CH2CH3); 2.029-2.143 (4 H, overlapping m, -CH2CH2CH2CH2SO3); 2.997 (2 H, t, J = 6.3, -CH2CH2CH2CH2SO3); 3.270 (4 H, t, J = 7.7, -CH2CH2CH2CH3); 4.680 (2 H, t, J = 7.4, -CH2CH2CH2CH2SO3); 6.616 (2 H, d, J = 8.6, Phe); 6.816 (1 H, d, J = 15.9, -CH=); 6.918-6.992 (2 H, overlapping m, -CH=, Phe); 7.259 – 7.312 (3 H, overlapping m, -CH=CH-, thiophene); 7.469 (1 H, t, J = 7.62, -CH=), 7.608 (1 H, t, J = 7.6, Benzth); 7.650 (1 H, d, J = 3.8, thiophene); 7.841-7.905 (3 H, m, overlapping, Benzth).
Potassium-2-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]-ethenyl}thiophen-2-yl)ethenyl]-3,3-dimethyl-1-(4-sulfonatobutyl)-3H-indolium-5-sulfonate (PGH-4)
The dye resulting from reacting (2) with intermediate (6) (8.11 mmol) was purified by chromatography over C18-reversed phase silica gel (175g) equilibrated with water and eluted with a methanol-water step gradient from 0 to 65%. Fractions containing pure dye (RPTLC, AcN-H2O 4:6, R f = 0.23) were combined and dried giving 2.2 g blue solid. This residue was dissolved in 70 ml dimethylsulfoxide and reprecipitated with ethyl acetate (700 mL). Pure PGH-4 dye was recovered by filtration and vacuum dried. Yield: 1.5 g (2.03 mmol, 25.2%). λmax(EtOH)/nm 690 (ε/dm3 mol−1 cm−1 55 172). 1H NMR (DMSO-d6, 300 mHz): 0.930 (6 H, t, J = 7.3, -CH2CH2CH2CH3), 1.338 (4 H, m, -CH2CH2CH2CH3), 1.525 (4 H, m, -CH2CH2CH2CH3), 1.770 (6 H, s, Ind-Me2), 1.814-1.910 (4 H, overlapping m, -CH2CH2CH2CH2SO3), 2.585 (2 H, m, -CH2CH2CH2CH2SO3), 3.35 (4 H, obscured by D2O, -CH2CH2CH2CH3), 4.539 (2 H, t, J = 6.8, -CH2CH2CH2CH2SO3), 6.679 (2 H, d, J = 9.1, Phe), 7.122 (1 H, d, J = 15.6, -CH=), 7.285 (2 H, overlapping m, -HC=CH-), 7.387 (1 H, d, J = 4.2 , thiophene), 7.498 (2 H, d, J = 8.6, Phe), 7.799 (2 H, s, Ind), 8.002 (1 H, s, Ind), 8.145 (1 H, d, J = 4.5, thiophene), 8.624 (1 H, d, J = 15.5, =CH-).
potassium iodide - 2-[(E)-2-(5-{(E)-2-[4-(dibutylamino)-phenyl]ethenyl}thiophen-2-yl)ethenyl]-1-ethyl-3,3-dimethyl-3H-indolium-5-sulfonate (1:1) (PGH-6)
Intermediates (2) and (8) (1.18 mmol) formed a dye which was purified over C18-reversed phase silica (50g) equilibrated with water and eluted with a 2-propanol-water gradient (0 to 75%). Fractions containing pure dye (C18-RPTLC (IPA-H2O, 1:1) Rf = 0.30) were combined and dried giving 0.49 g (0.66 mmol 56% yield) blue solid. λmax(EtOH)/nm 690 (ε/dm3 mol−1 cm−1 67 577). 1H NMR (DMSO-d6, 300 mHz): 0.934 (6 H, t, J = 7.4, -CH2CH2CH2CH3); 1.306-1.447 (7 H, m, overlapping -CH2CH2CH2CH3 , -CH2CH3); 1.480-1.558 (4 H, m, -CH2CH2CH2CH3); 1.785 (6 H, s, Ind-Me2); 3.365 (obscured by DMSO-d6 signal, -CH2CH2CH2CH3); 4.562 (2 H, m, -CH2CH3); 6.689 (2 H, d, J = 9.3, Phe), 7.030 (1 H, d, J = 15.3, -CH=); 7.207-1.339 (2 H, dd, J = 15.6 and 8.4, -CH=CH-); 7.389 (1 H, d, J = 4.2, thiophene); 7.490 (2 H, d, J = 9.0, Phe); 7.748-7.832 (2 H, dd, J = 8.2 and 8.0, Ind); 8.024 (1 H, s, Ind); 8.105 (1 H, d, J = 4.1, thiophene); 8.649 (1 H, d, J = 15.5, =CH-).
