

Preface
Cardiovascular effects of epoxyeicosatrienoic acids (EETs) include vasodilation, vascular smooth muscle cell anti-migratory actions, and anti-inflammatory actions. These endogenous lipid mediators are broken down to diols by soluble epoxide hydrolase (sEH), and so inhibiting this enzyme would be expected enhance the beneficial cardiovascular properties of EETs. The rapid development of 1,3-disubstituted urea based sEH inhibitors (sEHIs) has resulted in a number of studies demonstrating cardiovascular protection, and it has been shown that sEHIs are anti-hypertensive, anti-inflammatory, and protect the brain, heart and kidney from damage. Although challenges for the future exist — including improving the drug like properties of sEHIs and finding better ways to target sEHIs to specific tissues — the recent initiation of first in human clinical trials has highlighted the promise of sEHIs as a therapeutic target.
Many of the enzymes, receptors and the eicosanoid metabolites of the arachidonate cascade are major therapeutic targets, particularly for inflammatory disease. The first pathway to be targeted was cyclooxygenase (COX), which leads to the generation of prostaglandins (PG). Indeed, aspirin and non-steriodal anti-inflammatory drugs (NSAIDs), including COX-2 inhibitors, are effective drugs that treat pain and inflammation.1,2 These drugs also may be useful for treating or preventing cardiovascular diseases —inhibition of blood clotting by aspirin has been touted as a preventative for ischemic events such as heart attacks and stroke1 — and prostacylin analogs are used for the treatment of pulmonary hypertension.3,4 On the other hand, enthusiasm for the COX pathway was greatly decreased because of the increased incidence of acute renal failure, myocardial infarction and thrombotic stroke in patients treated with COX-2 inhibitors.1,2,5,6 The second eicosanoid and inflammatory pathway targeted for therapeutics was the lipoxygenase (LOX) generation of leukotrienes (LT). 5-LOX and LT receptor antagonists have been developed for the treatment of asthma and seasonal allergies.7,8 These two eicosanoid pathways continue as important therapeutic targets as novel receptors and metabolites have been identified and their roles in a myriad of diseases are being better defined [figure 1].
Figure 1. Therapeutic Targets of the Arachidonate Cascade.
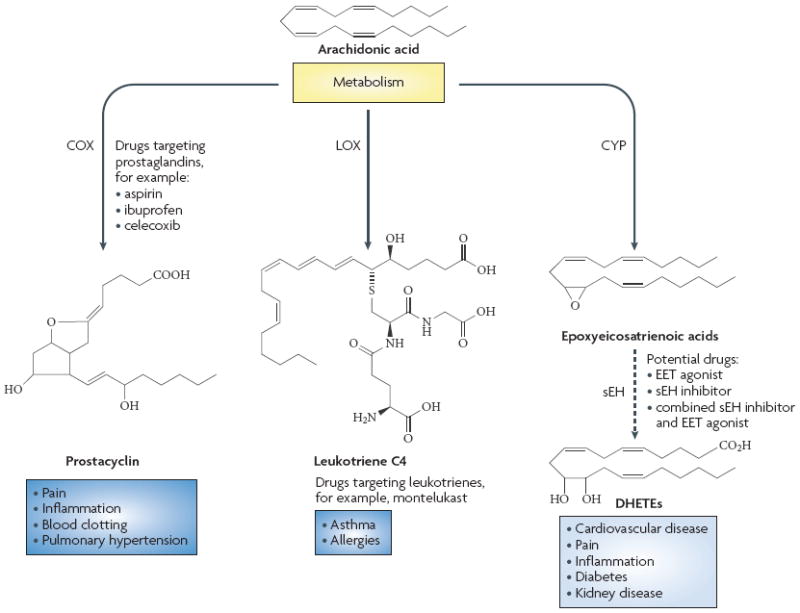
Three major pathways the cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP) pathways can metabolize arachidonic acid. Inhibitors of the COX-1 and COX-2 enzymes are used for the treatment of pain, inflammation and blood clotting and prostacyclin analogs are used to treat pulmonary hypertension. Leukotriene receptor antagonists that inhibit the cysteinyl leukotriene CysLT1 receptor are used to treat asthma and allergies. Soluble epoxide hydrolase inhibitors that increase epoxyeicosatrienoic acid levels are being developed for the treatment of cardiovascular diseases and inflammation.
A third eicosanoid pathway, the cytochrome P450 (P450) was first described in 1980 and is comprised of two enzymatic pathways9,10,11 - the hydroxylases and the epoxygenases. The hydroxylase P450 enzymes convert arachidonic acid into hydroxyeicosatetraenoic acids (HETEs). 20-HETE is the major metabolite of this pathway and has been determined to be pro-inflammatory and important to vascular function.12,13 This pathway and metabolite are currently being targeted for the treatment of cardiovascular diseases such as hypertension and stroke.13-16. The second pathway is the generation of epoxyeicosatrienoic acids (EETs) by P450 enzymes, which catalyze the epoxidation of arachidonic acid olefin bonds resulting in the production of four regioisomeric EETs: 5,6-EET; 8,9-EET; 11,12-EET; 14,15-EET. EETs or epoxyeicosanoids have been demonstrated to be endothelium-derived hyperpolarizing factors (EDHFs), protect from ischemic injury and possess anti-inflammatory actions in canine and rodent disease models.17-21 Conversion of EET epoxides to their corresponding diols (DHETs) by soluble epoxide hydrolase (sEH) enzyme are responsible for decreasing EET levels and thus diminishing their beneficial cardiovascular properties,20,21 and so inhibition of this enzyme would be a target for cardiovascular disease. Recently, sEH inhibitors (sEHIs) have been developed to enhance the cardiovascular actions offered by EETs. This article will highlight the development of sEHIs as cardiovascular therapeutics and discuss the potential for this treatment and challenges that lie ahead.
Biological Aspects of Epoxyeicosanoids
Since the first descriptions of the biological actions of EETs, which included increases in epithelial transport in the kidney and dilation of small mesenteric resistance arteries, there has been growing interest in these eicosanoid metabolites.22,23 Interest in EETs was greatly increased in 1996 with the identification of EETs as an EDHF.17 Over the past decade it has become increasingly apparent that EETs have a myriad of cardiovascular actions, the overwhelming majority of which appear to be cardiovascular protective.
The cellular signaling mechanisms responsible for the various EET biological actions have been and continue to be intensively investigated. There is ample evidence that supports the possibility that EETs bind to receptors that are coupled by a G-protein to intracellular signaling cascades;24,25 however, an EET receptor has yet to be identified. EETs could also function inside the cell by coupling to and activating ion channels, signaling proteins or transcription factors. Experimental evidence supports an intracellular mechanism of action in that EETs are incorporated into cell membrane phospholipids, bind to fatty acid binding proteins, and peroxisome proliferators-activated receptor (PPAR) γ.21,24,26,27 Please see the following reviews for more comprehensive descriptions of EET biological activities and cellular signaling mechanisms.28,29
As with other eicosanoid pathways, there is heterogeneity with respect to cellular signaling mechanisms and biological activities in various cell types and tissues. Other experimental issues have made investigation of EETs and the P450 enzymatic pathways difficult. One consistent concern is the quality and purity of regioisomeric EETs and their proper handling. Likewise, investigations in cell culture systems are limited by the fact that epoxygenase and epoxide hydrolase enzymes decrease rapidly following cell isolation. Experimental approaches to circumvent these issues include the generation of genetically manipulated mice, transfection of cell culture lines with P450 enzymes, and development of EET analogs and antagonists with better chemical properties and greater stability.18,30 Moreover, EET receptor identification could provide more clarity to the apparent biological heterogeneity much like the discovery of multiple PGE2 (EP) receptors helped explain away the apparent contradictions in biological and cell-signaling mechanisms for that eicosanoid.31,32 Even with these experimental concerns there has been tremendous progress in determining EET biological actions and EETs remain an attractive therapeutic target for cardiovascular diseases.
Vascular Actions of EETs
EETs as vasodilators and EDHFs are the most extensively examined cardiovascular actions. Vasodilation in response to EETs has been observed in a number of organs including the heart, brain, kidney, skeletal muscle and intestine.13,17,23,33,34 In contrast, EETs cause vasoconstriction in the lung - a finding that was not unexpected since PGs also have opposite effects in this vasculature when compared to other organs.35,36 All regioisomeric EETs have been demonstrated to be vasodilators with 11,12-EET and 14,15-EET consistently exhibiting the most vasodilator activity.21,34 These two regioisomeric EETs generated by endothelial cells dilate blood vessels by activating large-conductance calcium-activated K+ (KCa) channels on vascular smooth muscle cells,33,37,38,39 resulting in K+ efflux from the smooth muscle cell and subsequent membrane hyperpolarization.17,38 There is evidence for cAMP activation of protein kinase A (PKA) and ADP ribosylation of Gsα cell signaling mechanisms as being responsible for mediating EET activation of vascular smooth muscle cell KCa channels.39-42 The ability of EETs to activate KCa channels and dilate blood vessels can be regulated by sEH-mediated conversion to DHETs that exhibit diminished or absent vasorelaxation.34,38,43 In this regard, sEH inhibition impedes the conversion of EETs to DHETs and improves dilator activity in human blood vessels.43 EETs or sEHIs have been demonstrated to oppose the vasoconstrictor activities of the pro-hypertensive hormones endothelin-1 and angiotensin II.20 Thus decreased endothelial EET conversion to DHETs could be one mechanism responsible for the anti-hypertensive actions observed with the administration of sEHIs as well as for other cardioprotective properties.
