

Nitric oxide synthase (NOS) is the primary biological source of the ubiquitous signaling molecule, nitric oxide (•NO). The enzyme utilizes tetrahydrobiopterin (H4B) as an essential cofactor, where it plays a key role in the catalytic conversion of L-arginine to citrulline and •NO.1 The pterin has been shown to serve both structural and catalytic roles in the enzyme by affecting the monomer to dimer transition, promoting protein stability, and forming a radical cation during catalytic turnover.2
We have previously developed redox-active sensitizer wires to probe the active sites of heme enzymes.3 In this report, we describe a new class of redox-active wires that target the pterin binding site of NOS. The pterin lies adjacent to the heme, and directly interacts with the catalytic center through hydrogen bonds to the heme propionate group. It is close in space, but physically distinct from the face of the heme containing the arginine binding site, allowing both cofactor and substrate to bind simultanously.4,5 Thus, introducing a molecular wire at the pterin site may allow photochemical triggering of enzyme turnover, thereby offering an opportunity to study catalytic intermediates and to shed new light on the cofactor’s role in catalysis.6 Here we report the design and synthesis of a Ru(II)-pterin wire, and investigate its interaction with the heme domain of murine inducible nitric oxide synthase (iNOSheme).
The wire combines two essential moieties: an analogue of the cofactor to direct binding, and a redox-active sensitizer that can be used for light-induced charge injection to generate specific oxidation states of the heme (the pterin is believed to provide an electron to the heme during catalysis).7 In this study, the 6-phenyl pterin analogue was synthesized in the catalytically incompetent, fully oxidized form (1). This moiety has been shown to bind in a competitive manner with the fully reduced cofactor.8 The tether was attached to the pterin ring at the O-4 position, which allows it to extend through the iNOS active-site channel that is normally occupied by solvent or other small molecules;5 the linker length was chosen based on modeling studies. In addition, we incorporated a Ru(II)-diimine sensitizer that acts as a luminescent probe of binding, and as an excited-state oxidizing or reducing agent.9
The pterin-linked wire 6 was synthesized from the oxidized 6-phenyl pterin (1; see Scheme 1), which was prepared according to the procedure of Storm, et al., via an Isay condensation reaction.10 The 2-amino functionality was protected as the DMF-acetal group, to improve solubility and facilitating further synthetic modification.11 Alkylation of the 4-hydroxyl was performed under standard Williamson ether synthesis conditions. This chemistry led to a combination of O-4- and N-3-alkylated species, which could be resolved using silica gel flash chromatography.12 Cleavage of both the ester and DMF-acetal protecting groups was accomplished in a single step with 1M NaOH/DMF. The addition of the long chain alkane improved solubility, allowing the final conjugation to the Ru(II) complex via esterification under standard conditions; alternatively, it was converted to the amide 5 for use as a model compound.13
Scheme 1.
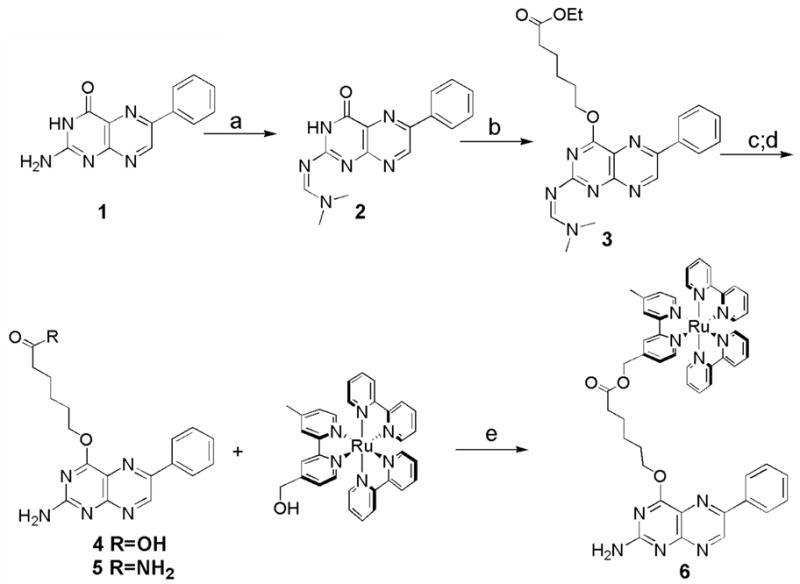
Synthesis of pterin probes. Reagents and conditions: a) DMF-acetal (10 eq.), DMF, 50° C, 2 h, 92%; b) ethyl 6-bromohexanoate (5 eq.), K2CO3 (2 eq.), DMF, 4 h, 68%; c) 1 M NaOH/DMF, 1 h, 82%; d) NH3/MeOH, 18 h, 85%; e) 4, EDCI (1.5 eq.), DMAP (1.5 eq), pyridine, 12 h, 68%.