4-{2-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-6-methoxyquinolinium-1-yl}butane-1-sulfonate (PGH-7)
The reaction products from intermediates (2) and (9) (0.589 mmol) with piperidine were separated by silica gel chromatography eluting with an acetonitrile-dichloromethane chloride gradient (0 to 100%). Pure PGH-7 dye (0.13 g, 0.205 mmol, 34.8%) was recovered showing Rf = 0.59 by silica gel TLC (MeOH/EtOAc, 1:1). λmax(EtOH)/nm 594 (ε/dm3 mol−1 cm−1 42 124). 1H NMR (1:3 MeOH-d4:CDCl3, 300 mHz): 1.046 (6 H, t, J = 7.2, -CH2CH2CH2CH3); 1.388-1.1.510 (4 H, m, -CH2CH2CH2CH3); 1.569-1.725 (4 H, m, -CH2CH2CH2CH3); 2.180-2.364 (4 H, broad m, -CH2CH2CH2CH2SO3); 3.150 (2 H, t, J = 6.0, -CH2CH2CH2CH2SO3); 3.378 (obscured by MeOH signal, t, J = 7.7, -CH2CH2CH2CH3); 3.928 (3 H, s, -OMe); 4.860 (2 H, broad, -CH2CH2CH2CH2SO3); 6.656 (2 H, d, J = 9.0, Phe); 6.806 (1 H, d, J = 16.4, -CH=); 6.916-6.972 (2 H, overlapping signals s+d, J = 16.6, thiophene, -CH=); 7.037 (1 H, d, J = 14.7, -CH=); 7.285 (1 H, s, Phe); 7.353 (2 H, d, J = 8.4, Phe); 7.548 (1 H, d, J = 3.2, thiophene); 7.661 (1 H, t, J = 10.2 Phe); 8.061 (1 H, d, J = 15.3 -CH=); 8.250 (2 H, d, J = 9.3 Phe); 8.488 (1 H, d, J = 8.5, Phe).
4-{4-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-6-methoxyquinolinium-1-yl}butane-1-sulfonate (PGH-8)
The dye from intermediates (2) and (10) (0.589 mmol) and piperidine was purified by silica gel chromatography eluting with a methanol-dichloromethane gradient yielding 95 mg PGH-8 dye (0.150 mmol, 25%) showing a single component by C18-RPTLC (IPA-H2O, 3:1; Rf = 0.67). λmax(EtOH)/nm 598 (ε/dm3 mol−1 cm−1 34 056). 1H NMR (10% MeOH-d4/90% CDCl3, 500 mHz): 0.893 (6 H, t, J = 7.0, -CH2CH2CH2CH3); 1.264-1.322 (4 H, m, -CH2CH2CH2CH3); 1.520 (4 H, broad, -CH2CH2CH2CH3); 1.923 (2 H, m, -CH2CH2CH2CH2SO3); 2.186 (2 H, m, -CH2CH2CH2CH2SO3); 2.919 (2 H, t, J = 7.0, -CH2CH2CH2CH2SO3); 3.247 (4 H, broad, -CH2CH2CH2CH3); 4.010 (3 H, s, -OMe); 4.885 (2 H, t, J = 7.3, -CH2CH2CH2CH2SO3); 6.564 (2 H, broad, Phe(2)); 6.928 (3 H, broad signal,-CH=, -CH=, thiophene); 7.261-7.299 (4 H, m (broad), Phe, Phe, thiophene -CH=); 7.570 (1 H, s, Phe); 7.709 (1 H, d, J = 9.7, Phe); 7.898 (1 H, d, J = 15.3 –CH=); 7.970 (1 H, d, J = 5.24 Pyr); 8.220 (1 H, d, J = 9.8 Phe); 8.985 (1 H, broad, Pyr).
