

Abstract
Exposure of newborn rats to a variety of anesthetics has been shown to induce apoptotic neurodegeneration in the developing brain. We investigated the effect of the general anesthetic propofol on the brain of 7-day-old (P7) Wistar rats during the peak of synaptic growth. Caspase and calpain protease families most likely participate in neuronal cell death. Our objective was to examine regional and temporal patterns of caspase-3 and calpain activity following repeated propofol administration (20 mg/kg). P7 rats were exposed for 2, 4 or 6 h to propofol and killed 0, 4, 16 and 24 h after exposure. Relative caspase-3 and calpain activities were estimated by Western blot analysis of the proteolytic cleavage products of α-II-spectrin, protein kinase C and poly(ADP-ribose) polymerase 1. Caspase-3 activity and expression displayed a biphasic pattern of activation. Calpain activity changed in a region- and time-specific manner that was distinct from that observed for caspase-3. The time profile of calpain activity exhibited substrate specificity. Fluoro-Jade B staining revealed an immediate neurodegenerative response that was in direct relationship to the duration of anesthesia in the cortex and inversely related to the duration of anesthesia in the thalamus. At later post-treatment intervals, dead neurons were detected only in the thalamus 24 h following the 6-hour propofol exposure. Strong caspase-3 expression that was detected at 24 h was not followed by cell death after 2- and 4-hour exposures to propofol. These results revealed complex patterns of caspase-3 and calpain activities following prolonged propofol anesthesia and suggest that both are a manifestation of propofol neurotoxicity at a critical developmental stage.
Key Words: Anesthesia-induced neurotoxicity, Apoptosis, Thalamus, Spectrin, Poly(ADP-ribose) polymerase 1, Protein kinase C, Neurodegeneration
Introduction
General anesthetics that are widely used in clinical practice can induce apoptotic cell death in the central nervous system of experimental animals when administered during synaptogenesis that occurs during the first 2 weeks of life [1]. Anesthetics are routinely combined in order to achieve adequate depth and duration of anesthesia. However, studies in animals have revealed detrimental effects of these agents when they are used simultaneously, as compared to when they are applied on their own [2]. This imposes a necessity to elucidate the underlying mechanisms of action and potential harmful effects of each anesthetic individually.
Propofol (2,6-diisopropylphenol) is an alkyl phenol derivate that has gained wide clinical use for induction and maintenance of general anesthesia. The greatest attributes of propofol are its pharmacokinetic properties that account for the clinical benefits of rapid onset of anesthesia and a short recovery time. Regarding the mechanism of action, several experimental studies have shown that propofol potentiates GABAA receptor functioning, while at higher concentrations, it causes opening of GABAA receptors [3]. A study of the effects of propofol in developing animals suggests that propofol increases neurodegeneration that leads to subsequent behavioral and learning impairments [2,4]. Propofol-induced neurotoxicity depends on the concentration and duration of treatment, as shown in in vitro studies, and on the dose, as revealed in in vivo studies in mice [2,5,6].
The cellular mechanisms and signaling pathways that underlie anesthesia-induced neurotoxicity are poorly understood. Activation of caspase-3 appears to be induced by general anesthetics in postnatal brains during maturation [6,7,8]. Of the identified caspases, caspase-3 stands out since it is activated by many cell death signals and cleaves a variety of important cellular proteins such as cytoskeletal proteins, kinases, and DNA repair enzymes [9].
Another protease that is involved in cell death is calpain. It is likely that calpain is activated by an initial insult via a rise in intracellular calcium from both extracellular and intracellular sources [10]. Under normal conditions, calpain activation is a controlled process that leads to a limited cleavage of substrates that have either regulatory or signaling functions. However, strong or persistent calpain activity underlies the pathophysiology of several conditions. Calpain is generally associated with excitotoxic neuronal injury and necrosis, although evidence for a role in apoptotic cell death has been presented [11]. A growing body of literature has revealed the existence of functional connections between the calpains and caspases in vitro and in vivo. The calpains have the potential to both positively and negatively modulate the caspase cascade during apoptosis. There is also evidence that caspases indirectly upregulate calpain activity by cleaving calpastatin, an endogenous calpain inhibitor [9].
While the involvement of caspase-3 in propofol-induced cell death in the rat and mouse postnatal brain was demonstrated in two studies [6,8], the contribution of calpain in propofol-mediated neuronal cell death and central nervous system injury has not been investigated. Caspase and calpain share many substrates. After activation, they generate fragments with different molecular weights representing the signature products of their respective proteolytic activities [9].
The objective of this study was to gain insight into the temporal and regional activity of caspase-3 and calpain activities following prolonged propofol anesthesia by assessing proteolytic degradation of α-II-spectrin and poly(ADP-ribose) polymerase 1 (PARP-1; substrates of both calpain and caspase-3), and protein kinase C (PKC, a calpain substrate). Fluoro-Jade B staining was performed at extended time points after 2-, 4- and 6-hour anesthesia to evaluate the possible consequences of the activities of these proteases on neuron survival.
Materials and Methods
Animals and Treatment
Seven-day-old (P7) male Wistar rats (average body weight 12–14 g) were used in all experiments. All experimental procedures were approved by the institutional committee, as well as by the University of Virginia Animal Care and Use Committee, and were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH). The procedures were designed to minimize the suffering of the animals and the number of rats used. Rat pups were placed in a temperature-controlled incubator set to an ambient temperature of 35–36°C. Animals not intended to be killed immediately after anesthesia were allowed to recover in the incubator for 1 h and were returned to their mothers to feed. Animals were administered propofol manufactured for intravenous human use (Recofol®; Schering Oy, Turku, Finland). In addition to the active substance, the formulation contained soybean oil, purified egg phosphatide, glycerol and water. The drug was used according to the manufacturer's instructions. Loss of the righting reflex served as an indicator of anesthetic-induced unconsciousness and sleeping time. We used a dose of 20 mg/kg that impaired the righting reflex for 43 ± 5 min.