Several spectroscopic methods were utilized to quantify wire binding to iNOSheme, Upon binding H4B, the iNOS heme undergoes a conversion from low- to high-spin, with a complete shift obtained after incubation with both pterin and L-arginine. Addition of wire 6 to iNOSheme effects a change in the UV/vis spectrum, consistent with partial conversion from the DTT-ligated bis-thiolate complex to a high-spin state (Figure 1). This spectral change is more subtle than observed with the natural cofactor, but exhibits the same difference spectral features with a maximum at 400 nm and a minimum at 459 nm (see Figure S1). These results are consistent with other synthetic pterin analogues that have been found to produce partial low- to high-spin shifts in optical spectra.14
Figure 1.
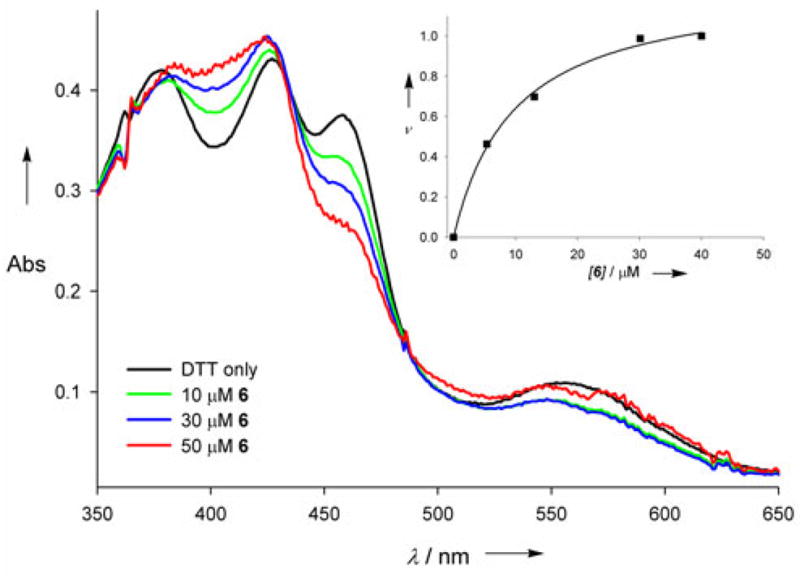
Reconstitution of substrate- and tetrahydrobiopterin-free iNOSheme with wire 6. iNOSheme (~10 μM) was incubated at 25 °C with the pterin analogue at the specified concentration in the presence of 1 mM DTT, and the absorbance spectrum was taken after 2 hours. The spectrum of the DTT-equilibrated sample (without wire) shows the formation of a bis-thiolate heme. Inset shows the binding curve obtained from titration data, plotting the fractional difference in absorption (ΔA/ΔAtotal) as a function of concentration.
The affinity (Kd ~8 μM) of 6 for the protein was obtained from a plot of ν=ΔA/ΔAtotal vs. concentration (see Figure 1, inset). Binding of the natural cofactor causes several structural and electronic modifications in the enzyme, including adjustments in the dimerization interface as well as the substrate and pterin binding pockets, and is known to be quite slow.15 The time dependence of the absorption changes in the difference spectra for wire 6 was analyzed, and the spectral changes were found to be complete within ~60 minutes, in good agreement with the rate of binding of the natural cofactor (see Figure S2). Thus the structural and electronic modifications within the enzyme upon binding wire 6 are fully consistent with those observed upon binding H4B.