Vascular homeostasis is controlled by endothelial cell and vascular smooth muscle cell proliferation and migration, and EETs and sEH appear to be important regulators of these cellular processes.18,30,44-49 EETs promote endothelial cell proliferation and migration and are angiogenic, and it has been demonstrated that the epoxides and not the corresponding diols resulted in proliferative effects.45 In murine and human cell lines, EETs or overexpression of CYP2C epoxygenases leads to proliferative responses,30,46 that have been attributed to activation of two cell-signaling pathways; the p38 mitogen-acitvated protein kinase (MAPK) pathway and the phosphatidylinositol 3-kinase-Akt (PI3K/Akt) pathway.30 11,12-EET activates MAPK, which upregulates cyclin D, and Akt, which phosphorylates forkhead factors (FOXO) and decreases cyclin-dependent kinase inhibitor p27kip1 in endothelial cells.46,47 More recently, 11,12-EET-mediated proliferation, migration and tube formation in human umbilical vein cells (HUVECs) was demonstrated to be dependent on activation of sphingosine kinase 1 (SK1) that phosphorylates sphingosine to generate spingosine-1-phosphate (S1P).49 On the other hand, EETs exhibit antimigratory actions in vascular smooth muscle cells. 11,12-EET and 14,15-EET moderately attenuated the migration of aortic smooth muscle cells in response to platelet-derived growth factor,50 and CYP2J epoxygenase overexpression or inhibition of sEH also reduced smooth muscle cell proliferation and migration.50 Activation of the cAMP-dependent PKA pathway and decreased cyclin D levels have been implicated in the antimigratory actions of EETs and sEHIs in vascular smooth muscle cells.30 Although these findings suggest a vascular smooth muscle cell effect of EETs and sEHIs, other reports have failed to demonstrate that EETs or sEHIs result in vascular smooth muscle proliferation.51,52 More importantly, in vivo angiogenesis is stimulated by EETs in a subcutaneous sponge model and inhibition of sEH enhanced these pro-angiogenic and neo-vascularization responses.48 Effects of EETs and sEHIs on proliferation and migration of endothelial and vascular smooth muscle cells reveal the possible importance of targeting this pathway in angiogenesis, atherosclerosis and other cardiovascular diseases.
Anti-inflammatory Actions of EETs
Inflammation and inflammatory diseases contribute significantly to vascular and end organ damage and cardiovascular disease progression.53,54 Likewise, interactions between inflammation and the epoxygenase pathway, that can affect cardiovascular function in disease states, have been clearly established.55-58 Cytokines can decrease CYP2C expression and oppose epoxygenase-mediated vasodilation.18,59 Conversely, TNFα and CCR2 inhibition result in an increase in kidney CYP2C expression and decrease renal injury in hypertension.55,56 Experimental evidence supports the postulate that EETs interfere with activation of the transcription factor, nuclear factor κB (NF-κB) to exert their vascular anti-inflammatory effects.57,58 11,12-EET but not other regioisomeric EETs prevent TNFα induced activation of NF-κB and increased vascular cell adhesion molecule-1 (VCAM-1) expression in endothelial cells.57 Likewise, CYP2J epoxygenase overexpression in endothelial cells also decreases NF-κB activation.57 Although additional studies will be required to determine the exact cellular signaling mechanisms responsible for the anti-inflammatory actions of EETs, there is considerable evidence that EETs decrease inflammation. Further anti-inflammatory actions attributed to EETs include decreased aggregation of human polymorphonuclear leukocytes and leukocyte adhesion to endothelial cells,58-61 and attenuation of IL-1β induced fever. In the latter case 11,12-EET (administered to the brain) had a greater antipyretic action than other EETs.62,63 Studies using sEHIs also support the notion that EETs have anti-inflammatory actions.64-67 Inhibition of sEH decreased cigarette smoke-induced lung inflammation and significantly reduces neutrophils, alveolar macrophages and lymphocytes in the bronchial fluid.68 These findings have provided sufficient evidence that sEHIs could be protective against the deleterious effects of inflammation associated with cardiovascular diseases as well as a treatment for other inflammatory diseases.
Epoxygenase Pathway Polymorphisms in Human Disease
The notion that both the EETs and the sEH enzyme are therapeutic targets for human disease is supported by genetic studies. There are a number of polymorphisms in EPHX2 — the gene responsible for sEH production —that result in amino acid substitutions that influence sEH enzymatic activity.69-73 Two studies have linked genetic variation in EPHX2 to increased risk for coronary artery disease, and smoking further increased the risk associated with this genetic variation.69,72,74 Other cardiovascular diseases associated with genetic variation in EPHX2 include ischemic stroke and hypercholesterolemia.71,75 Genetic variations in the epoxygenase CYP2J2, CYP2C8 and CYP2C9 can effect transcription or enzymatic activity.73,76,77 CYP2C8 and CYP2C9 variants have been associated with myocardial infarction and cardiovascular disease.76,77 Taken together, these findings in the patient population provide evidence that sEHIs could have potential therapeutic value in a large variety of cardiovascular diseases and that they may be of particular benefit for patients of certain genotypes.
Design of Soluble Epoxide Hydrolase Inhibitors
The rapid development of sEHIs for in vivo use and clinical testing in the past decade is quite remarkable. A landmark study published in 200078 demonstrated that injection of a sEHI to a spontaneously hypertensive rats (SHR) lowered blood pressure. This study was followed up by the first demonstration that chronic inhibition of sEH lowered blood pressure in angiotensin-induced hypertension.79 Another breakthrough came in 2005 when it was shown that an orally administered sEHI was anti-hypertensive and slowed the progression of renal damage.80 Following this, a number of studies have provided exciting findings on the broad potential for sEHIs as a cardiovascular therapeutic, and as further evidence of the rapid development of this therapeutic, a first in class sEHI began clinical phase IIa testing in humans this year (http://www.aretetherapeutics.com/news/2009/021009.html). In this section we will describe the evolution of selective sEHIs from enzyme inhibition in vitro to oral administration into rodents and subsequently humans.
There are two well studied α/β-hydrolase fold epoxide hydrolase enzymes that differ by subcellular localization and substrate selectivity.56,81,82 The microsomal epoxide hydrolase (mEH) is involved in the metabolism of environmental contaminants, and it has been extensively studied in this role,81,82 whereas the sEH was first discovered while studying the metabolism of terpenoid epoxide that mimicked insect juvenile hormone.81,82 At the same time EETs were being established as endogenous lipid mediators with biological activity. Subsequently it was discovered that arachidonic acid and linoleic acid epoxides are metabolized by sEH and sEH coverts these epoxides to diols with high Vm and low Km.81,82,83 Since this finding some three decades ago, the message and gene have been cloned and the sEH catalytic mechanism determined. The mammalian sEH is a homodimer with monomers arranged in an anti-parallel form.81,82,83 Each monomer is composed of two domains; the C-terminal domain contains epoxide hydrolase activity while the N-terminal domain hydrolyzes phosphates on lipophilic backbones.84,85,86 This highly conserved enzyme is widely distributed in tissues including the liver and kidney where sEH specific activity is highest.85,87,88,89 The functional significance of the N-terminal domain of the mammalian sEH remains highly speculative. The N-terminal domain possesses phosphatase activities that dephophorylate polyisoprenyl phosphates known to regulate cholesterol levels.84,86,90 It has been speculated that the N-terminal domain could stabilize the epoxide hydrolase activity because expression of the human sEH C-terminal domain alone has reduced activity.90 The N-terminal domain might promote dimerization of the sEH enzyme.89,91 To date, there are no known selective inhibitors of this N-terminal domain, so determination of the functions of the N-terminal domain awaits further experimental studies. Current sEHIs inhibit the epoxide hydrolase activity of the C-terminal domain without affecting the phosphatase activity of the N-terminal domain.