Experimental Procedures
P7 rat pups (n = 92) were administered either 2, 4 or 6 bolus injections of 20 mg/kg of propofol intraperitoneally at 1-hour intervals in order to achieve propofol anesthesia designated as 2-, 4- and 6-hour anesthesia, respectively. The animals were decapitated either immediately after cessation of the exposure times (designated as the 0-hour time point), or after the recovery periods that lasted 4, 16 or 24 h following termination of exposure (referred to as 4-, 16- and 24-hour time points, respectively; schematically presented in fig. 1). This schedule was chosen in order to assess a range of postexposure effects. We were unable to measure ventilation, oxygenation and perfusion in pups due to technical difficulties. However, the animals were closely monitored throughout the experiment [2]. The pups appeared pink, well perfused and with no visible signs of cyanosis. Though others investigating propofol reported that prolonged propofol anesthesia did not change the physiological parameters such as pO2, pCO2, lactate and blood sugar concentrations in rats and mice [4,12], the lack of these parameters might be a limitation of the present study. The control animals were decapitated immediately after maternal separation. Control sham-treated animals received intraperitoneal injections of physiological saline [2,12] at 60-min intervals. It has to be noted that we did not examine the effect of the lipid emulsion as a vehicle in this study. It could be a limiting factor in the interpretation of the results, though it was shown that lipid vehicle does not have any effect in untreated animals [5,13,14,15,16]. The cortex and the thalamus from both hemispheres were isolated and frozen for protein analysis (n = 4 per group). For histological studies, whole brains were isolated (n = 3 per group).
Fig. 1.

Timeline of the experiments. Each rectangle marks a 2-hour time period, the gray color represents the duration of the anesthesia. Time point 0 indicates termination of anesthesia. Small arrows point to propofol injections (20 mg/kg), and the arrowheads indicate the postexposure intervals at which the animals were killed.
Tissue Extracts
To obtain whole-cell extracts, the tissue was homogenized with a Dounce homogenizer in 10 vol (w/v) of lysis buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM EDTA pH 8.0, 1 mM EGTA pH 7.2, 0.5% Triton X-100) containing a complete protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitors (20 mM β-glycerophosphate, 5 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 250 mM sodium fluoride). The homogenates were sonicated and centrifuged at 16 000 g at 4°C for 30 min. The supernatants were collected and stored at −70°C until use. Protein concentrations were determined by the bicinchonic acid micro-protein assay (Micro BCA Protein Assay Kit; Pierce Inc., Rockford, Ill., USA) with albumin as standard.
Western Blot Analysis
Proteins (20–40 pg per lane) were separated by 10% SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham Bioscience, Otelfingen, Switzerland). The membranes were blocked at room temperature for 1 h in 3% bovine serum albumin in Tris-buffered saline/0.1% Tween 20 (TBS-T), followed by incubation for 2 h or overnight with primary antibodies. The following antibodies were used: mouse anti-α-spectrin (1:8,000, Chemicon International, Tamecula, Calif., USA), rabbit anti-conventional PKC (1:2,500, gift from Dr. Andy Czernik, Rockefeller University, New York, N.Y., USA), mouse anti-PARP-1 (1:4,000, PharMingen International, San Diego, Calif., USA) and rabbit anti-caspase-3 antibody that recognizes the 17-kDa cleavage product (1:850, Cell Signaling Technology, Beverly, Mass., USA). All antibodies were diluted in 1% bovine serum albumin/TBS-T. The immunoblots were processed with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies (1:5,000, Santa Cruz Biotechnology, Inc., Santa Cruz, Calif., USA) in TBS-T for 1 h at room temperature. All blots were reprobed with rabbit anti-β-actin antibody (1:100,000; MP Biomedicals, Santa Ana, Calif., USA) that served as an endogenous control. The signal was detected by enhanced chemiluminescence (Amersham Bioscience) after exposure of an X-ray film.
Fluoro-Jade B Staining and Image Analysis
Rat brains (n = 3 per group) used for immunohistochemical analysis were fixed in 4% paraformaldehyde for 24 h at 4°C and cryoprotected in a sucrose gradient in 0.01 M phosphate-buffered saline (in 10, 20 and 30% sucrose for 24 h at 4°C). The brains were frozen in isopentane, cooled on dry ice and stored at −70°C. Cortical areas (retrosplenial, parietal, cingulated, occipital and piriform regions) and the anterior thalamus were used on the 7th postnatal day when they are very vulnerable to anesthesia [7]. The brain regions were identified by the size and shape of the lateral ventricle and hippocampus. The brains were cut, and every 5th coronal section (18 μm thick) was mounted on a slide, allowed to dry overnight and stored at −20°C. The slides were first immersed in a basic alcohol solution consisting of 1% NaOH in 80% ethanol, distilled water and incubated in 0.06% KMnO4 solution for 10 min. The slides were transferred for 10 min to a 0.0001% solution of Fluoro-Jade B (Chemicon International) dissolved in 0.1% acetic acid. The slides were rinsed by three changes of distilled water for 1 min per change. The slides were then immersed in 0.01% Hoechst 33258 (Acros Organics, Fair Lawn, N.J., USA) staining solution for 10 min and coverslipped with glycerol. The sections were examined with an Axio Observer Microscope Z1 (Zeiss, Jena, Germany) using a filter system suitable for visualizing fluorescein isothiocyanate. Fluoro-Jade B stains degenerating neuronal somata and their processes, presenting results that are comparable to other histological stains [2]. Cells labeled with Fluoro-Jade B were observed as individual shiny green spots that were clearly discernible from the background. Three rats per group were analyzed by a researcher who was unaware of the treatment. The number of degenerating neurons labeled by Fluoro-Jade B was counted in 3 fields under the area of the screen (0.38 mm2) in 5 sections of the regions of interest per animal. Sections from all groups were run in the same assay. The Fluoro-Jade B assay was run with MK-801-treated P7 rats as a positive control for neurodegeneration (data not shown).
Statistical Analysis
Semi-quantitative evaluation of protein levels detected by immunoblotting was performed by densitometric scanning using the computerized image analysis program ImageQuant 5.0. The data are presented as percentages (means ± SEM) relative to the control samples assumed to be 100%. Differences between the experimental groups were tested using Kruskal-Wallis nonparametric ANOVA with Mann-Whitney's post hoc U test (Statistica version 5.0; StatSoft, Tulsa, Okla., USA). Significance was reported at p < 0.05.