1-(5-carboxypentyl)-4-[(E)-2-(5-{(E)-2-[4-(dibutylamino)-phenyl]ethenyl}thiophen-2-yl)ethenyl]quinolinium bromide (PGH-9)
(Method A- COOH form)
Solutions of Carboxaldehyde (2) and quaternized lepidine (11A) (2.51 mmol) and were mixed with piperidine and stirred overnight at ambient temperature. When the A612 / A398 ratio remained constant, the mixture was concentrated to dryness, and purified by silica chromatography, eluting with a methanol–dichloromethane gradient [TLC MeOH-EtOAc (3:1), Rf = 0.25]. Yield: 0.335 g (0.506 mmol, 34%).
(Method B- DIPEA salt and conversion to COOH)
Carboxaldehyde (2) and quaternized lepidine (11B) (3 mmol) solutions were stirred with piperidine. Product was purified by chromatography as described above giving 2.08 g blue solid. The DIPEA salt was converted to the free carboxylic acid by partitioning between dichloromethane (130ml) and 10% hydrobromic acid (125 ml). The organic layer was separated, washed with water and dried giving 0.66 g PGH 9 pure by TLC (methanol-ethyl acetate, 3:1, Rf 0.25) (0.997 mmol, 33%).
1H NMR (AcN-d3, 300 mHz): 0.952 (6 H, t, J = 7.3, -CH2CH2CH2CH3); 1.318-1.393 (4 H, m, -CH2CH2CH2CH3); 1.417-1.465 (2 H, m, -CH2CH2CH2CH2CH2COOH); 1.532-1.644 (6 H, m, -CH2CH2CH2CH3,-CH2CH2CH2CH2CH2COOH); 2.010 (obscured by AcN signal, -CH2CH2CH2CH2CH2COOH); 2.240 (2 H, t, J = 7.0 -CH2CH2CH2CH2CH2COOH); 3.318 (obscured by MeOH signal, -CH2CH2CH2CH3); 4.791 (2 H, t, J = 7.5, -CH2CH2CH2CH2CH2COOH); 6.654 (2 H, d, J = 9.0, Phe); 6.918-7.083 (3 H, overlapping d +dd, J = 4.0, thiophene, J = 15.8 and 15.9, -CH=CH-); 7.341 (2 H, d, J = 9.0, Phe); 7.499 (1 H, d, J = 4.0, thiophene); 7.625 (1 H, d, J = 15.6, -CH=); 7.951 (1 H, t, J = 7.3, Phe); 8.126-8.190 (3 H, m, Phe, -CH=, Pyr); 8.280 (1 H, d, J = 8.8 Phe); 8.687 (1 H, d, J = 8.9 Phe); 8.983 (1 H, d, J = 6.7, Pyr). MALDI-TOF m/z 581 (M+, 100%), 583 (50), 525 (19).
1-(5-carboxypentyl)-2-[(E)-2-(5-{(E)-2-[4-(dibutylamino)-phenyl]ethenyl}thiophen-2-yl)ethenyl]-3,3-dimethyl-3H-indolium-5-sulfonate (PGH-10)
Intermediates (2) and (12) (1.18 mmol) produced a blue dye which was isolated by normal phase chromatography eluted with a methanol in dichloromethane gradient. NP-TLC 20% MeOH/CH2Cl2 Rf ~ 0.50. Yield 0.87 g (1.09 mmol, 93%). 1H NMR (DMSO-d6, 300 mHz): 0.936 (6 H, t, J = 7.4, -CH2CH2CH2CH3); 1.283-1.615 (12 H, m, overlapping -CH2CH2CH2CH3, -CH2CH2CH2CH2CH2COOH); 1.788-1.862 (8 H, m, overlapping -CH2CH2CH2CH2CH2COOH, Ind-Me2); 2.229 (2 H, t, J = 7.3, CH2CH2CH2CH2CH2COOH); 3.342 (obscured by DMSO-d6 signal, -CH2CH2CH2CH3); 4.540 (2 H, t, J = 6.7, -CH2CH3); 6.690 (2 H, d, J = 8.6, Phe), 7.029 (1 H, d, J = 15.2, -CH=); 7.211-7.341 (2 H, dd, J = 16.0 and 7.0, -CH=CH-); 7.396 (1 H, d, J = 4.3, thiophene); 7.495 (2 H, d, J = 9.0, Phe); 7.747-7.822 (2 H, dd, J = 8.3 and 4.8, Ind); 8.023 (1 H, s, Ind); 8.101 (1 H, d, J = 4.0, thiophene); 8.654 (1 H, d, J = 15.8, =CH-). MALDI-TOF m/z 677 (M+, 100%), 678 (64), 679 (40).