The first generation sEHIs were potent inhibitors acting as alternative substrates and included chalcone oxides and glycidols.21,85,92 Unfortunately, these alternative substrates were quickly inactivated by glutathione and glutathione transferases making them difficult to use in tissues and in vivo.21,85 A major breakthrough came when amides, ureas and carbamates were found to be potent and stable transition state inhibitors of sEH, because these tools facilitated experiments to search for endogenous roles for this enzyme.85,93 The design of these transition state mimics was based on the knowledge of the catalytic mechanism of the enzyme.85,93 X-ray structures of the murine and human enzyme with these urea sEHIs suggested that the urea is the central pharmacophore and that hydrogen bond stabilized salt bridges between urea and residues of the C-terminal sEH active site were formed.85,93 This supports the hypothesis that ureas are imitating features present in transient intermediates or transition states encountered along the reaction coordinate of the epoxide ring opening by the sEH. These 1,3-disubstituted ureas, carbamates and amides inhibit the C-terminal epoxide hydrolase activity of the sEH enzyme with nanomolar Ki’s but do not dramatically alter phosphatase activity of the N-terminal domain.85,93 The urea pharmacophore appeared to be the most potent, however with adequate substituents amides and carbamates of equal potencies can be obtained. Subsequent modifications to improve in vivo stability allowed evaluation of the role of the sEH enzyme in cardiovascular diseases.65,85,93,94 Although the mEH has the same catalytic mechanism as the sEH, selection of substituents on the amides and ureas allow one to design inhibitors with greater than 1000 × selectivity for one hydrolase over the other.85,95 The sEHIs tested appear to be very selective for the sEH and the 300+ positive hits from a NIH screen on sEH have no consistent inhibition of other enzymes.96 The anticancer drug sorafenib — a potent inhibitor of several kinases is a very potent sEH inhibitor. This joint inhibition appears limited to sEHI closely related to sorafenib and possibly the sEH inhibition of sorafenib reduces some of the side effects associated with this drug class when used at high doses.97
The first report of a sEHI to demonstrate in vivo biological effects used a single bolus dose N,N’-dicyclohexylurea (DCU), which lowered blood pressure in hypertensive rats.78 Chronic administration was first achieved with 1-cyclohexyl-3-dodecyl-urea (CDU), which when injected intraperitoneally for four consecutive days had anti-hypertensive actions.79
These high melting ureas have limited solubility in either water or organic solvents so careful formulation is needed to demonstrate in vivo efficacy.81,85,98 Incorporation of functional polar groups into one of the alkyl chains of 1,3-disubstituted urea sEHIs resulted in compounds that were weak structural mimics of EETs with improved physical properties.94,99 One example was 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA, figure 2) that has been widely used in cultured cells and animals.21,65,81,85 Although AUDA can be orally administered, it requires DMSO for in vitro experiments or a sizable amount of 2-hydroxylpropyl β-cyclodextrin for it to be administered in drinking water for in vivo studies.21,65,81 If lipophilic compounds fall out of solution bioavailability decreases dramatically. As expected for an enzyme with a largely hydrophobic catalytic tunnel, addition of polar groups in general results in a dramatic reduction in potency. However, a polar group — termed – a secondary pharmacophore — such as an ether, ester, amide, sulfonamide, alcohol or ketone roughly 7-8Ǻ from the polar group of the central pharmacophore increased water solubility with out sacrificing potency.81,94,98,100 The application of this concept resulted in more drug-like sEHI molecules including t-AUCB, TPAU (figure 2), and others that have both excellent potency and efficacy in many species.
Figure 2. Soluble Epoxide Hydrolase Inhibitor (sEHI) Structures and Binding to the Enzymatic Pocket.

Left Panel: A.) The early compound AUDA contains the central pharmacophore C that forms multiple hydrogen bonds in the enzyme catalytic site. The R1 or left side of the molecule rests in a hydrophobic pocket of the sEH catalytic tunnel. The hydrophobic right side of AUDA was designed to mimic 14,15-EET. AUDA is a highly potent sEHI but must be formulated carefully for use in vivo. B.) TPAU is a potent sEHI illustrating that a polar secondary pharmacophore P 7-8 Å from from C increases solubility while maintaining potency. It uses a piperidine as a L or linker group between C and P. C.) AUCB (both trans and cis isomers are active) has an ether as P with a R2 on the right side reaching toward the enzyme surface and mimicking the carboxylate of EETs. Both TPAU and AUCB are highly potent and have good oral availability and pharmacokinetic characteristics. AUCB is more generally potent across multiple species. Right Panel: Structure of sEH enzymatic pocket with bound sEHI.
In parallel to the development of sEHIs better suited for experimental studies, development of sEHIs for use in humans has advanced. These sEHIs are initially being developed for the treatment of hypertension. Arête Therapeutics commenced phase 1 clinical trials in healthy volunteers with the first in class sEH inhibitor AR9281 in October 2007 and phase 2 trials are in progress (http://www.aretetherapeutics.com/news/2009/021009.html and http://www.aretetherapeutics.com/news/2008/121808.html). Amazingly, in less than a decade the sEH has gone from obscurity to a recognized therapeutic target, and sEHIs have gone from their first demonstration of anti-hypertensive actions to being tested for the treatment of diseases in humans.
Cardiovascular Therapeutic Effects of Soluble Epoxide Hydrolase Inhibitors
sEHIs have cardiovascular protective effects in hypertension, cerebral ischemia, cardiac ischemia, cardiac hypertrophy and atherosclerosis,79,101-105 suggesting that these agents have broad potential for the treatment of many cardiovascular diseases and associated morbidity.21,65,106 The progression of end organ damage, inflammation, and endothelial dysfunction associated with cardiovascular disease are also attenuated by sEH inhibition.67,103,104,107 Studies conducted in mice with Ephx2 gene deficiency a support the notion that sEHIs effects are a consequence of inhibition of the C-terminal epoxide hydrolase domain.35,101,103,104,108,109,110 Although these Ephx2 -/- mice have the potential to unveil the function of the N-terminal domain of the sEH enzyme, a role for this domain has remained elusive.
Antihypertensive effects
sEHIs have hypertensive properties in numerous animal models of hypertension.20,21,65 (figure 3) In the SHR, the urea DCU lowered blood pressure and decreased urinary DHET excretion,78 and CDU (given once daily) lowered blood pressure in hypertension driven by angiotensin infusion in the rat.79 The first sEHI to be successfully administered orally to hypertensive animals,80 AUDA, lowered blood pressure in rat and mouse models of hypertension.80,102,111 Blood pressure was consistently lowered by 25 to 30 mmHg in the rat models of hypertension, however; in mice that had angiotensin dependent hypertension, blood pressure was lowered to that of control mice.80,102,111 The mechanism for lowering blood pressure appears to be dependent on decreased vascular resistance and enhanced sodium excretion by the kidney.67,80,102 These findings are in line with the biological actions of EETs to dilate blood vessels and inhibit renal tubular sodium reabsorption.20,21,22,111 It was also in these initial hypertension studies that the first evidence for end organ protection by sEHIs was recognized.65,67
Figure 3. Soluble Epoxide Hydrolase Inhibitor (sEHI) Anti-hypertensive and End Organ Protective Actions.
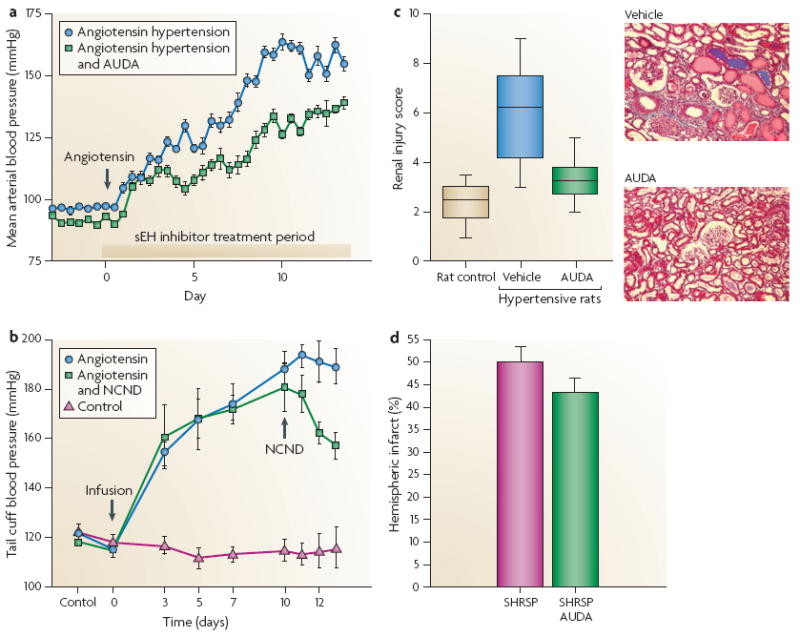
A.) sEHI treatment given orally at the onset of hypertension induced by angiotensin infusion lowers blood pressure. B.) sEHI treatment when given after the development of hypertension induced by angiotensin infusion lowers blood pressure. C.) sEHI decreases renal injury in diabetic hypertensive Goto-Kakizaki rats independent of lowering blood pressure. D.) sEHI decreases brain injury associated with cerebral ischemia in stroke-prone spontaneously hypertensive rats (SHRSP) independent of lowering blood pressure. Figure adapted from data in references 79,80,109,117.
There have been some conflicting reports on sEHi-mediated blood pressure lowering in rats and mice. These species differences may be because many tissues in some rat strains including liver and kidney have very low sEH activity and rat strains vary dramatically in their sEH levels. Although the first demonstration of an sEHI to lower blood pressure was in the SHR, subsequent studies have shown variable levels of blood pressure lowering in this model113-115 which could be in part due to polymorphisms in the Ephx2 gene between SHR strains.113,116 There is also conflicting results on sEHi-mediated changes in blood pressure in Ephx2-/- mice. The initial Ephx2-/- male mice had decreased blood pressures that could not be confirmed when these mice were back bred into a C57/BL6 background or in an independently generated Ephx2-/- mice colony.117,118 More recently studies in a second Ephx2-/- C57/BL6 back bred colony did not demonstrate lower blood pressures in males at baseline but DOCA-salt induced hypertension was attenuated.119 Interestingly, each Ephx2-/- mouse colony demonstrated high EET levels and high 20-HETE levels that could have offset some of the blood pressure effects.118 Even though the anti-hypertensive actions of sEHIs have been variable, the ability of sEHIs to protect from end organ damage associated with cardiovascular diseases has been much more consistent.