Results
Repeated Propofol Administration Leads to Different α-II-Spectrin Cleavage Profiles in the Cortex and Thalamus
To study the vulnerability of the immature rat brain to propofol neurotoxicity, P7 pups were subjected to propofol anesthesia that lasted 2, 4 or 6 h, and subsequently decapitated, either immediately after termination of the anesthesia (at the 0-hour time point), or after 4, 16 and 24 h. To examine whether repeated propofol administration influenced calpain activity, we performed Western blot analysis of α-II-spectrin in the cortex and thalamus. Cleavage of the 280-kDa full-length spectrin by calpain led to the formation of the 145-kDa fragment which served as a relative measure of calpain activity, while the appearance of an additional 120-kDa fragment pointed to caspase-3 activity, as shown on representative blots after 4 h of propofol exposure (fig. 2a, b). Quantitative analysis showed that the cortex and thalamus of untreated controls contained abundant amounts of the 145-kDa fragment. Repeated propofol treatment changed calpain activity in time-dependent and brain structure-specific manners (fig. 2c, d). Spectrin proteolysis in the cortex was increased only after the 6-hour treatment. A decrease in the spectrin 145-kDa fragment (to about 60–76% of the control value) was observed at 16 and 24 h after all 3 exposures. In the thalamus, the amount of the 145-kDa protein fragment significantly increased immediately after termination of propofol anesthesia and remained elevated for the next 4 h (fig. 2d). Processing of full-length spectrin to the 145-kDa peptide returned to the baseline level 24 h after treatments. A change in the caspase-3-generated 120-kDa fragment was detected only after termination of the 6-hour anesthesia in the cortex, where a 30% increase was observed. At other time points, it was near the control value (fig. 2e). A 200% increase in the 120- kDa protein fragment was observed in the thalamus following 2-, 4- and 6-hour exposures to propofol anesthesia (fig. 2f). The kinetics of the changes differed depending on the length of the propofol treatment. It is worth noting the biphasic profile of the 120-kDa product following the 2- and 6-hour propofol treatments.
Fig. 2.

Time course of spectrin breakdown products after the 2-, 4- and 6-hour exposures to propofol in the cortex and thalamus of P7 rats. Whole-cell extracts were used to detect the presence of the 280-kDa intact spectrin protein; calpain produced a 145-kDa fragment and caspase-3 produced a 120-kDa fragment in the cortex (a) and the thalamus (b). Bars represent a quantitative densitometric evaluation of the 145-kDa spectrin fragment in the cortex (c) and the thalamus (d), and the 120-kDa spectrin fragment in the cortex (e) and the thalamus (f). Results are presented for animals at different recovery time points (0, 4, 16 and 24 h) after exposure to propofol for 2, 4 and 6 h. Representative immunoblots of the 4-hour treatment are shown. β-Actin was run as an internal standard for equal loading. The results are the means ± SEM. a p < 0.05 vs. control value presented as a black line, b p < 0.05 between treatments.
Changes in PARP-1 Proteolysis Induced by Repeated Propofol Administration
Calpain and caspase-3 activities were further evaluated by examining the PARP-1 cleavage pattern. Intact PARP-1 is a 116-kDa protein. After propofol treatment, different degradation products were detected in the cortex and thalamus at all of the examined times, as shown on representative blots obtained after 2-hour anesthesia (fig. 3a, b). Major approximately 50-, 40- and approximately 20-kDa cleavage products and minor approximately 62- and approximately 35-kDa bands were observed. The 3 abundant bands were quantified (fig. 3c–h). Quantification of the 50- and 20-kDa cleavage products in the cortex revealed slight changes over time, and increases of both 50- and 20-kDa bands at the end of the treatment (fig. 3c, g), while the level of the 40-kDa polypeptide changed only after the 2-hour anesthesia. In the thalamus, we observed the accumulation of 50-, 40- and 20-kDa protein fragments up to the end of the period of observation (fig. 3d–h). The most pronounced changes were observed in the intensity of the approximately 20-kDa PARP-1 fragment that exhibited a 400% increase in the thalamus 24 h after cessation of the 2-, 4- and 6-hour anesthesia. Significantly higher amounts of the approximately 50- and approximately 20-kDa products were observed early after the 4-hour exposure compared to the 2- and 6-hour exposures.
Fig. 3.

Time course of appearance of PARP-1 cleavage fragments in the cortex and thalamus of P7 rats after exposure to propofol for 2, 4 and 6 h. Whole-cell extracts were used to detect the presence of 50-, 40- and 20-kDa PARP-1 fragments in the cortex (a) and the thalamus (b). Bars represent quantitative densitometric evaluation of the approximately 50-kDa PARP-1 fragment in the cortex (c) and the thalamus (d), the approximately 40-kDa PARP- 1 fragment in the cortex (e) and the thalamus (f), and the approximately 20-kDa PARP-1 fragment in the cortex (g) and the thalamus (h). Results are presented for animals at different recovery time points (0, 4, 16 and 24 h) after exposure to propofol for 2, 4 and 6 h. Representative immunoblots of the 2-hour treatment are shown. β-Actin was run as an internal standard for equal loading. Results are presented as means ± SEM. a p < 0.05 vs. control value presented as a black line, b p < 0.05 between treatments.
Cleavage of PARP-1 by caspase-3 gives rise to the 24- and 89-kDa signature products. The used C-II-10 antibody recognizes the N-terminus of the 89-kDa protein fragment; this band was not observed.
Different Outcomes of PKC Proteolysis by Calpain after Repeated Propofol Administration
To further analyze calpain activity, PKC, the substrate only for calpain, was examined. Proteolysis of PKC by calpain yields major 46-kDa catalytic and 36-kDa regulatory subunits. Immunoblotting revealed the presence of 82- and 36-kDa bands that correspond to native PKC and the regulatory subunit of PKC, respectively. Representative blots on figure 4a and b show PKC proteolysis after the 6-hour propofol treatment. Marked calpain activity, as judged by the appearance of the 36-kDa fragment, was observed in the control samples. In the cortex, a distinct increase was detected immediately after termination of the 2-, 4- and 6-hour anesthesia (by 68, 50 and 100%, respectively; fig. 4a, c). The abundance of the 36-kDa fragment decreased considerably over time. The most pronounced decline (by about 50%) was observed 24 h after termination of the 6-hour anesthesia. In the thalamus, propofol increased calpain proteolysis immediately after termination of the anesthesia. Increased proteolysis was maintained for 24 h after the 2- and 6-hour anesthesia (fig. 4b, d). In marked contrast to the protracted increase of the 36-kDa fragment, exposure to propofol for 4 h was followed by an 80% increase above the basal value at the 0-hour time point, after which it decreased, and at the end of the recovery period it was about 60% of the control value.
Fig. 4.