4-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-1-{6-[(2,5-dioxopyrrolidin-1-yl)oxy]-6-oxohexyl}quinolinium bromide (PGH-9-OSu)
N,N,N′N′-Tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (TSTU; 275 mg, 0.913 mmol) was added to a solution of PGH-9 dye (0.40 g, 604 mmol) dissolved in dimethylformamide (30 ml) and diisopropylethylamine (300 μl) and stirred for 1 hour at room temperature. Normal phase silica TLC analysis (EtOAc-MeOH, 1:3; Rf = 0.75) showed the quantitative formation of the active NHS ester, which was used directly for the preparation of amino-PEG conjugates.
4-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-1-[6-( methoxypolyethylene glycol-750-amino)-6-oxohexyl]quinolinium bromide (PGH-9-PEG750)
mPEG750-NH2 (224 mg 0.300 mmol) was added to previously prepared PGH-9-OSu in DMF/DIPEA solution (14 ml, 0.142 mmol) and mixed for 5 days. Thin layer chromatography [EtOAc-MeOH (1:3)] confirmed product formation by the loss of active ester at the solvent front and increase of PEG product at the origin. The product was isolated by precipitation with 200 ml diethyl ether and collection of the blue solid. Chromatography of this product over alumina eluted with a methanol-dichloromethane gradient caused the dye to change color from deep blue to a light greenish blue. This material was rechromatographed over silica gel, eluting with increasing solvent polarity from dichloromethane, 2-propanol, acetonitrile, methanol, and water. The PEG derivative finally eluted with methylsulfoxide and dimethylformamide followed by 1% acetic acid in toluene-ethyl acetate (1:2). These fractions were concentrated and the residual material was partitioned between ethyl acetate and aqueous potassium chloride (4M). The organic layer was collected and concentrated to an oil, which precipitated upon addition of ether yielding 377 mg of blue solid. MALDI-TOF m/z 1211 (98%), 1255 (100), 1299 (92). Mass spectrum contained a series of peaks ranging from 1123-1607 spaced at 44 m/z increments due to polydispersity of PEG group.
Repeat Synthesis (w/o chromatography)
mPEG750-NH2 (250 mg 0.333 mmol) was added to previously prepared PGH-9-OSu in DMF/DIPEA solution (0.302 mmol). The reaction was stirred at room temperature until complete as described above. Solvent removed by rotary evaporation using an acetonitrile azeotrope. This product showed sufficient purity by silica and reversed phase TLC (1% acetic acid in methanol-acetonitrile, 1:3) and required no further purification; yield: 540 mg.
4-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-1-[6-(methoxypolyethylene glycol-2000-amino)-6-oxohexyl]-quinolinium bromide (PGH-9-PEG2000)
mPEG2000-NH2 (444 mg 0.222 mmol) was added to previously prepared PGH-9-OSu in DMF/DIPEA solution (0.202 mmol) and mixed until reaction was complete (5 days), as described. The reaction was diluted with diethyl ether (30 ml), and the blue product was recovered after decanting the solvents and dried. Chromatography over alumina eluting with a methanol-dichloromethane gradient resulted in a complete change in color from blue to brown-yellow. However, C-2 reversed phase silica eluted with a gradient from 1% acetic acid in water to methanol yielded 130 mg blue solid (0.051 mmol, 23%). MALDI-TOF m/z 2531 (M+, 14%), 2575 (13), 2487 (13). Polydispersity of the PEG reagent resulted in a series of product peaks ranging from m/z 2004-3104 with 44 m/z increments.
4-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-1-[6-( methoxypolyethylene glycol-5000-amino)-6-oxohexyl]quinolinium bromide (PGH-9-PEG5000)
mPEG5000-NH2 (1.11 g 0.222 mmol) and PGH-9-OSu in DMF/DIPEA solution (0.202 mmol) were mixed until reaction was complete (5 days), as described. Product was purified as described for PGH-9-PEG2000 yielding 104.5 mg (0.0188 mmol, 9%). MALDI-TOF m/z 5572 (M+, 89%), 5528 (93), 5616 (84). Polydispersity of the PEG reagent resulted in a series of product peaks ranging from m/z 5044-5968 with 44 m/z increments.
Repeat Synthesis (w/o chromatography)
mPEG5000-NH2 (1.66 mg 0.333 mmol) and PGH 9-OSu in DMF/DIPEA solution were reacted as before. Product was isolated by precipitation with ethyl ether (50 ml); centrifuged and solvent decanted, then dried under vacuum yielding 1.2 g (0.215 mmol, 71%).