Kidney Protective Properties
Chronic sEHI treatment attenuated renal vascular and glomerular injury in rats with angiotensin-induced hypertension, demonstrating that sEHIs provided protection from end organ damage associated with cardiovascular disease.67,80 In this model, decreases in collagen expression in glomeruli and tubular cells as well as decreased vascular hypertrophy were observed. Moreover, urinary albumin excretion was decreased and macrophage infiltration was reduced. These studies also determined that sEHI treatment starting either at the onset or after the establishment of hypertension provided similar protection to the kidney. Although in these hypertensive animal models, the renal protection afforded by sEHIs could have been a result of the decrease in blood pressure, a more recent study in diabetic Goto-Kakizaki rats clearly demonstrated that AUDA provides renal protection independent of lowering blood pressure.107 Moreover, the elevated plasma cholesterol and triglyceride levels observed in the diabetic Goto-Kakizaki rats were not lowered by AUDA treatment.107 In addition to the studies animal models of chronic progressive kidney disease, sEHIs can provide protection from acute renal injury induced by the chemotherapeutic agent cisplatin in mice.120 Inhibition of sEH decreased blood urea nitrogen levels for up to 96 hours and reduced tubular damage associated with cisplatin.120 Overall, these studies have consistently found improved renal vascular function, decreased glomerular injury and a decrease in renal inflammation that demonstrate the promise of sEHIs as a treatment for acute and chronic kidney disease.
Cardiac Protective Properties
A major therapeutic potential for sEHIs is the cardiac protective properties, especially from myocardium ischemic events. Ephx2 gene deficient mice have improved recovery of left ventricular developed pressure (LVDP) and reduced infarct size following ischemia and reperfusion and are also protected from developing pressure overload induced heart failure and cardiac arrhythmias.104 The ability of sEHIs to improve cardiac function has been established in various experimental models and species.19,104,108,121 AUDA reduces the cardiac infarct size in dogs and this protection is similar to that observed with 14,15-EET administration.19 Similar findings were observed in mice that were administered AUDA-BE and subjected to left coronary artery occlusion followed by reperfusion.104 Furthermore, in dogs and mice the EET antagonist, 14,15-EEZE inhibits the protection to the heart provided by sEHIs.19,104 Acute myocardial infarction hypertension can result in cardiac hypertrophy due to ventricular remodeling.101,104,111,121 The first evidence that sEHIs could attenuate cardiovascular hypertrophy was the observation that heart weight and collagen were decreased in sEHI-treated deoxycorticosterone (DOCA) salt hypertensive rats.111 Likewise, cardiac hypertrophy in stroke-prone SHR and angiotensin infused rats was prevented by inhibition of sEH.101,122 The cardiac protective actions of sEHIs have also been found in mice with pressure overload induced myocardial hypertrophy, where sEHIs prevented the development or reversed left ventricular hypertrophy,104,121 which was linked to the ability of sEHIs to block NF-κB activation.121 Although there is overwhelming evidence that Ephx2 deficiency and sEHIs provide cardiac protection, Ephx2 knockout mice had reduced survival from cardiac arrest and cardiopulmonary resuscitation.123 Future experimental evidence is required to determine the potential for sEHIs as therapies for various heart ailments.
Ischemic stroke protection & Vascular Disease
Another potential therapeutic use for sEHIs is protection from ischemic brain damage that accompanies stroke. Chronic treatment with AUDA to stroke-prone SHR decreases cerebral infarct size after middle cerebral artery occlusion.103,114,115,124 Interestingly, blood pressure was not lowered in these hypertensive rats supporting the notion that the cerebral protective effects were independent of blood pressure.114 Ephx2 deficient mice have decreased infarct size following a cerebral ischemia.103,124 Ischemic stroke protection has also been determined in a mouse model of focal ischemia reperfusion injury, where administration of AUDA-BE or exogenous EETs resulted in at least a 50% reduction in infarct volume.124,125 Moreover, administration of sEHIs one hour prior to the onset or at the start of reperfusion provided cerebral protection.124,125
The mechanisms by which sEHIs protect the brain from ischemic damage appear to be multi-modal and involve the cerebral vasculature and neurons.124,125 EETs and sEHIs can protect the neurons through anti-apoptotic and anti-inflammatory actions and vasodilator EETs mediate cerebral blood flow regulation and could contribute to brain protection.124,125 Angiogenic and attenuated vascular remodeling that allow for enhanced perfusion of the ischemic area have been observed in the stroke-prone SHR treated with sEHIs.115 However, these vascular changes do not occur in normotensive animals that also demonstrate decreased infarct volume when treated with sEHIs.115 Taken together, these findings indicate that sEHIs have broad pharmacological potential for treating ischemic stroke.
Other areas that are beginning to be explored include effects of sEHIs on vascular remodeling, angiogenesis, diabetes, and atherosclerosis. Inhibition of sEH decreased vascular hypertrophy in hypertension and decreases vascular smooth muscle cell proliferation.51,52,67 Angiogenic actions of EETs have been demonstrated in mice that were enhanced in the presence of a sEHI.48 Increased microvascular densities and increased middle cerebral artery compliance were associated with AUDA treatment in the stroke-prone SHR.114,115 More recent studies demonstrate that sEHIs or Ephx2 deletion antagonizes neointimal formation in vivo by mechanisms that are endothelium dependent.105,126 Along these lines, atherosclerosis in apolipoprotein e knockout mice was reduced by sEHI treatment.105 Thus, sEHIs may have therapeutic potential for specific types of vascular remodeling and atherosclerosis.
Anti-inflammatory Properties
The anti-inflammatory actions of sEHIs have been demonstrated to be a key component to end organ protection in cardiovascular disease models.18,65 There is also strong evidence that sEHIs have efficacy for the treatment of inflammatory diseases.64,66,109,127 AUDA-BE reduced the production of cytokines and pro-inflammatory lipid mediators and diminished lipopolysaccharide-induced mortality in mice.127 Furthermore, topical application of sEHIs reduced lipopolysaccharide-induced thermal hyperalgesia and mechanical allodynia inflammatory pain in rats.66 Although there is ample evidence that sEH inhibition is anti-inflammatory, Ephx2 deficient or sEHI administration to wild-type mice did not reduce lipopolysaccharide-induced inflammatory gene expression or neutrophil accumulation in the liver. 109
Lung inflammation is another area where sEHIs could have therapeutic value. Mice exposed to tobacco smoke had only three aortic endothelial cell genes that demonstrated a 3-fold or greater increase in expression, one of which was Ephx2.68 Inhibition of sEH reduces macrophage infiltration into the rat lung exposed to tobacco smoke and was further reduced by the combination of AUDA-BE and EET treatment.68 On the whole, these experimental findings reveal that sEHIs provide beneficial anti-inflammatory and analgesic actions.
Therapeutic Potential and Challenges
Based on studies in a wide variety of experimental animal models of cardiovascular disease, significant interest has been generated in the therapeutic potential that can be afforded by sEHIs. Human studies are also providing evidence for a contribution of sEH to cardiovascular as well as other disease states. One consideration is that the long-term treatment of many cardiovascular diseases requires a very high safety profile. To date the very large therapeutic index makes sEHIs very attractive in this respect. However, the requirement to perform clinical trials in addition to currently accepted treatments and the high bar for safety of new therapies makes the route to the clinic very expensive. It could be that other disease indications will be more attractive routes for the first sEHI phase 3 trials. The findings from animal studies and the initial clinical trials will undoubtedly be expanded on in the future and clinical translational studies will ultimately determine the best therapeutic uses and limitations for sEHIs. What are these therapeutic potentials and what challenges lie ahead?
Future for Novel Therapeutic Applications
The therapeutic potential for sEHIs for treating cardiovascular diseases appears to be exceptionally promising. Patients with cardiovascular diseases are treated many times with multiple medications for conditions such as high blood pressure, hypercholesterolemia, high blood glucose and hyperlipidemia to name just a few. There is ever increasing evidence that sEHIs can synergize with existing medications and could be designed as combinational drugs.27,64,128 COX-2 protein is decreased by sEHIs resulting in decreased PGE2 levels while maintaining the PGI2 to TXA2 ratio, suggesting that low dose COX-2 inhibition in combination with sEHIs have additive to synergistic anti-hyperalgesic and anti-inflammatory effects without a decrease in the cardiotoxic PGI2 to TX ratio.64 The complexity of the arachidonate cascade suggests that sEHIs will have interactions with other NSAIDs, 5-LOX and LT receptor antagonists currently on the market, which could be beneficial or detrimental as therapies for cardiovascular diseases. Further evaluation of sEHIs and interactions with other eicosanoid pathways will undoubtedly provide some surprises with the potential for improving cardiovascular therapeutics.