Time course of PKC levels in the cortex and thalamus of P7 rats after 2-, 4- and 6-hour exposures to propofol. Whole-cell extracts were used to detect the presence of the 82-kDa intact PKC protein and the calpain-produced 36-kDa PKC fragment in the cortex (a) and the thalamus (b). Bars represent quantitative densitometric evaluation of the intact 36-kDa protein in the cortex (c) and the thalamus (d). Results are presented for animals at different recovery time points (0, 4, 16 and 24 h) after exposure to propofol for 2, 4 and 6 h. Representative immunoblots of the 6-hour treatment are shown. β-Actin was run as an internal standard for equal loading. The results are the means ± SEM. a p < 0.05 vs. control value presented as a black line, b p < 0.05 between treatments.
Propofol Induced the Formation of Active Fragments of Caspase-3 in the Cortex and Thalamus
Activation of caspase-3 requires proteolytic processing of the intact proenzyme (32 kDa) and formation of a heterodimer containing the large (17 kDa) and small subunits (12 kDa). Caspase-3 active fragments of 19 and 17 kDa were detected in both structures (fig. 5a, b). In the cortex, 2 h of propofol exposure caused the most prominent generation of protein fragments resulting from caspase-3 activity 24 h after the treatment (96% more than the control), while 4 h of exposure produced the highest increase, observed 16 h after termination of the anesthesia (78% more than the control; fig. 5a, c). Exposure to propofol for 6 h produced a biphasic increase of active fragments. The first peak (a 174% increase) was observed immediately after cessation of anesthesia. A maximal increase of 195% relative to the control value was observed 16 h after anesthesia. In the thalamus, a significant and persistent effect of propofol was established after all 3 anesthesia times (fig. 5d). The animals that were anesthetized for 2 and 4 h exhibited an early peak at 4 h after termination of the anesthesia (760 and 500% increases, respectively). The 6-hour anesthesia produced a sharp 10-fold rise 16 h after anesthesia. At the end of the recovery period, the 19/17-kDa subunits increased 13- and 14-fold after all 3 anesthetic regimens.
Fig. 5.

Time course of appearance of active caspase-3 subunits in the cortex and thalamus of P7 rats after exposure to propofol for 2, 4 and 6 h. Whole-cell extracts were used to detect the appearance of active 19/17-kDa caspase-3 subunits in the cortex (a) and the thalamus (b). Bars represent quantitative densitometric evaluation of 19/17-kDa subunits in the cortex (c) and the thalamus (d). Results are presented for animals at different recovery time points (0, 4, 16 and 24 h) after exposure to propofol for 2, 4 and 6 h. Representative immunoblots are shown. β-Actin was run as an internal standard for equal loading. Results are presented as means ± SEM. a 0 < p.05 vs. control value presented as a black line, b p < 0.05 between treatments.
Propofol-Induced Neurodegeneration
The biochemical parameters presented above indicate that propofol administration to postnatal P7 rat pups increased calpain and caspase-3 activities that could result in neuronal damage of the cortex and thalamus. We next examined potential propofol neurotoxicity at the histological level using Fluoro-Jade B, which stains degenerating neuronal somata and their processes. Fluoro-Jade B-stained sections of the cortex and thalamus obtained immediately after termination of exposure to propofol lasting 2, 4 and 6 h are shown in figure 6. The control samples exhibited a very low degree of developmental neuroapoptosis, as revealed by a minimal number of stained cells (fig. 6a, left and right panels). The propofol treatment induced the appearance of degenerating neurons in both structures. Fluoro-Jade B-labeled cells were detected in the retrosplenial cortex after the 4-hour anesthesia; their numbers peaked after the 6-hour anesthesia (fig. 6c, d, left panel). In the thalamus, the most pronounced increase in the number of Fluoro-Jade B-labeled cells was detected in the laterodorsal thalamic nucleus following the 2-hour exposure to propofol (a 17-fold increase compared to the control). The number of degenerating neurons in the thalamus declined proportionately with the duration of the anesthesia (fig. 6b–d, right panel), as quantification of the degenerating neurons in these brain regions shows (fig. 6e). When brain sections were examined 4 and 16 h after the termination of all 3 exposures, no increases in dead cells were detected (data not shown). When neurodegeneration was evaluated 24 h after exposures, no dead cells were detected in either the cortex or the thalamus of the animals that were maintained in propofol anesthesia for 2 and 4 h (data not shown). Only the 6-hour anesthesia caused the appearance of dying neurons at the late phase of observation (fig. 7). An increased number of Fluoro-Jade B-positive cells was observed in the laterodorsal thalamic nucleus (fig. 7b). No apparent changes in the number of Fluoro-Jade B-stained cells were observed in the cortex (fig. 7a). To investigate the mode of cell death in these neurons, sections were stained with Hoechst 33258, a blue fluorescent DNA stain, alongside Fluoro-Jade B. Many cells contained 1 or more foci of compact, Hoechst-staining chromatin, an indication that they were undergoing apoptosis.
Fig. 6.
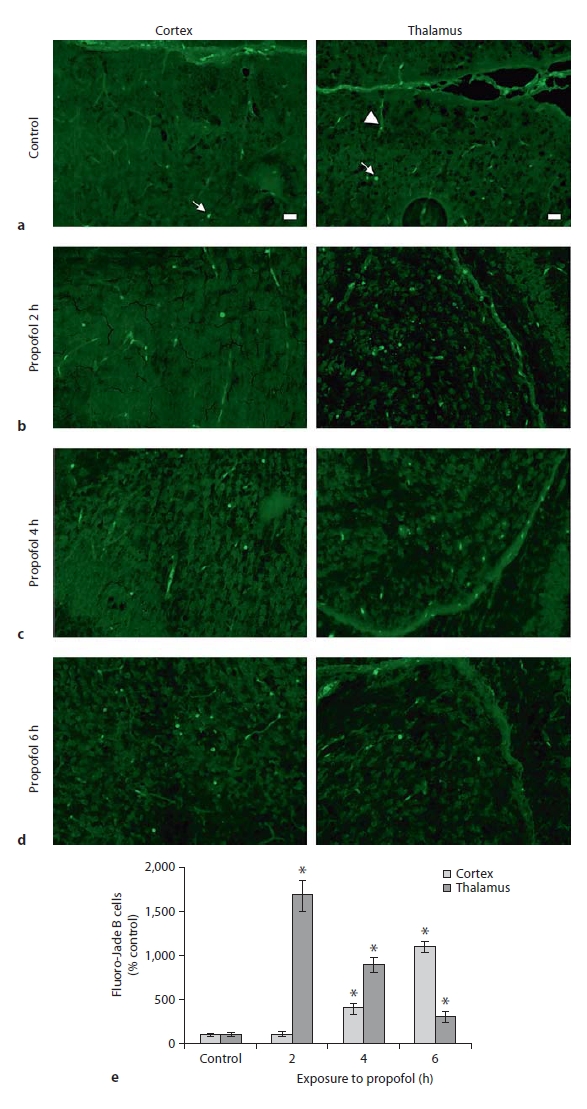
Fluoro-Jade B staining of P7 rat brains immediately (0 h) after termination of the 2-, 4- and 6-hour exposures to propofol. An increase in the number of Fluoro- Jade B-positive cells was apparent in the retrosplenial cortex following 4 and 6 h of exposure (c, d, left panel), and in the laterodorsal thalamic nucleus following 2-, 4- and 6-hour exposure (b, c, d, respectively, right panel). Quantitative analysis of Fluoro-Jade B staining at the retrosplenial cortex and the laterodorsal thalamic nucleus is shown (e). For each condition, 3 animals were randomly assigned. Data are presented as means ± SEM. A probability of * 0 < p.05 was considered significant. Degenerating neurons are marked with arrows, arrowheads point to blood vessels. Scale bar = 20 μm.