The binding of wire 6 to iNOSheme was confirmed by analysis of changes in Ru(II) emission. As shown in Figure 2, the emission centered at 610 nm is dramatically quenched upon addition of iNOSheme, owing to Förster energy transfer. Further, the Ru(II) emission is restored upon addition of the natural cofactor, H4B, or the higher affinity pterin analogue, 4-amino-H4B (4AH4B), indicating that the interaction of 6 with the enzyme is competitive with pterin cofactors.
Figure 2.
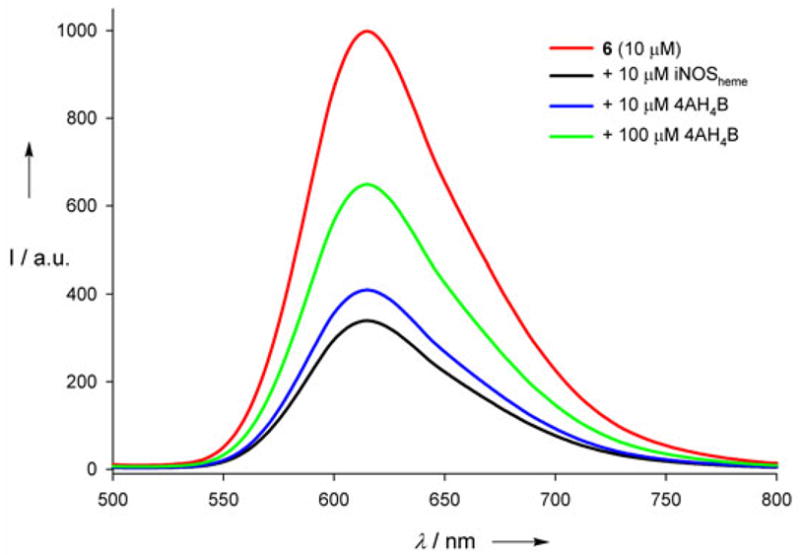
The steady-state Ru(II) luminescence spectrum of 6 (10 μM, red) is quenched in the presence of equimolar iNOSheme (black); it recovers upon addition of 10 μM (blue) and 100 μM (green) 4AH4B.
Time-resolved emission experiments also confirmed Ru(II)-pterin binding to the iNOS heme (see Figure S4).3 The emission decay of the wire is monoexponential in the absence of protein (τ=368±2 ns), but becomes biexponential (τ1=370 ns, τ2=40±4 ns) in the presence of iNOSheme. Fits of the decays provided estimates of the ratio of enzyme-bound to free Ru(II), giving a dissociation constant of approximately 4 μM (see Figure 3). As in the steady-state experiment, addition of the cofactor analogue 4AH4B triggers dissociation of the wire from the protein, and recovery of the longer-lived, unquenched species (see Figure S7).
Figure 3.
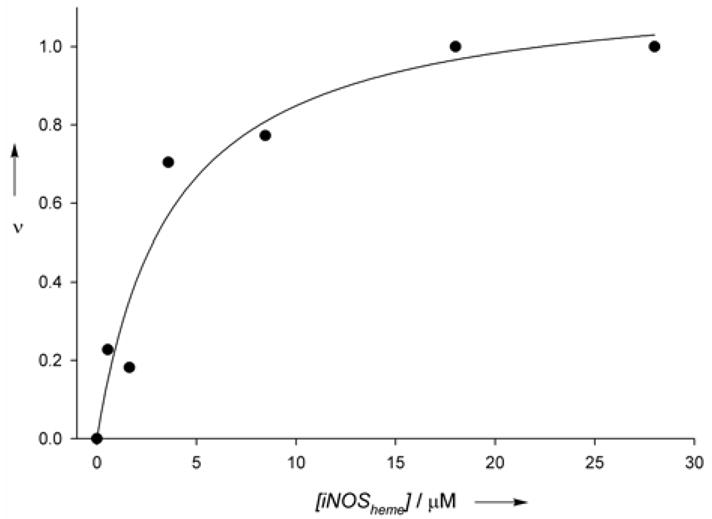
The relative fraction of the fast phase of luminescence decay for compound 6 (taken as fraction bound) as a function of [iNOSheme]free. An estimate for the dissociation constant of 6 to iNOSheme was obtained by fitting the curve to a single-site binding model (Kd=3.8±1.4 μM), assuming that the saturation value of the fractional fast phase represents a 1:1 complex.