Another interesting finding has been that AUDA has weak PPARα agonistic activity, suggesting that one could design combinational drugs from this sEHI,128 that could be beneficial for patients with hyperlipidemia and hypertension. Although AUDA failed to lower blood pressure, cholesterol or triglyceride levels in hypertensive and diabetic Goto-Kakizaki rats,107 urea based alkanoic acid sEHIs can transactivate PPARα and this PPARα activity attenuates vascular smooth muscle cell proliferation.27 One could also envision designing a sEHI with PPARγ agonistic activity for the treatment of cardiometabolic syndrome. This is an interesting prospect since PPARγ agonists have the unwanted effect of causing fluid retention that could be detrimental to patients with heart failure and other cardiovascular diseases.129 Interestingly, sEHIs and EETs are natriuretic, i.e. they increase sodium and water excretion, and could lessen the fluid retaining state during PPARγ agonist treatment.20,65,79,102 Overall there is great potential for sEHIs as anti-hypertensive treatment that could be used in combination with other medications for patients with poor cardiovascular health, and they may be particularly valuable in patients with co-morbidities.
In addition, sEHIs could have broad neural protective actions. There is now mounting evidence that sEHIs provide protection from brain damage following cerebral ischemia by means that are independent of vascular actions, possibly due to sEHI-induced increase in the neuronal expression of pro-survival anti-apoptotic genes.115 This is further support by data demonstrating that ischemic preconditioning in the brain involves a hypoxic inducible factor-α mediated increase in the CYP2c11 epoxygenase enzyme in astrocytes.130
Recent studies also indicate that 14,15-EET activates opioid receptors in the ventrolateral periaqueductal gray area of the brain to produce antinociception.131 Interestingly, topical application of sEHIs can reduce inflammatory-induced pain and demonstrates the promise for sEHIs as analgesics.66,132 On the other hand, the potential for sEHIs to treat neurological disorders such as Alzheimer’s or multiple sclerosis has yet to be explored.
Potential for Unwanted Effects
The potential of unwanted effects must also be considered when developing sEHIs for the treatment of cardiovascular diseases. sEHIs can promote angiogenesis and this could result in acceleration of tumorogenesis in patients with some types of cancer.48,115 EETs are potent angiogenic lipids that promote vascularization of tumors in vivo.48,133,134 Epoxygenase metabolites have been demonstrated to be a component of the VEGF-induced angiogenic ERK1/2, Akt and STAT-3 endothelial cell signaling pathways.134,135 Though angiogenesis is a potential unwanted effect with sEHI treatment, there are cardiovascular diseases where angiogenesis would be beneficial. Additionally, these findings have led to the postulate that enhancing sEH activity or inhibiting EET production and/or actions could be a therapeutic target for various cancers.
Another concern is that sEHIs exacerbate hypoxic pulmonary vasoconstriction and hypoxia-induced pulmonary vascular remodeling.35,36 Chronic hypoxia elicits pulmonary hypertension and vascular remodeling that is associated with increased EET generation and epoxygenase inhibition reduced the hypoxic pulmonary vasoconstriction.35,36 Ephx2-/- mice also demonstrate an increased pulmonary vasoconstriction in response to hypoxia.35 The concern of pulmonary hypertension may be limited to that induced by hypoxia since in monocrotaline-induced pulmonary hypertension, sEHI reduced vascular remodeling and the development of pulmonary hypertension.135 Another possible concern in the lungs is that EETs can increase endothelial cell permeability that could result in an unwanted increase in alveolar fluid volume.136,137 On the other hand, 14,15-EET combats TNF-α induced hyperreactivity in human airway smooth muscle cells.138 These findings suggest that sEHIs have the potential unwanted effect of pulmonary vasoconstriction but could be beneficial to combat bronchial inflammation.
There are also potential unwanted cardiovascular effects that have the potential to limit the therapeutic utility for sEHIs. Although sEHIs can improve cardiac function following ischemia, Ephx2 deletion or sEHIs delayed blood pressure recovery and resulted in higher mortality after cardiopulmonary resuscitation in mice.123 The effect of sEHI on blood clotting is also complex. Platelet aggregation could be slowed or inhibited, resulting in enhanced bleeding and hemorrhaging in patients taking sEHIs.59,61,139 However, PGI2 to TXA2 ratios and other data provide evidence that sEHIs would speed clotting in animals treated with aspirin but delay clotting in animals treated with rofecoxib.64,80 This observation seems to be in agreement with the trend that EETs appear to move a variety of biological functions back toward steady state.
Epoxyeicosanoids as a Therapeutic Target
The fact that sEHIs and EETs are angiogenic and have the potential for increasing tumor growth means that inhibiting epoxygenase enzymes or EETs could be a treatment for tumor growth. Interestingly, the CYP2J2 epoxygenase enzyme has been found to be up-regulated in many tumors.140 A recent study that explored the possibility that selective inhibition of the CYP2J2 epoxygenase enzyme would repress tumor growth showed that selective CYP2J2 inhibitors that decreased EET production had marked antitumor properties in in vitro and in vivo settings including various human cancer cells.141 One could also imagine that EET antagonists such as 14,15-EEZE or even the use of sEH protein as potential cancer therapeutics.
On the other hand, epoxyeicosanoids and EET analogs are being pursued as a potential therapeutic target for cardiovascular diseases. Increasing EET levels or overexpressing epoxygenase enzymes are cardioprotective,21,65,104,142 first demonstrated when 11,12-EET was added to transplant preservation solutions resulting in improved coronary artery endothelial function.143 Sulfonamide analogs of EET were developed 15 years ago and other EET analogs and antagonists have been designed and utilized in in vitro perfused vascular and organ experimental conditions.31,58,144-146 These EET analogs and antagonists have determined structure activity requirements for the biological EET actions.58,145,146 Ultimately determination of binding sites or receptors for EETs could provide new targets for the treatment of cardiovascular diseases. Recent evidence suggests that EET analogs can be effectively designed to administer chronically to SHR and have anti-hypertensive actions.147 A combinational drug that has EET mimetic actions and sEHI activity is another possibility. Since the finding that certain sEHIs can vasodilate mesenteric resistance arteries there has been progress in attempts to design EET analogs that can also inhibit the sEH enzyme.148 One could envision the possible utility for EET analogs being used for acute myocardial infarction and drug eluting stents. On the whole, there is great potential with pursuing epoxyeicosanoids as a cardiovascular therapeutic target.
Conclusion
There has been striking rapid progress made with the evaluation of sEHIs as a therapy for cardiovascular diseases since the first description of their anti-hypertensive actions in 2000. Future research will be to further explore other non-cardiovascular diseases that could potentially be treated with sEHIs. There is strong evidence that inflammatory diseases, neurological diseases such as Alzheimer’s disease and diseases associated with pain may benefit from sEHI treatment. Therefore, the potential for sEHIs for the treatment of cardiovascular diseases is great and other potential therapeutic possibilities appear to be on the horizon.