Fig. 7.
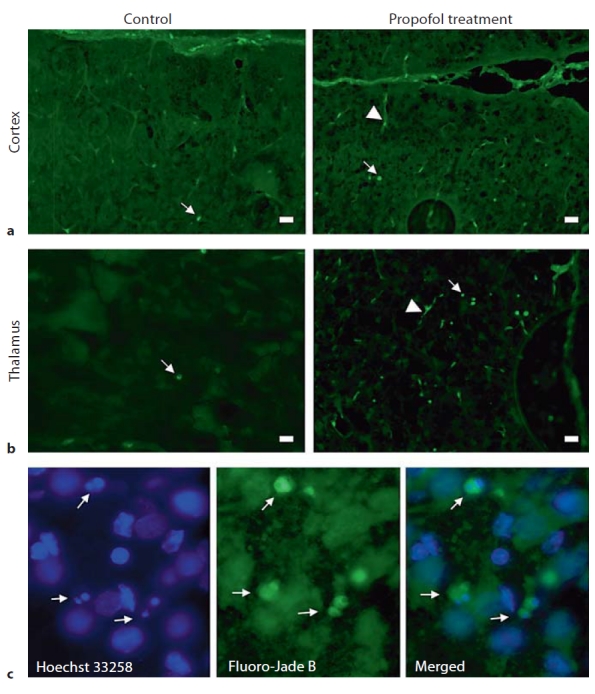
Fluoro-Jade B staining of P7 rat brains at the 24-hour time point following exposure to propofol for 6 h. An increase in the number of Fluoro-Jade B-positive cells was apparent in the thalamus (b) but not in the cortex (a). c The laterodorsal thalamic nucleus was stained with Hoechst 33258, a blue fluorescent DNA stain, alongside Fluoro-Jade B. Some neurons were double-labeled; the same neuron revealed bright clumps of fragmented and condensed chromatin, typical for apoptotic nuclei, as well as positive green staining for Fluoro-Jade B (×40). Degenerating neurons are marked with arrows, arrowheads point to blood vessels. Scale bar = 20 μm.
Discussion
In this work, we examined the regional and temporal profiles of calpain and caspase-3 activities in the cortex and thalamus of P7 rats after repeated propofol administration that resulted in anesthesia lasting 2, 4 and 6 h. The main finding is that the propofol treatment produced effects on the activities of both proteases that depended on the duration of the anesthesia and were time- and brain region-specific. We assumed that both calpain and caspase-3 contributed to cell death, as observed by Fluoro-Jade B staining of the cortex and thalamus following prolonged propofol anesthesia.
The relative activities of calpain and caspase-3, manifested as changes in the levels of major proteolytic fragments of their substrates, α-II-spectrin, PKC and PARP-1, were detected and quantified by Western immunoblotting analysis. α-II-Spectrin is a structural component of the cytoskeleton. Calpain-mediated proteolysis of spectrin is involved in the process of synaptogenesis and neurite extension [10]. Repeated propofol administration to P7 rats produced a different effect on calpain activity in the examined structures. In the thalamus, an increase in the 145-kDa fragment was detected. In contrast to studies which used other models [17,18], instead of an accumulation of the 145-kDa spectrin fragments over time, we detected a transient increase immediately after cessation of anesthesia. Although increased spectrin proteolysis may not be a critical event in the pathway to neuronal cell death, it is undoubtedly one of the hallmarks of the initial stage of cell death. In the cortex, the most remarkable finding after repeated propofol administration was the decrease below the control level of calpain activity at the later post-treatment time points. Given that calpain-mediated proteolysis is associated with normal cell functioning and brain development, the question arises whether a decrease as well as an increase of spectrin proteolysis had detrimental effects at this vulnerable period during the brain growth spurt. Spectrin is an actin-binding protein, and changes in spectrin dynamics have considerable consequences for the cytoskeleton. It was previously demonstrated that propofol induces phosphorylation of actin [13,19]. Changes in the cytoskeleton could trigger a chain of biochemical events which lead to long-term modifications that are related to structural changes and synaptic plasticity, but that also activate cell death processes [14,20].
The pattern of PKC proteolysis confirmed our finding of brain structure-dependent calpain activity following propofol treatment. The transient increase in the calpain-generated PKC 36-kDa fragment that was observed in the cortex immediately upon termination of anesthesia may be the consequence of the ability of propofol to switch the intact 82-kDa PKC protein to an activated, open conformation which represents a more susceptible target for calpain [21]. Proteolysis of PKC by calpain is one of the mechanisms of PKC activation that cause the release of constitutively active catalytic subunits. This mode of PKC activation has a crucial regulatory effect on neurite outgrowth [22]. Overactivated catalytic PKC subunits could have access to and therefore phosphorylate substrates that are normally unavailable to intact PKC and have some detrimental effects. PKC activation directly or indirectly impacts spectrin-actin filaments, causing destabilization of the cytoskeleton and changes in dendritic branches and postsynaptic spines [20].