The affinity of 6 for iNOSheme appears to be determined by the pterin and its linker rather than the Ru(II) sensitizer. Both the pterin core 1 and intermediate wire 5, lacking the Ru(II) complex, are inherently fluorescent, and display marked quenching in the presence of protein (see Figure S3). The efficient quenching of pterin fluorescence by the heme was used to characterize wire binding to the protein. The 4 μM dissociation constant for 6 is in agreement with that for compound 5 (5.1 ± 1 μM), which was obtained by steady-state fluorescence quenching (see Figure S8); it is significantly lower than the value (26 μM) reported for pterin 1.8 Thus, the addition of the tether rather than the Ru(II) complex is likely responsible for the increased binding affinity. Indeed, other pterin analogues have been reported that show enhanced binding upon alkylation at the 4-position,8a,16 and wires designed for P450cam show higher affinities than substrate alone.3b It is likely that the additional hydrophobic contacts provided by the linker in the access channel are responsible for each of these enhanced affinities.
A model for the interaction of wire 6 with iNOSheme is shown in Figure 4. The pterin portion of the wire was overlaid with the H4B bound at the dimer interface and the linker and Ru(II) conformations were adjusted to minimize steric conflicts. The distance between the heme edge and the nearest diimine ring of the Ru(II) complex is 17 Å in this model, which agrees well with the value of 15.7 ± 1.4 Å obtained by assuming that the quenched (40 ns) component of the emission decay is attributable solely to Förster energy transfer to the heme in the wire-enzyme conjugate.
Figure 4.
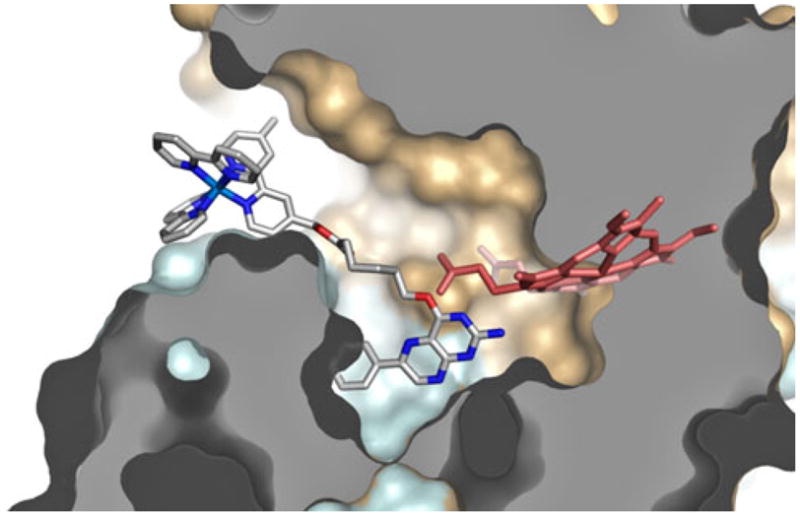
Structural model of wire 6 docked into iNOSheme.
Upon steady state illumination at 450 nm, we observe reduction of the wire bound enzyme in the presence of CO and the reductive quencher TMPD (N,N,N’,N’-tetramethylphenylenediamine). Changes in the UV/vis spectra are dependent both on the concentration of the wire and the irradiation time. Photoreduction yields a species with an absorption maximum at 420 nm, rather than one with a peak at 450 nm associated with the [PCysFeII (CO)] form of the enzyme (see Figure 5). In a control experiment, reduction of the wire-bound enzyme with sodium dithionite produces a combination of P-450 and P-420 species, with the P-420 species predominating.18 In addition, experiments performed on the enzyme in the presence of 50 μM H4B demonstrate instability of the P-450 form, and show conversion to the P-420 form with time or exposure to 450 nm light (see Figure S9). It thus appears that photolysis of the CO-bound heme in the presence of the natural cofactor results in conversion to the 5-coordinate form; it follows that any P-450 species formed upon irradiation would be converted to the P-420 form.
Figure 5.