References
- 1.Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–11. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- 2.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puri A, McGoon MD, Kushwaha SS. Pulmonary arterial hypertension: current therapeutic strategies. Nat Clin Pract Cardiovasc Med. 2007;4:319–29. doi: 10.1038/ncpcardio0890. [DOI] [PubMed] [Google Scholar]
- 4.Steiropoulos P, Trakada G, Bouros D. Current pharmacological treatment of pulmonary arterial hypertension. Curr Clin Pharmacol. 2008;3:11–9. doi: 10.2174/157488408783329887. [DOI] [PubMed] [Google Scholar]
- 5.Braden GL, O’Shea MH, Mulhern JG, Germain MJ. Acute renal failure and hyperkalaemia associated with cyclooxygenase-2 inhibitors. Nephrol Dial Transplant. 2004;19:1149–53. doi: 10.1093/ndt/gfg622. [DOI] [PubMed] [Google Scholar]
- 6.Fries S, Grosser T. The Cardiovascular Pharmacology of COX-2 Inhibition. Hematology Am Soc Hematol Educ Program. 2005:445–51. doi: 10.1182/asheducation-2005.1.445. [DOI] [PubMed] [Google Scholar]
- 7.Capra V, et al. Cysteinyl-leukotrienes and their receptors in asthma and other inflammatory diseases: critical update and emerging trends. Med Res Rev. 2007;27:469–527. doi: 10.1002/med.20071. [DOI] [PubMed] [Google Scholar]
- 8.Ribeiro JD, Toro AA, Baracat EC. Antileukotrienes in the treatment of asthma and allergic rhinitis. J Pediatr (Rio J) 2006;82:S213–21. doi: 10.2223/JPED.1553. [DOI] [PubMed] [Google Scholar]
- 9.Capdevila J, Marnett LJ, Chacos N, Prough RA, Estabrook RW. Cytochrome P-450-dependent oxygenation of arachidonic acid to hydroxyicosatetraenoic acids. Proc Natl Acad Sci U S A. 1982;79:767–70. doi: 10.1073/pnas.79.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chacos N, Falck JR, Wixtrom C, Capdevila J. Novel epoxides formed during the liver cytochrome P-450 oxidation of arachidonic acid. Biochem Biophys Res Commun. 1982;104:916–22. doi: 10.1016/0006-291x(82)91336-5. [DOI] [PubMed] [Google Scholar]
- 11.Oliw EH, Lawson JA, Brash AR, Oates JA. Arachidonic acid metabolism in rabbit renal cortex. Formation of two novel dihydroxyeicosatrienoic acids. J Biol Chem. 1981;256:9924–31. [PubMed] [Google Scholar]
- 12.Ishizuka T, et al. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther. 2008;324:103–10. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- 13.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 14.Sarkis A, Lopez B, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid and epoxyeicosatrienoic acids in hypertension. Curr Opin Nephrol Hypertens. 2004;13:205–14. doi: 10.1097/00041552-200403000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Renic M, et al. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2009;29:629–39. doi: 10.1038/jcbfm.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyata N, et al. Beneficial effects of a new 20-hydroxyeicosatetraenoic acid synthesis inhibitor, TS-011 [N-(3-chloro-4-morpholin-4-yl) phenyl-N’-hydroxyimido formamide], on hemorrhagic and ischemic stroke. J Pharmacol Exp Ther. 2005;314:77–85. doi: 10.1124/jpet.105.083964. [DOI] [PubMed] [Google Scholar]
- 17.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–23. doi: 10.1161/01.res.78.3.415. This is the original report that revealed that epoxyeicosatrienoic acids EETs were endothelium-derived hyperpolarizing factors EDHFs and placed them as important regulators of vascular function
- 18.Fleming I. DiscrEET regulators of homeostasis: epoxyeicosatrienoic acids, cytochrome P450 epoxygenases and vascular inflammation. Trends Pharmacol Sci. 2007;28:448–52. doi: 10.1016/j.tips.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Gross GJ, et al. Effects of the selective EET antagonist 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am J Physiol Heart Circ Physiol. 2008;294:H2838–44. doi: 10.1152/ajpheart.00186.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:F496–503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 21.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson HR, Corona S, Capdevila JH, Chacos N, Manna S, Womack A, Falck JR. Effects of epoxyeicosatrienoic acids on ion transport in the rabbit cortical collecting tubule. In: Braquet P, Garay RP, Frohlich JC, Nicosia S, editors. Prostaglandins, and Membrane Ion Transport. Raven Press; New York: 1985. pp. 311–318. [Google Scholar]
- 23.Proctor KG, Falck JR, Capdevila J. Intestinal vasodilation by epoxyeicosatrienoic acids: arachidonic acid metabolites produced by a cytochrome P450 monooxygenase. Circ Res. 1987;60:50–9. doi: 10.1161/01.res.60.1.50. [DOI] [PubMed] [Google Scholar]
- 24.Fleming I. Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins Other Lipid Mediat. 2007;82:60–7. doi: 10.1016/j.prostaglandins.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–90. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 26.Hao CM, Breyer MD. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol. 2008;70:357–77. doi: 10.1146/annurev.physiol.70.113006.100614. [DOI] [PubMed] [Google Scholar]
- 27.Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. 2009;50:S52–6. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W, et al. Characterization of 14,15-epoxyeicosatrienoyl-sulfonamides as 14,15-epoxyeicosatrienoic acid agonists: use for studies of metabolism and ligand binding. J Pharmacol Exp Ther. 2007;321:1023–31. doi: 10.1124/jpet.107.119651. This study describes the development of EET agonists that could possibly be used for finding EET receptors
- 29.Widstrom RL, Norris AW, Van Der Veer J, Spector AA. Fatty acid-binding proteins inhibit hydration of epoxyeicosatrienoic acids by soluble epoxide hydrolase. Biochemistry. 2003;42:11762–7. doi: 10.1021/bi034971d. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, et al. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102:16747–52. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michaelis UR, Fleming I. From endothelium-derived hyperpolarizing factor (EDHF) to angiogenesis: Epoxyeicosatrienoic acids (EETs) and cell signaling. Pharmacol Ther. 2006;111:584–95. doi: 10.1016/j.pharmthera.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 33.Gebremedhin D, et al. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol. 1992;263:H519–25. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- 34.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol. 1996;7:2364–70. doi: 10.1681/ASN.V7112364. [DOI] [PubMed] [Google Scholar]
- 35.Keseru B, et al. Epoxyeicosatrienoic acids and the soluble epoxide hydrolase are determinants of pulmonary artery pressure and the acute hypoxic pulmonary vasoconstrictor response. Faseb J. 2008;22:4306–15. doi: 10.1096/fj.08-112821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pokreisz P, et al. Cytochrome P450 epoxygenase gene function in hypoxic pulmonary vasoconstriction and pulmonary vascular remodeling. Hypertension. 2006;47:762–70. doi: 10.1161/01.HYP.0000208299.62535.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Archer SL, et al. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation. 2003;107:769–76. doi: 10.1161/01.cir.0000047278.28407.c2. [DOI] [PubMed] [Google Scholar]
- 38.Fisslthaler B, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–7. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 39.Li PL, Zhang DX, Ge ZD, Campbell WB. Role of ADP-ribose in 11,12-EET-induced activation of K(Ca) channels in coronary arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. 2002;282:H1229–36. doi: 10.1152/ajpheart.00736.2001. [DOI] [PubMed] [Google Scholar]
- 40.Imig JD, Inscho EW, Deichmann PC, Reddy KM, Falck JR. Afferent arteriolar vasodilation to the sulfonimide analog of 11, 12-epoxyeicosatrienoic acid involves protein kinase A. Hypertension. 1999;33:408–13. doi: 10.1161/01.hyp.33.1.408. [DOI] [PubMed] [Google Scholar]
- 41.Li PL, Chen CL, Bortell R, Campbell WB. 11,12-Epoxyeicosatrienoic acid stimulates endogenous mono-ADP-ribosylation in bovine coronary arterial smooth muscle. Circ Res. 1999;85:349–56. doi: 10.1161/01.res.85.4.349. [DOI] [PubMed] [Google Scholar]
- 42.Node K, et al. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem. 2001;276:15983–9. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 43.Larsen BT, et al. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BK(Ca) channels: implications for soluble epoxide hydrolase inhibition. Am J Physiol Heart Circ Physiol. 2006;290:H491–9. doi: 10.1152/ajpheart.00927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medhora M, et al. Emerging mechanisms for growth and protection of the vasculature by cytochrome P450-derived products of arachidonic acid and other eicosanoids. Prostaglandins Other Lipid Mediat. 2007;82:19–29. doi: 10.1016/j.prostaglandins.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 45.Medhora M, et al. Epoxygenase-driven angiogenesis in human lung microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2003;284:H215–24. doi: 10.1152/ajpheart.01118.2001. [DOI] [PubMed] [Google Scholar]
- 46.Potente M, Fisslthaler B, Busse R, Fleming I. 11,12-Epoxyeicosatrienoic acid-induced inhibition of FOXO factors promotes endothelial proliferation by down-regulating p27Kip1. J Biol Chem. 2003;278:29619–25. doi: 10.1074/jbc.M305385200. [DOI] [PubMed] [Google Scholar]
- 47.Potente M, Michaelis UR, Fisslthaler B, Busse R, Fleming I. Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J Biol Chem. 2002;277:15671–6. doi: 10.1074/jbc.M110806200. [DOI] [PubMed] [Google Scholar]
- 48.Pozzi A, et al. Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J Biol Chem. 2005;280:27138–46. doi: 10.1074/jbc.M501730200. [DOI] [PubMed] [Google Scholar]
- 49.Yan G, Chen S, You B, Sun J. Activation of sphingosine kinase-1 mediates induction of endothelial cell proliferation and angiogenesis by epoxyeicosatrienoic acids. Cardiovasc Res. 2008;78:308–14. doi: 10.1093/cvr/cvn006. [DOI] [PubMed] [Google Scholar]
- 50.Sun J, et al. Inhibition of vascular smooth muscle cell migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ Res. 2002;90:1020–7. doi: 10.1161/01.res.0000017727.35930.33. [DOI] [PubMed] [Google Scholar]