PARP-1 was cleaved to its calpain-specific pattern, revealing fragments ranging from 20 to 62 kDa. A similar pattern of cleavage was observed in cells treated with necrotic inducers, which are known to trigger the calcium-calpain pathway [23,24]. PARP-1 proteolysis was higher in the thalamus compared to the cortex where it was in agreement with spectrin and PKC proteolysis. The PARP-1 fragments that appeared later were probably the consequence of the different localization of PARP-1 (in the nucleus), compared to the other 2 monitored calpain substrates which were located in the cytosol (PKC) and cytoskeleton (spectrin) [25]. Activation of PARP-1 participates in neurite outgrowth and differentiation of PC12 cells following NGF treatment. This effect is at least in part mediated by PKC activation [26]. However, the increase in calpain-mediated cleavage products detected herein pointed to the proteolytic inactivation of PARP-1 and the silencing of its enzymatic activity that plays a role in survival. PARP-1 is a target for PKC phosphorylation [27]. It would be interesting to establish whether phosphorylation can change PARP-1 sensitivity to calpain, as observed for spectrin phosphorylation [28]. Overall, calpain activity affects many parameters of neuronal growth, function and morphology. It can interfere with neuronal activity that is critical for establishing proper connections at the peak of brain synaptogenesis.
The relative proteolytic activity of caspase-3 was estimated through the generation of the spectrin 120-kDa fragments only, while the 89-kDa PARP-1 fragment characteristic of caspase-3 cleavage was not detected. It is possible that its absence was due to further degradation of the 89-kDa subunit by calpain [24] which recognizes 4 specific motifs in PARP-1 [23]. To further assess the role of caspase-3 in the cascade triggered by the propofol treatment, we measured the appearance of active caspase-3 subunits (fig. 5). Repeated propofol application produced changes in the patterns of released 19/17-kDa caspase-3 subunits that were anesthesia-, time- and region-specific. In the thalamus, a more pronounced activation of caspase-3 was observed in comparison to the cortex. It correlated with the higher level of the caspase-generated 120-kDa spectrin fragment. It showed an almost biphasic temporal profile. This confirmed that propofol-mediated caspase-3 activation was functional, though the extent of caspase-3 activation was much higher than the degree of spectrin proteolysis (fig. 2f).
Evaluation of propofol toxicity at the histological level revealed that the number of Fluoro-Jade B-labeled dead cells depended on the duration of propofol anesthesia and that it was a distinct feature of a specific brain structure. In the laterodorsal thalamic nucleus, the highest number of dead cells was detected immediately after the shortest, 2-hour anesthesia. To the best of our knowledge, the detection of dying cells at such an early time point after a relatively short treatment has not been reported, as most studies depict dead cells at delayed periods after treatments [2,4]. In the retrosplenial cortex, the opposite tendency was observed, and the level of dead cells was proportional to the duration of the anesthesia. These experiments revealed that the thalamus was affected by the propofol anesthesia much earlier than the cortical neurons. Our findings point to different vulnerabilities of different brain regions, since neuronal populations undergo synaptogenesis at different times during brain development [7,29,30]. Also, immature GABA receptors are excitatory early during development and convert to an inhibitory role in more mature neurons [3]. The conversion of GABA receptors occurs during the first week of postnatal development, and at day 7, some regions have either completed the switch or are well along in the process of switching. Thus, GABAergic excitation is most likely region-dependent and should be considered as a contributor to damage [31]. A further possibility is that the expression of GABAA subunits is different in these regions at this stage of development. Propofol induces tyrosine phosphorylation of GABAA receptor β2 and β3 subtypes. It mediates diverse calcium-dependent signaling mechanisms that could lead to neuronal injury or death [13].
Taking into account the observed changes in calpain and caspase-3 activities, the effects of the treatments at subsequent time points were also examined by Fluoro-Jade B staining. Unexpectedly, dying cells were seen only 24 h after the 6-hour propofol exposure in the thalamus and hippocampus (observed as widespread neurodegeneration in the CA1 region; data not shown). This result is in agreement with reports that describe cell death 24 h after the propofol treatment [4,6,8]. However, the levels of caspase-3 active subunits in the thalamus following the 2 shorter exposures to propofol were the same as after the 6-hour anesthesia, although no cell death was detected. In view of the observed trend of increasing caspase-3 activation following shorter treatments, we cannot exclude the possibility that caspase-3 activation increased further after 24 h. In the neonatal brain that has a great potential for activation of the apoptotic pathways in response to different insults, procaspase-3 is several-fold more abundant than in the mature brain [30]. The discrepancy between caspase-3 cleavage and Fluoro-Jade B labeling during the delayed phase may be a consequence of the activation of protective processes [32,33] that oppose the execution of the death program, except in a more severe insult such as exposure to anesthesia lasting 6 h. Recent studies have shown that propofol can induce retraction of neurites and long-term changes in dendritic arbor development that could potentially lead to an impairment of neuronal networks [5,14]. As numerous data support the participation of caspase-3 in nonapoptotic cellular events, such as axon guidance and cytoskeletal remodeling, we could not exclude the possibility that shorter propofol treatments mostly contributed to morphological changes without associated cell death [32,34]. Further investigations will be necessary to evaluate all of the factors that impact the functionality and role of activated caspase-3 in the absence of cell death following shorter propofol exposure.
Some limitations of this study should be mentioned. The dose of propofol used in our study is much higher than those usually applied in practice as a bolus (3 mg/kg i.v.). However, the dosing paradigms used in animal studies typically do not reflect doses used for pediatric patients, and can be 20–50 times higher in animals than in typical clinical practice [1]. A continuous infusion of propofol is the clinically relevant way of administration. However, in small rodents, continuous infusion is very often avoided as venous puncture can be technically difficult, so anesthetics are acceptable to be injected via the intraperitoneal, subcutaneous or intramuscular way [2,6,35]. Continuous infusion would also be valuable to circumvent pain during repeated propofol injecting. Repetitive pains per se could impact cell death in the developing brain [36,37], and the thalamocortical complex is essential for consciousness and pain processing. This has to be taken into consideration when interpreting different scores of degenerating neurons in the cortex and thalamus between shorter and longer anesthesia regimens. We were unable to perform arterial blood analyses. Others who have investigated propofol in rats did not detect significant differences in arterial blood parameters between control and experimental groups [4]. Based on their observation and on close monitoring of pups during experiments revealing their well-being, we presume that neurodegeneration is not a manifestation of cardiorespiratory deficiency but of propofol action.