1,1′-{polyoxyethylene(3,340 MW)-α,ω-diylbis[imino(6-oxohexane-6,1-diyl)]}bis{4-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}thiophen-2-yl)-ethenyl]quinolinium} dibromide (bis-PGH-9-PEG3400)
Polyoxyethylene bisamine (3,350 MW) (372 mg 0.111 mmol) was reacted with PGH-9-OSu and purified as described for PGH-9-PEG2000, yielding 65 mg (0.0151 mmol, 7.5%). MALDI-TOF m/z 4329 (M+, 84%), 4373 (77), 4241 (81). Mass spectrum contained a series of peaks ranging from 3935-4683 spaced at 44 m/z increments due to polydispersity of PEG group.
2-[(E)-2-(5-{(E)-2-[4-(dibutylamino)phenyl]ethenyl}-thiophen-2-yl)ethenyl]-3,3-dimethyl-1-(6-(methoxypolyethylene glycol-5000-amino)-6-oxohexyl)-3H-indolium-5-sulfonate (PGH 10-PEG5000)
TSTU (300 mg, 0.999 mmol) was added to a solution of PGH-10 (0.53 g, 0.666 mmol) in dimethylformamide (39 ml) and diisopropylethylamine (0.4 ml). After 30 min, O-(2-Aminoethyl)-O′methoxypolyethylene glycol – 5,000 (3.55 g, 0.710 mmol) was added and the mixture was mixed for 3 days. Product remained at the origin with both C18-RPTLC (2-propanol-water, 1:1) and silica TLC (methanol-dichloromethane, 1:4). Precipitation and purification of this product by C2-reversed phase chromatography were as described before, yielding 739.5 mg (0.133 mmol, 20%). λmax(MeOH)/nm 690 (ε/dm3 mol−1 cm−1 119 321). MALDI-TOF m/z 5580 (M+, 99%), 5536 (93), 5624 (92). Polydispersity of the PEG reagent gave in a series of product peaks ranging from m/z 5096-6419 with m/z 44 increments.
Supplementary Material
Acknowledgements
This work was supported by the National Heart, Lung and Blood Institute with RO1 HL057929, HL70722 and HL69097 to Guy Salama. Thanks are due to Sujata Emani for synthesis and purification of styryl intermediates, to Dr. Frederick Lanni and Patrick Byrne for spectral analysis of dyes in frog hearts; to Ghassan Azour and Dean Tai for evaluation of PEG dyes in cardiac cells; to Jonathan Douds for spectral analysis method development; to Eric Lanni for mass spectroscopy expertise; to Roberto Gil for help with NMR measurements. The NMR spectrometers of the Department of Chemistry NMR Facility at Carnegie Mellon University (USA) were purchased in part with funds from NSF (CHE-0130903). MALDI-TOF mass spectrometer was funded by NSF (CHE-9808188).
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See http://dx.doi.org/10.1039/b000000x/
References
- 1.Davila HV, Salzberg BM, Cohen LB, Waggoner AS. Nature: New biology. 1973;241:159–160. doi: 10.1038/newbio241159a0. [DOI] [PubMed] [Google Scholar]
- 2.Morad M, Salama G. Journal of Physiology-London. 1979;292:267–295. doi: 10.1113/jphysiol.1979.sp012850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross WN, Salzberg BM, Cohen LB, Grinvald A, Davila HV, Waggoner AS, Wang CH. The Journal of membrane biology. 1977;33:141–183. doi: 10.1007/BF01869514. [DOI] [PubMed] [Google Scholar]
- 4.Salama G, Choi BR, Azour G, Lavasani M, Tumbev V, Salzberg BM, Patrick MJ, Ernst LA, Waggoner AS. The Journal of membrane biology. 2005;208:125–140. doi: 10.1007/s00232-005-0826-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waggoner AS. Annual review of biophysics and bioengineering. 1979;8:47–68. doi: 10.1146/annurev.bb.08.060179.000403. [DOI] [PubMed] [Google Scholar]
- 6.Waggoner AS, Grinvald A. Annals of the New York Academy of Sciences. 1977;303:217–241. [PubMed] [Google Scholar]
- 7.Salama G. In: Spectroscopic Probes of Membrane Potential. Loew LM, editor. CRC Uniscience Pub; Boca Raton, FL: 1988. pp. 137–199. [Google Scholar]