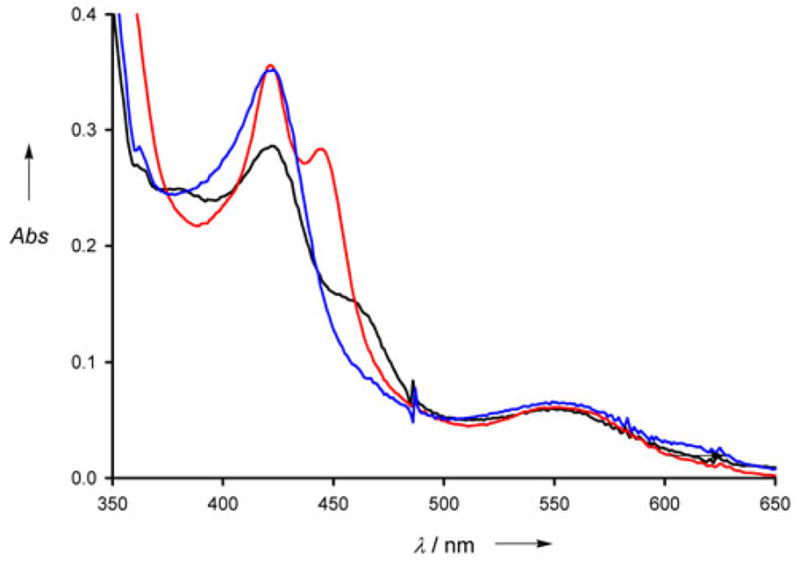
Photoreduction of iNOSheme using wire 6. Illumination of the iNOSheme:wire complex under an atmosphere of CO causes an increase in absorption at 420 nm, consistent with the formation of the reduced CO-bound P-420 species (black line, 0 min, blue line, 30 min). 1 mM TMPD was used for the experiment; no spectral change was observed without TMPD, or without light. Chemical reduction with Na2S2O4 in the dark results in production of a combination of P-450 and P-420 species (red line).
In summary, our pterin-based photoactive wire binds in competition with native pterins to the heme domain of murine iNOS. Charge injection from the metal complex should facilitate mechanistic investigations of the enzyme, and may help determine how the pterin acts as an essential redox-active cofactor at specific points of the catalytic cycle. Importantly, the wire also can be used as a fluorescent probe for rapid screening of small molecule inhibitors that target the pterin binding site.
Supplementary Material
Footnotes
We thank Yitzhak Tor, Doug Magde and M.G. Finn for synthetic resources and many helpful discussions. Supported by NIH grant GM070868 (to DBG and HBG), and NRSA fellowship GM074406 (to ECG) and the Ellison Medical Foundation (Senior Scholar Award in Aging to HBG).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Edith C. Glazer, Department of Molecular Biology, The Scripps Research Institute, La Jolla, CA 92037 (USA), Department of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125 (USA)
Dr. Yen Hoang Le Nguyen, Department of Chemistry and Chemical Engineering California Institute of Technology Pasadena, CA 91125 (USA)
Prof. Harry B. Gray, Department of Chemistry and Chemical Engineering California Institute of Technology Pasadena, CA 91125 (USA)
Prof. David B. Goodin, Department of Molecular Biology, The Scripps Research Institute, La Jolla, CA 92037 (USA).