In this study, we have shown that repeated propofol administration as a single anesthetic induces cell death in the cortex and thalamus during the most vulnerable periods of rat brain development. The conclusion that propofol-induced neurotoxicity is at least in part mediated by caspase-3 and the calpain-dependent pathways is based on our findings that these enzymes exhibit complex regional and temporal patterns of activation. The activation of caspase-3 is a widely accepted sign of apoptotic cell death [9]. Calpain activation can execute the apoptotic process by itself; however, its activation suggests that other cell death processes might also be under way. These potential interactions could contribute to the hybrid morphology of neuronal death usually observed in vivo [17,18]. To resolve whether calpain activity which coincides with caspase-3 activity participates in apoptotic and/or necrotic cell death will need additional data and ultrastructural analyses. Propofol treatment is known to change calcium homeostasis [13,31] that can modulate the activity of calpain, a calcium-sensitive protease. This was confirmed in our study. Recent data have revealed that volatile anesthetics, which are also GABA agonists, induce cell damage by disrupting intracellular calcium homeostasis, and that their toxicity depends on their different ability to increase the Ca2+ level [38]. Moreover, caspase-3 activation induced by isoflurane also depends on the cytosolic calcium levels [39]. Thus, the duration of the propofol anesthesia-specific effects observed in our study may be the consequence of changed calcium signaling in the cortex and thalamus, which in turn triggers initial reversible neuronal changes and irreversible loss of neuronal cells. Complex interactions between caspases and calpains during the process of cell death must be considered when evaluating the mechanisms and efficacy of therapeutic interventions aimed at reducing anesthesia-induced neuronal death.
Acknowledgements
This work was supported by grant 143004B from the Ministry of Sciences and Technological Development, Republic of Serbia, and the Fogarty International Research Award, NIH Award GC11479-128322 (to V.J.-T., Principal Investigator; S.R., Awardee; parent grant R01 HD 44517 from the NIH/NICHD to V.J.-T.).
References
- 1.Loepke AW, Soriano SG. An assessment of the effects of general anesthetics on developing brain structure and neurocognitive function. Anesth Analg. 2008;106:1681–1707. doi: 10.1213/ane.0b013e318167ad77. [DOI] [PubMed] [Google Scholar]
- 2.Fredriksson A, Ponten E, Gordh T, Eriksson P. Neonatal exposure to a combination of N-methyl-D-aspartate and γ-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology. 2007;107:427–435. doi: 10.1097/01.anes.0000278892.62305.9c. [DOI] [PubMed] [Google Scholar]
- 3.Henschel O, Gipson KE, Bordey A. GABAA receptors, anesthetics and anticonvulsants in brain development. CNS Neurol Disord Drug Targets. 2008;7:211–224. doi: 10.2174/187152708784083812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bercker S, Bert B, Bittigau P, Felderhoff-Muser U, Buhrer C, Ikonomidou C, Weise M, Kaisers UX, Kerner T. Neurodegeneration in newborn rats following propofol and sevoflurane anesthesia. Neurotox Res. 2009;16:140–147. doi: 10.1007/s12640-009-9063-8. [DOI] [PubMed] [Google Scholar]
- 5.Vutskits L, Gascon E, Tassonyi E, Kiss JZ. Clinically relevant concentrations of propofol but not midazolam alter in vitro denditic development of isolated γ-aminobutiric acid-positive interneurons. Anesthesiology. 2005;102:970–976. doi: 10.1097/00000542-200505000-00016. [DOI] [PubMed] [Google Scholar]
- 6.Cattano D, Young C, Straiko MMW, Olney JW. Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth Analg. 2008;106:1712–1714. doi: 10.1213/ane.0b013e318172ba0a. [DOI] [PubMed] [Google Scholar]
- 7.Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–827. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 8.Pesic V, Milanovic D, Tanic N, Popic J, Kanazir S, Jevtovic-Todorovic V, Ruzdijic S. Potential mechanism of cell death in the developing rat brain induced by propofol anesthesia. Int J Dev Neurosci. 2009;27:279–287. doi: 10.1016/j.ijdevneu.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang KK. Calpain and caspase: can you tell the difference? Trends Neurosci. 2000;23:20–26. doi: 10.1016/s0166-2236(99)01536-2. [DOI] [PubMed] [Google Scholar]
- 10.Czogalla A, Sikorski AF. Spectrin and calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell Mol Life Sci. 2005;62:1913–1924. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCollum AT, Nasr P, Estus S. Calpain activates caspase-3 during UV-induced neuronal death but only calpain is necessary for death. J Neurochem. 2002;82:1208–1220. doi: 10.1046/j.1471-4159.2002.01057.x. [DOI] [PubMed] [Google Scholar]
- 12.De Roo M, Klauser P, Briner A, Nikonenko I, Mendez P, Dayer A, Kiss JZ, Muller D, Vutskits L. Anesthetics rapidly promote synaptogenesis during a critical period of brain development. PLoS One. 2009;4:e7043. doi: 10.1371/journal.pone.0007043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjornstrom K, Turina D, Loverock A, Lundgren S, Wijkman M, Lindroth M, Eintrei CH. Characterization of the signal transduction cascade caused by propofol in rat neurons: from the GABAA receptor to the cytoskeleton. J Physiol Pharmacol. 2008;59:617–632. [PubMed] [Google Scholar]
- 14.Turina D, Loitto VM, Bjornstrom K, Sundqvist T, Eintrei C. Propofol causes neurite retraction in neurons. Br J Anesth. 2008;101:374–379. doi: 10.1093/bja/aen185. [DOI] [PubMed] [Google Scholar]
- 15.Nakao S, Nagata A, Miyamoto E, Masuzawa M, Murayama T, Shingu K. Inhibitory effect of propofol on ketamine-induced c-Fos expression in the rat posterior cingulated and retrosplenial cortices is mediated by GABAA receptor activation. Acta Anesthesiol Scand. 2003;47:284–290. doi: 10.1034/j.1399-6576.2003.00040.x. [DOI] [PubMed] [Google Scholar]
- 16.Oscarsson A, Massoumi R, Sjolander A, Eintrei C. Reorganization of actin in neurons after propofol exposure. Acta Anesthesiol Scand. 2001;45:1215–1220. doi: 10.1034/j.1399-6576.2001.451007.x. [DOI] [PubMed] [Google Scholar]