- 8.Loew LM, Simpson LL. Biophysical Journal. 1981;34:353–365. doi: 10.1016/S0006-3495(81)84854-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wuskell JP, Boudreau D, Wei MD, Jin L, Engl R, Chebolu R, Bullen A, Hoffacker KD, Kerimo J, Cohen LB, Zochowski MR, Loew LM. Journal of Neuroscience Methods. 2006;151:200–215. doi: 10.1016/j.jneumeth.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Cohen LB, Salzberg BM, Davila HV, Ross WN, Landowne D, Waggoner AS, Wang CH. The Journal of membrane biology. 1974;19:1–36. doi: 10.1007/BF01869968. [DOI] [PubMed] [Google Scholar]
- 11.Gupta RK, Salzberg BM, Grinvald A, Cohen LB, Kamino K, Lesher S, Boyle MB, Waggoner AS, Wang CH. The Journal of membrane biology. 1981;58:123–137. doi: 10.1007/BF01870975. [DOI] [PubMed] [Google Scholar]
- 12.Efimov IR, Biermann M, Zipes D. In: Cardiac Mapping. 2nd edn. Shenasa S, Borggrefe M, Breithardt G, editors. Blackwell Publishing; VT: 2003. pp. 131–156. [Google Scholar]
- 13.Grinvald A, Hildesheim R, Farber IC, Anglister L. Biophysical Journal. 1982;39:301–308. doi: 10.1016/S0006-3495(82)84520-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Obaid AL, Loew LM, Wuskell JP, Salzberg BM. Journal of Neuroscience Methods. 2004;134:179–190. doi: 10.1016/j.jneumeth.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Fluhler E, Burnham VG, Loew LM. Biochemistry. 1985;24:5749–5755. doi: 10.1021/bi00342a010. [DOI] [PubMed] [Google Scholar]
- 16.Kozlowski A, Charles SA, Harris JM. Biodrugs. 2001;15:419–429. doi: 10.2165/00063030-200115070-00001. [DOI] [PubMed] [Google Scholar]
- 17.Jen AKY, Rao VP, Wong KY, Drost KJ. Journal of the Chemical Society-Chemical Communications. 1993:90–92. [Google Scholar]
- 18.Hu ZY, Fort A, Barzoukas M, Jen AKY, Barlow S, Marder SR. Journal of Physical Chemistry B. 2004;108:8626–8630. [Google Scholar]
- 19.Itsuno S, Sawada T, Hayashi T, Ito K. Journal of Inorganic and Organometallic Polymers. 1994;4:403–414. [Google Scholar]
- 20.Raimundo JM, Blanchard P, Gallego-Planas N, Mercier N, Ledoux-Rak I, Hierle R, Roncali J. The Journal of organic chemistry. 2002;67:205–218. doi: 10.1021/jo010713f. [DOI] [PubMed] [Google Scholar]
- 21.Matiukas A, Mitrea BG, Pertsov AM, Wuskell JP, Wei M.-d., Watras J, Millard AC, Loew LM. Am J Physiol Heart Circ Physiol. 2006;290:H2633–2643. doi: 10.1152/ajpheart.00884.2005. [DOI] [PubMed] [Google Scholar]
- 22.Fromherz P. Journal of Physical Chemistry. 1995;99:7188–7192. [Google Scholar]
- 23.Roberts MJ, Bentley MD, Harris JM. Advanced Drug Delivery Reviews. 2002;54:459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 24.Hassner A, Birnbaum D, Loew LM. Journal of Organic Chemistry. 1984;49:2546–2551. [Google Scholar]
- 25.Chen CT, Marder SR. Advanced Materials. 1995;7:1030–&. [Google Scholar]
- 26.Loew LM, Scully S, Simpson L, Waggoner AS. Nature. 1979;281:497–499. doi: 10.1038/281497a0. [DOI] [PubMed] [Google Scholar]
- 27.Johnsson M, Hansson P, Edwards K. Journal of Physical Chemistry B. 2001;105:8420–8430. [Google Scholar]
- 28.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS. Bioconjugate Chemistry. 1993;4:105–111. doi: 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- 29.Baker LC, London B, Choi BR, Koren G, Salama G. Circulation research. 2000;86:396–407. doi: 10.1161/01.res.86.4.396. [DOI] [PubMed] [Google Scholar]
- 30.Ernst LA, Gupta RK, Mujumdar RB, Waggoner AS. Cytometry. 1989;10:3–10. doi: 10.1002/cyto.990100103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.