References
- 1.Alderton WK, Cooper CE, Knowles RG. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei CC, Crane BR, Stuehr DJ. Chem Rev. 2003;103:2365–2383. doi: 10.1021/cr0204350. [DOI] [PubMed] [Google Scholar]
- 3.a) Wilker JJ, Dmochowski IJ, Dawson JH, Winkler JR, Gray HB. Angew Chem. 1999;111:93–96. [Google Scholar]; Angew Chem Int Ed. 1999;38:90–92. [Google Scholar]; b) Dunn AR, Dmochowski IJ, Winkler JR, Gray HB. J Am Chem Soc. 2003;125:12450–12456. doi: 10.1021/ja0294111. [DOI] [PubMed] [Google Scholar]; c) Dunn AR, Hays AMA, Goodin DB, Stout CD, Chiu R, Winkler JR, Gray HB. J Am Chem Soc. 2002;124:10254–10255. doi: 10.1021/ja0271678. [DOI] [PubMed] [Google Scholar]; d) Dunn AR, Belliston-Bittner W, Winkler JR, Getzoff ED, Stuehr DJ, Gray HB. J Am Chem Soc. 2005;127:5169–5173. doi: 10.1021/ja046971m. [DOI] [PubMed] [Google Scholar]; e) Belliston-Bittner W, Dunn AR, Nguyen YHL, Stuehr DJ, Winkler JR, Gray HB. J Am Chem Soc. 2005;127:15907–15915. doi: 10.1021/ja0543088. [DOI] [PubMed] [Google Scholar]
- 4.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 5.Raman CS, Li H, Martasek P, Kral V, Masters BSS, Poulos TL. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 6.a) Stuehr DJ, Wei CC, Wang Z, Hille R. Dalton Trans. 2005:3427–3435. doi: 10.1039/b506355h. [DOI] [PubMed] [Google Scholar]; b) Stuehr DJ, Wei CC, Santolini J, Wang Z, Aoyagi M, Getzoff ED. Biochem Soc Symp. 2004:39–49. doi: 10.1042/bss0710039. [DOI] [PubMed] [Google Scholar]
- 7.a) Hurshman AR, Krebs C, Edmondson DE, Huynh BH, Marletta MA. Biochemistry. 1999;38:15689–15696. doi: 10.1021/bi992026c. [DOI] [PubMed] [Google Scholar]; b) Hurshman AR, Marletta MA. Biochemistry. 2002;41:3439–3456. doi: 10.1021/bi012002h. [DOI] [PubMed] [Google Scholar]
- 8.a) Kotsonis P, et al. J Biol Chem. 2001;276:49133–49141. doi: 10.1074/jbc.M011469200. [DOI] [PubMed] [Google Scholar]; b) Matter H, Kotsonis P, Klingler O, Strobel H, Froehlich LG, Frey A, Pfleiderer W, Schmidt HHHW. J Med Chem. 2002;45:2923–2941. doi: 10.1021/jm020074g. [DOI] [PubMed] [Google Scholar]
- 9.Berglund J, Pascher T, Winkler JR, Gray HB. J Am Chem Soc. 1997;119:2464–2469. [Google Scholar]
- 10.Storm CB, Shiman R, Kaufman S. J Org Chem. 1971;36:3925–3927. [Google Scholar]
- 11.Yao Q, Pfleiderer W. Helv Chim Acta. 2003;86:1–12. Pterins are notoriously insoluble; we have found that the DMF-acetal protecting group is far superior to many others in terms of making the compound amenable to further chemistry, while still being sufficiently labile, in contrast to amide and thiol protecting groups. [Google Scholar]
- 12.A more direct approach to the O-4-species was attempted using Mitsunobu chemistry (see Ref. 11); however, it was found that this route also led to a combination of O- and N-alkylated species, but with an overall lower yield.
- 13.The amide was used instead of the carboxylic acid to prevent any undesired reactions with the protein.
- 14.Presta A, Siddhanta U, Wu C, Sennequier N, Huang L, Abu-Soud HM, Erzurum S, Stuehr DJ. Biochemistry. 1998;37:298–310. doi: 10.1021/bi971944c. [DOI] [PubMed] [Google Scholar]
- 15.Hurshman AR, Krebs C, Edmondson DE, Marletta MA. Biochemistry. 2003;42:13287–13303. doi: 10.1021/bi035491p. [DOI] [PubMed] [Google Scholar]
- 16.Froehlich LG, Kotsonis P, Traub H, Taghavi-Moghadam S, Al-Masoudi N, Hofmann H, Strobel H, Matter H, Pfleiderer W, Schmidt HHHW. J Med Chem. 1999;42:4108–4121. doi: 10.1021/jm981129a. [DOI] [PubMed] [Google Scholar]
- 17.Förster energy transfer calculations were performed as described in Ref. 3d. See SI for further details.
- 18.The instability of the P-450 form of NOS is well documented; it is clear that the pterin plays a central role in determining the degree of conversion to the P-420 form. Pterin-free NOS is converted over time completely to the P-420 form, and variable fractions of the two forms are observed with sub-saturating levels of H4B or synthetic pterin analogues. See ref. 7b, 15, and Wang J, Stuehr DJ, Rousseau DL. Biochemistry. 1995;34:7080–7087. doi: 10.1021/bi00021a020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.