- 17.Northington FJ, Zelaya ME, O'Riordan DP, Blomgren K, Flock DL, Hagberg H, Ferriero DM, Martin LJ. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as ‘continuum’ phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience. 2007;149:822–833. doi: 10.1016/j.neuroscience.2007.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carloni S, Carnevali A, Cimino M, Balduini W. Extended role of necrotic cell death after hypoxia-ischemia-induced neurodegeneration in the neonatal rat. Neurobiol Dis. 2007;27:354–361. doi: 10.1016/j.nbd.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Jensen AG, Lindroth M, Sjolander A, Eintrei C. Propofol induces changes in the cytosolyc free calcium concentrations and the cytoskeletal organization of cultured human glial cells and primary embryonic rat brain cells. Anesthesiology. 1994;81:1220–1229. doi: 10.1097/00000542-199411000-00016. [DOI] [PubMed] [Google Scholar]
- 20.Rao A, Craig AM. Signaling between the actin cytoskeleton and the postsynaptic density of dendritic spines. Hippocampus. 2000;10:527–541. doi: 10.1002/1098-1063(2000)10:5<527::AID-HIPO3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 21.Gomez RS, Guatimosim C, Gomez MV. Mechanism of action of volatile anesthetics: role of protein kinase C. Cell Mol Neurobiol. 2003;23:877–885. doi: 10.1023/b:cemn.0000005316.38838.71. [DOI] [PubMed] [Google Scholar]
- 22.Shea TB, Cressman CM, Spencer MJ, Beermann ML, Nixon RA. Enhancement of neurite outgrowth following calpain inhibition is mediated by protein kinase C. J Neurochem. 1995;65:517–527. doi: 10.1046/j.1471-4159.1995.65020517.x. [DOI] [PubMed] [Google Scholar]
- 23.Buki KG, Bauer PI, Kun E. Isolation and identification of a proteinase from calf thymus that cleaves poly(ADP-ribose) polymerase and histone H1. Biochim Biophys Acta. 1997;1338:100–106. doi: 10.1016/s0167-4838(96)00189-6. [DOI] [PubMed] [Google Scholar]
- 24.Shah GM, Shah RG, Poirier GG. Different cleavage pattern for poly(ADP-ribose) polymerase during necrosis and apoptosis in HL-60 cells. Biochem Biophys Res Commun. 1996;229:838–844. doi: 10.1006/bbrc.1996.1889. [DOI] [PubMed] [Google Scholar]
- 25.Chiarugi A. Poly(ADP-ribose) polymerase: killer or conspirator? The ‘suicide hypothesis’ revisited. Trends Pharmacol Sci. 2002;23:122–129. doi: 10.1016/S0165-6147(00)01902-7. [DOI] [PubMed] [Google Scholar]
- 26.Visochek L, Steingart RA, Vulih-Shultzman I, Klein R, Priel E, Gozes I, Cohen-Armon M. PolyADP-rybosylation is involved in neurotrophic activity. J Neurosci. 2005;25:7420–7428. doi: 10.1523/JNEUROSCI.0333-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka Y, Koide S, Yoshihara K, Kamiya T. Poly (ADP-ribose) synthetase is phosphorylated by protein kinase C in vitro. Biochem Biophys Res Commun. 1987;148:709–717. doi: 10.1016/0006-291x(87)90934-x. [DOI] [PubMed] [Google Scholar]
- 28.Nicolas G, Fournier CM, Galand C, Malbert-Colas L, Bournier O, Kroviarski Y, Bourgeois M, Camonis JH, Dhermy D, Grandchamp B, Lecomte MC. Tyrosine phosphorylation regulates alpha II spectrin cleavage by calpain. Molec Cell Biol. 2002;22:3527–3536. doi: 10.1128/MCB.22.10.3527-3536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Tenkova TI, Stefanovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;283:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 30.Heaton MB, Paiva M, Madorsky I, Shaw G. Ethanol effects on neonatal rat cortex: comparative analyses of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Dev Brain Res. 2003;145:249–262. doi: 10.1016/j.devbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Kahraman S, Zup SL, McCarthy MM, Fiskum G. GABAergic mechanisms of propofol toxicity in immature neurons. Neurosurg Anesthesiol. 2008;20:233–240. doi: 10.1097/ANA.0b013e31817ec34d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLaughlin B. The kinder side of killer proteases: caspase activation contributes to neuroprotection and CNS remodeling. Apoptosis. 2004;9:111–121. doi: 10.1023/B:APPT.0000018793.10779.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villapol S, Acarin L, Faiz M, Castellano B, Gonzales B. Survivin and heat shock protein 25/75 colocalize with cleaved caspase-3 in surviving reactive astrocytes following excitotoxicity to the immature brain. Neuroscience. 2008;153:108–119. doi: 10.1016/j.neuroscience.2008.01.054. [DOI] [PubMed] [Google Scholar]
- 34.Acarin L, Villapol S, Faiz M, Rohn TT, Castellano B. Caspase-3 activation in astrocytes following postnatal excitotoxic damage correlates with cytoskeletal remodeling but not with cell death or proliferation. Glia. 2007;55:954–965. doi: 10.1002/glia.20518. [DOI] [PubMed] [Google Scholar]
- 35.Bazin JE, Constantin JM, Gindre G. Laboratory animal anesthesia: influence of anesthetic protocols on experimental models. Ann Fr Anesth Reanim. 2004;23:811–818. doi: 10.1016/j.annfar.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 36.Anand KJS, Coskun V, Thrivikraman KV, Nemeroff CB, Plotsky PM. Long-term behavioral effects of repetitive pain in neonatal rat pups. Physiol Behav. 1999;66:627–637. doi: 10.1016/s0031-9384(98)00338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anand KJS, Garg S, Rovnaghi CR, Narsinghani U, Bhutta AT, Hall RW. Ketamine reduces the cell death following inflammatory pain in newborn rat brain. Pediatr Res. 2007;62:283–290. doi: 10.1203/PDR.0b013e3180986d2f. [DOI] [PubMed] [Google Scholar]
- 38.Yang H, Liang G, Hawkins B, Madesh M, Pierwola A, Wei H. Inhalational anesthetics induce cell damage by disruption of intracellular calcium homeostasis with different potencies. Anesthesiology. 2008;109:243–250. doi: 10.1097/ALN.0b013e31817f5c47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang G, Dong Y, Zhang B, Ichinose F, Wu X, Culley JD, Crosby G, Tanzi RE, Xie Z. Isoflurane-induced caspase-3 activation is dependent on cytosolic calcium and can be attenuated by memantine. J Neurosci. 2008;28:4551–4560. doi: 10.1523/JNEUROSCI.5694-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]