- 51.Davis BB, et al. Attenuation of vascular smooth muscle cell proliferation by 1-cyclohexyl-3-dodecyl urea is independent of soluble epoxide hydrolase inhibition. J Pharmacol Exp Ther. 2006;316:815–21. doi: 10.1124/jpet.105.091876. [DOI] [PubMed] [Google Scholar]
- 52.Davis BB, et al. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A. 2002;99:2222–7. doi: 10.1073/pnas.261710799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foley RN, Collins AJ. End-stage renal disease in the United States: an update from the United States Renal Data System. J Am Soc Nephrol. 2007;18:2644–8. doi: 10.1681/ASN.2007020220. [DOI] [PubMed] [Google Scholar]
- 54.Zoccali C, Mallamaci F, Tripepi G. Traditional and emerging cardiovascular risk factors in end-stage renal disease. Kidney Int Suppl. 2003:S105–10. doi: 10.1046/j.1523-1755.63.s85.25.x. [DOI] [PubMed] [Google Scholar]
- 55.Elmarakby AA, et al. Chemokine receptor 2b inhibition provides renal protection in angiotensin II - salt hypertension. Hypertension. 2007;50:1069–76. doi: 10.1161/HYPERTENSIONAHA.107.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension. 2006;47:557–62. doi: 10.1161/01.HYP.0000198545.01860.90. [DOI] [PubMed] [Google Scholar]
- 57.Node K, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–9. doi: 10.1126/science.285.5431.1276. This study is the first description of vascular anti-inflammatory properties of epoxyeicosatrienoic acids (EETs)
- 58.Falck JR, et al. 11,12-epoxyeicosatrienoic acid (11,12-EET): structural determinants for inhibition of TNF-alpha-induced VCAM-1 expression. Bioorg Med Chem Lett. 2003;13:4011–4. doi: 10.1016/j.bmcl.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 59.Fitzpatrick FA, et al. Inhibition of cyclooxygenase activity and platelet aggregation by epoxyeicosatrienoic acids. Influence of stereochemistry. J Biol Chem. 1986;261:15334–8. [PubMed] [Google Scholar]
- 60.Pratt PF, Rosolowsky M, Campbell WB. Effects of epoxyeicosatrienoic acids on polymorphonuclear leukocyte function. Life Sci. 2002;70:2521–33. doi: 10.1016/s0024-3205(02)01533-3. [DOI] [PubMed] [Google Scholar]
- 61.Heizer ML, McKinney JS, Ellis EF. 14,15-Epoxyeicosatrienoic acid inhibits platelet aggregation in mouse cerebral arterioles. Stroke. 1991;22:1389–93. doi: 10.1161/01.str.22.11.1389. [DOI] [PubMed] [Google Scholar]
- 62.Kozak W, Kluger MJ, Kozak A, Wachulec M, Dokladny K. Role of cytochrome P-450 in endogenous antipyresis. Am J Physiol Regul Integr Comp Physiol. 2000;279:R455–60. doi: 10.1152/ajpregu.2000.279.2.R455. [DOI] [PubMed] [Google Scholar]
- 63.Nakashima T, Yoshida Y, Miyata S, Kiyohara T. Hypothalamic 11,12-epoxyeicosatrienoic acid attenuates fever induced by central interleukin-1beta in the rat. Neurosci Lett. 2001;310:141–4. doi: 10.1016/s0304-3940(01)02115-2. [DOI] [PubMed] [Google Scholar]
- 64.Schmelzer KR, et al. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:13646–51. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Imig JD. Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovasc Drug Rev. 2006;24:169–88. doi: 10.1111/j.1527-3466.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- 66.Inceoglu B, et al. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006;79:2311–9. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao X, et al. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–53. Description of the end organ protective properties of sEHI and the first demonstration that sEHI decreased renal inflammation associated with hypertension
- 68.Smith KR, et al. Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc Natl Acad Sci U S A. 2005;102:2186–91. doi: 10.1073/pnas.0409591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fornage M, et al. Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation. 2004;109:335–9. doi: 10.1161/01.CIR.0000109487.46725.02. This study is the original description of the association of sEH polymorphisms with cardiovascular disease in the human population
- 70.Fornage M, et al. The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet. 2005;14:2829–37. doi: 10.1093/hmg/ddi315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koerner IP, et al. Polymorphisms in the human soluble epoxide hydrolase gene EPHX2 linked to neuronal survival after ischemic injury. J Neurosci. 2007;27:4642–9. doi: 10.1523/JNEUROSCI.0056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee CR, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet. 2006;15:1640–9. doi: 10.1093/hmg/ddl085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srivastava PK, Sharma VK, Kalonia DS, Grant DF. Polymorphisms in human soluble epoxide hydrolase: effects on enzyme activity, enzyme stability, and quaternary structure. Arch Biochem Biophys. 2004;427:164–9. doi: 10.1016/j.abb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 74.Wei Q, et al. Sequence variation in the soluble epoxide hydrolase gene and subclinical coronary atherosclerosis: interaction with cigarette smoking. Atherosclerosis. 2007;190:26–34. doi: 10.1016/j.atherosclerosis.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 75.Sato K, et al. Soluble epoxide hydrolase variant (Glu287Arg) modifies plasma total cholesterol and triglyceride phenotype in familial hypercholesterolemia: intrafamilial association study in an eight-generation hyperlipidemic kindred. J Hum Genet. 2004;49:29–34. doi: 10.1007/s10038-003-0103-6. [DOI] [PubMed] [Google Scholar]
- 76.Dreisbach AW, et al. The Prevalence of CYP2C8, 2C9, 2J2, and soluble epoxide hydrolase polymorphisms in African Americans with hypertension. Am J Hypertens. 2005;18:1276–81. doi: 10.1016/j.amjhyper.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 77.Spiecker M, et al. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation. 2004;110:2132–6. doi: 10.1161/01.CIR.0000143832.91812.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu Z, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–8. doi: 10.1161/01.res.87.11.992. This is the first experimental evidence that sEHI can increase epoxide levels and lower blood pressure in an animal model of hypertension
- 79.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–4. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 80.Imig JD, et al. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46:975–81. doi: 10.1161/01.HYP.0000176237.74820.75. This study was the first to administer a sEHI orally and demonstrate anti-hypertensive and end organ protective effects
- 81.Morisseau C, Hammock BD. Gerry Brooks and epoxide hydrolases: four decades to a pharmaceutical. Pest Manag Sci. 2008;64:594–609. doi: 10.1002/ps.1583. [DOI] [PubMed] [Google Scholar]
- 82.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 83.Oesch F, Schladt L, Hartmann R, Timms C, Worner W. Rat cytosolic epoxide hydrolase. Adv Exp Med Biol. 1986;197:195–201. doi: 10.1007/978-1-4684-5134-4_16. [DOI] [PubMed] [Google Scholar]
- 84.EnayetAllah AE, et al. Opposite regulation of cholesterol levels by the phosphatase and hydrolase domains of soluble epoxide hydrolase. J Biol Chem. 2008;283:36592–8. doi: 10.1074/jbc.M806315200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–33. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 86.Newman JW, Morisseau C, Harris TR, Hammock BD. The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc Natl Acad Sci U S A. 2003;100:1558–63. doi: 10.1073/pnas.0437724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem. 2004;52:447–54. doi: 10.1177/002215540405200403. [DOI] [PubMed] [Google Scholar]
- 88.Yu Z, et al. Vascular localization of soluble epoxide hydrolase in the human kidney. Am J Physiol Renal Physiol. 2004;286:F720–6. doi: 10.1152/ajprenal.00165.2003. [DOI] [PubMed] [Google Scholar]
- 89.Harris TR, et al. Identification of two epoxide hydrolases in Caenorhabditis elegans that metabolize mammalian lipid signaling molecules. Arch Biochem Biophys. 2008;472:139–49. doi: 10.1016/j.abb.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tran KL. Lipid sulfates and sulfonates are allosteric competitive inhibitors of the N-terminal phosphatase activity of the mammalian soluble epoxide hydrolase. Biochemistry. 2005;44:12179–87. doi: 10.1021/bi050842g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harris TR, Aronov PA, Hammock BD. Soluble epoxide hydrolase homologs in Strongylocentrotus purpuratus suggest a gene duplication event and subsequent divergence. DNA Cell Biol. 2008;27:467–77. doi: 10.1089/dna.2008.0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mullin CA, Hammock BD. Chalcone oxides--potent selective inhibitors of cytosolic epoxide hydrolase. Arch Biochem Biophys. 1982;216:423–39. doi: 10.1016/0003-9861(82)90231-4. [DOI] [PubMed] [Google Scholar]
- 93.Morisseau C, et al. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci U S A. 1999;96:8849–54. doi: 10.1073/pnas.96.16.8849. This is the original description of the development of urea compounds as sEHI
- 94.Kim IH, et al. Optimization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J Med Chem. 2005;48:3621–9. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morisseau C, et al. Development of metabolically stable inhibitors of mammalian microsomal epoxide hydrolase. Chem Res Toxicol. 2008;21:951–7. doi: 10.1021/tx700446u. [DOI] [PubMed] [Google Scholar]
- 96.Xie Y, et al. Discovery of potent non-urea inhibitors of soluble epoxide hydrolase. Bioorganic & Medicinal Chemistry Letters. 2009;19:2354–9. doi: 10.1016/j.bmcl.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu J-Y, et al. Sorafenib has sEH inhibitory activity which contributes to its effect profile in vivo. Molecular Cancer Therapeutics. doi: 10.1158/1535-7163.MCT-09-0119. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ghosh S, et al. Oral delivery of 1,3-dicyclohexylurea nanosuspension enhances exposure and lowers blood pressure in hypertensive rats. Basic Clin Pharmacol Toxicol. 2008;102:453–8. doi: 10.1111/j.1742-7843.2008.00213.x. [DOI] [PubMed] [Google Scholar]
- 99.Morisseau C, et al. Structural refinement of inhibitors of urea-based soluble epoxide hydrolases. Biochem Pharmacol. 2002;63:1599–608. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 100.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–40. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ai D, et al. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:564–9. doi: 10.1073/pnas.0811022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jung O, et al. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–65. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 103.Koerner IP, et al. Soluble epoxide hydrolase: regulation by estrogen and role in the inflammatory response to cerebral ischemia. Front Biosci. 2008;13:2833–41. doi: 10.2741/2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seubert JM, et al. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99:442–50. doi: 10.1161/01.RES.0000237390.92932.37. This study used the combination of genetic and pharmacological manipulation of sEHI and epoxides and demonstrated cardiac protective effects from ischemic events
- 105.Ulu A, et al. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol. 2008;52:314–23. doi: 10.1097/FJC.0b013e318185fa3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marino JP., Jr Soluble epoxide hydrolase, a target with multiple opportunities for cardiovascular drug discovery. Curr Top Med Chem. 2009;9:452–63. doi: 10.2174/156802609788340805. [DOI] [PubMed] [Google Scholar]
- 107.Olearczyk JJ, et al. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin Sci (Lond) 2009;116:61–70. doi: 10.1042/CS20080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Motoki A, et al. Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H2128–34. doi: 10.1152/ajpheart.00428.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fife KL, et al. Inhibition of soluble epoxide hydrolase does not protect against endotoxin-mediated hepatic inflammation. J Pharmacol Exp Ther. 2008;327:707–15. doi: 10.1124/jpet.108.142398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Katragadda D, et al. Epoxyeicosatrienoic acids limit damage to midochondrial function following stress in cardiac cells. J Mol Cell Cardiol. 2009;46:867–75. doi: 10.1016/j.yjmcc.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 111.Loch D, Hoey A, Morisseau C, Hammock BO, Brown L. Prevention of hypertension in DOCA-salt rats by an inhibitor of soluble epoxide hydrolase. Cell Biochem Biophys. 2007;47:87–98. doi: 10.1385/cbb:47:1:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fleming I. Vascular cytochrome p450 enzymes: physiology and pathophysiology. Trends Cardiovasc Med. 2008;18:20–5. doi: 10.1016/j.tcm.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 113.Fornage M, et al. Polymorphism in soluble epoxide hydrolase and blood pressure in spontaneously hypertensive rats. Hypertension. 2002;40:485–90. doi: 10.1161/01.hyp.0000032278.75806.68. [DOI] [PubMed] [Google Scholar]
- 114.Dorrance AM, et al. An epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2005;46:842–8. doi: 10.1097/01.fjc.0000189600.74157.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Simpkins AN, et al. Soluble epoxide hydrolase inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol. 2009;174:2086–95. doi: 10.2353/ajpath.2009.080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Corenblum MJ, et al. Altered soluble epoxide hydrolase gene expression and function and vascular disease risk in the stroke-prone spontaneously hypertensive rat. Hypertension. 2008;51:567–73. doi: 10.1161/HYPERTENSIONAHA.107.102160. [DOI] [PubMed] [Google Scholar]
- 117.Sinal CJ, et al. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–10. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 118.Luria A, et al. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem. 2007;282:2891–8. doi: 10.1074/jbc.M608057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Manhiani M, et al. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009 doi: 10.1152/ajprenal.00098.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Parrish AR, et al. Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol Toxicol. 2008 doi: 10.1007/s10565-008-9071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xu D, et al. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:18733–8. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li J, et al. Soluble epoxide hydrolase inhibitor, AUDA, prevents early salt-sensitive hypertension. Front Biosci. 2008;13:3480–7. doi: 10.2741/2942. [DOI] [PubMed] [Google Scholar]
- 123.Hutchens MP, et al. Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation. 2008;76:89–94. doi: 10.1016/j.resuscitation.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang W, et al. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39:2073–8. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang W, et al. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27:1931–40. doi: 10.1038/sj.jcbfm.9600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ai D, et al. Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc Natl Acad Sci U S A. 2007;104:9018–23. doi: 10.1073/pnas.0703229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schmelzer KR, et al. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102:9772–7. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fang X, et al. Activation of peroxisome proliferator-activated receptor alpha by substituted urea-derived soluble epoxide hydrolase inhibitors. J Pharmacol Exp Ther. 2005;314:260–70. doi: 10.1124/jpet.105.085605. [DOI] [PubMed] [Google Scholar]
- 129.Buckingham RE, Hanna A. Thiazolidinedione insulin sensitizers and the heart: a tale of two organs? Diabetes Obes Metab. 2008;10:312–28. doi: 10.1111/j.1463-1326.2006.00700.x. [DOI] [PubMed] [Google Scholar]
- 130.Liu M, Alkayed NJ. Hypoxic preconditioning and tolerance via hypoxia inducible factor (HIF) 1alpha-linked induction of P450 2C11 epoxygenase in astrocytes. J Cereb Blood Flow Metab. 2005;25:939–48. doi: 10.1038/sj.jcbfm.9600085. [DOI] [PubMed] [Google Scholar]
- 131.Terashvili M, et al. Antinociception produced by 14,15-epoxyeicosatrienoic acid is mediated by the activation of beta-endorphin and met-enkephalin in the rat ventrolateral periaqueductal gray. J Pharmacol Exp Ther. 2008;326:614–22. doi: 10.1124/jpet.108.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Iceoglu B, et al. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A. 2008;105:18901–6. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang S, Wei S, Pozzi A, Capdevila JH. The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Arch Biochem Biophys. 2009 doi: 10.1016/j.abb.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cheranov SY, et al. An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis. Blood. 2008;111:5581–91. doi: 10.1182/blood-2007-11-126680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Revermann M, et al. Inhibition of the soluble epoxide hydrolase attenuates monocrotaline-induced pulmonary hypertension in rats. J Hypertens. 2009;27:322–31. doi: 10.1097/hjh.0b013e32831aedfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Alvarez DF, Gjerde EA, Townsley MI. Role of EETs in regulation of endothelial permeability in rat lung. Am J Physiol Lung Cell Mol Physiol. 2004;286:L445–51. doi: 10.1152/ajplung.00150.2003. [DOI] [PubMed] [Google Scholar]
- 137.Jian MY, King JA, Al-Mehdi AB, Liedtke W, Townsley MI. High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. Am J Respir Cell Mol Biol. 2008;38:386–92. doi: 10.1165/rcmb.2007-0192OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Morin C, Sirois M, Echave V, Gomes MM, Rousseau E. EET displays anti-inflammatory effects in TNF-alpha stimulated human bronchi: putative role of CPI-17. Am J Respir Cell Mol Biol. 2008;38:192–201. doi: 10.1165/rcmb.2007-0232OC. [DOI] [PubMed] [Google Scholar]
- 139.Krotz F, et al. Membrane-potential-dependent inhibition of platelet adhesion to endothelial cells by epoxyeicosatrienoic acids. Arterioscler Thromb Vasc Biol. 2004;24:595–600. doi: 10.1161/01.ATV.0000116219.09040.8c. [DOI] [PubMed] [Google Scholar]
- 140.Jiang JG, et al. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 2005;65:4707–15. doi: 10.1158/0008-5472.CAN-04-4173. [DOI] [PubMed] [Google Scholar]
- 141.Chen C, et al. Selective inhibitors of CYP2J2 related to terfenadine exhibit strong activity against human cancers in vitro and in vivo. J Pharmacol Exp Ther. 2009;329:908–18. doi: 10.1124/jpet.109.152017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Seubert J, et al. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–14. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 143.Yang Q, Zhang RZ, Yim AP, He GW. Effect of 11,12-epoxyeicosatrienoic acid as an additive to St. Thomas’ cardioplegia and University of Wisconsin solutions on endothelium-derived hyperpolarizing factor-mediated function in coronary microarteries: influence of temperature and time. Ann Thorac Surg. 2003;76:1623–30. doi: 10.1016/s0003-4975(03)00735-5. [DOI] [PubMed] [Google Scholar]
- 144.Falck JR, et al. Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am J Physiol Heart Circ Physiol. 2003;284:H337–49. doi: 10.1152/ajpheart.00831.2001. [DOI] [PubMed] [Google Scholar]
- 145.Gauthier KM, Falck JR, Reddy LM, Campbell WB. 14,15-EET analogs: characterization of structural requirements for agonist and antagonist activity in bovine coronary arteries. Pharmacol Res. 2004;49:515–24. doi: 10.1016/j.phrs.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 146.Imig JD, Dimitropoulou C, Reddy DS, White RE, Falck JR. Afferent arteriolar dilation to 11, 12-EET analogs involves PP2A activity and Ca2+-activated K+ Channels. Microcirculation. 2008;15:137–50. doi: 10.1080/10739680701456960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Imig JD, Falck JR. Compositions and methods for the treatment of renal and cardiovascular disease. #20080153889. U.S Patent Application. 2008
- 148.Olearczyk JJ, et al. Substituted adamantyl-urea inhibitors of the soluble epoxide hydrolase dilate mesenteric resistance vessels. J Pharmacol Exp Ther. 2006;318:1307–14. doi: 10.1124/jpet.106.103556. [DOI] [PubMed] [Google Scholar]