

Abstract
Large-scale profiling methods have uncovered numerous gene and protein expression changes that correlate with tumorigenesis. However, determining the relevance of these expression changes and which biochemical pathways they affect has been hindered by our incomplete understanding of the proteome and its myriad functions and modes of regulation. Activity-based profiling platforms enable both the discovery of cancer-relevant enzymes and selective pharmacological probes to perturb and characterize these proteins in tumour cells. When integrated with other large-scale profiling methods, activity-based proteomics can provide insight into the metabolic and signalling pathways that support cancer pathogenesis and illuminate new strategies for disease diagnosis and treatment.
Cells with fundamental metabolic alterations commonly arise during tumorigenesis, and it is these types of changes that help to establish a biochemical foundation for disease progression and malignancy1, 2. A seminal example of this was discovered in the 1920s when Otto Warburg found that cancer cells consume higher levels of glucose and secrete most of the glucose carbon as lactate rather than oxidizing it completely3, 4. Since then, studies by multiple groups have uncovered a diverse array of metabolic changes in cancer, including alterations in glycolytic pathways3, 4, 5, 6, the citric acid cycle7, glutaminolysis8, 9, lipogenesis10, lipolysis11 and proteolysis12. These in turn modulate the levels of cellular building blocks (lipids, nucleic acids and amino acids), cellular energetics, oncogenic signalling molecules and the extracellular environment to confer pro-tumorigenic and malignant properties.
Despite these advances, our current understanding of cancer metabolism is far from complete and would probably benefit from experimental strategies that are capable of profiling enzymatic pathways on a global scale. To this end, conventional genomic13, 14 and proteomic15, 16, 17, 18 methods, which comparatively quantify the expression levels of transcripts and proteins, respectively, have yielded many useful insights. These platforms are, however, limited in their capacity to identify changes in protein activity that are caused by post-translational mechanisms19. Annotating biochemical pathways in cancer is further complicated by the potential for enzymes to carry out distinct metabolic activities in tumour cells that might not be mirrored in normal physiology. In addition, a substantial proportion of the human proteome remains functionally uncharacterized, and it is likely that at least some of these poorly understood proteins also have roles in tumorigenesis. These challenges require new proteomic technologies that can accelerate the assignment of protein function in complex biological systems, such as cancer cells and tumours. In this Review, we discuss one such proteomic platform, termed activity-based protein profiling (ABPP)20, 21, 22 and its implementation in the discovery and functional characterization of deregulated enzymatic pathways in cancer. We discuss the evidence that, when coupled with other large-scale profiling methods, such as metabolomics23, 24 and proteomics15, 16, 17, 18, ABPP can provide a compelling, systems-level understanding of biochemical networks that are important for the development and progression of cancer.
ABPP for enzyme discovery in cancer
ABPP uses active site-directed chemical probes to directly assess the functional state of large numbers of enzymes in native biological samples (Fig. 1). Activity-based probes consist of at least two key elements: a reactive group for binding and covalently labelling the active sites of many members of a given enzyme class (or classes), and a reporter tag for the detection, enrichment and identification of probe-labelled enzymes in proteomes. Activity-based probes can be adapted for in situ or in vivo labelling by substituting the reporter tag with a bio-orthogonal chemical handle, such as an alkyne. Probe-labelled enzymes are then detected by subsequent click chemistry conjugation to various azide-modified reporter tags25, 26. There are currently activity-based probes for a multitude of enzyme classes, including many that have central roles in cancer, such as hydrolases and proteases20, 27, 28, 29, 30, 31, 32, 33, 34, kinases35, 36, 37, 38, phosphatases39, histone deacetylases40, 41, glycosidases42, 43 and various oxidoreductases44, 45. ABPP can be applied to virtually any cell or tissue (assuming that the genome of the parental organism has been sequenced) and can be combined with a range of analytical methods for data acquisition, including gel- and mass spectrometry (MS)-based methods21. Although the specificity of ABPP probes is not absolute, and these probes can be toxic and disrupt biochemical pathways when applied to living systems, they are of great value for characterizing deregulated enzymatic activities in various cancer models and specimens, as discussed below. Examples of activity-based probes that have been used in cancer studies are provided in Table 1.
Figure 1. Activity-based protein profiling.
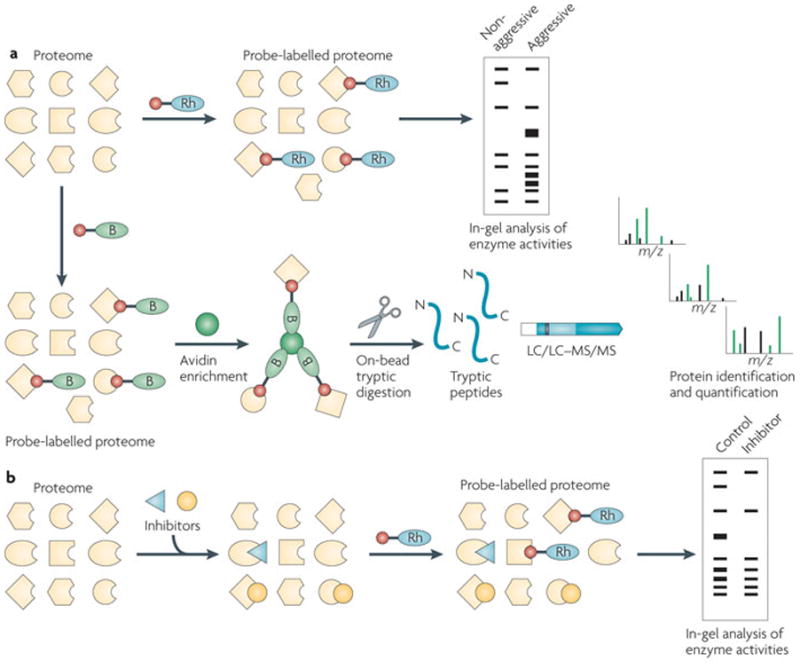
a) Activity-based protein profiling (ABPP) uses active site-directed chemical probes to assess the functional state of large numbers of enzymes in native biological systems. Activity-based probes consist of a reactive group (red ball) for targeting a specific set of enzymes and a detection handle (a fluorophore, such as a rhodamine (Rh) or biotin (B)). In a typical ABPP experiment, a proteome is reacted with the activity-based probe and probe-labelled proteins detected by either in-gel fluorescence scanning (for fluorophore-conjugated probes; top) or avidin enrichment, on-bead tryptic digest and liquid chromatograpy mass spectrometry (LC–MS) analysis (for biotinylated probes; bottom). b) ABPP can also be used in a competitive format to evaluate the potency and selectivity of enzyme inhibitors in native biological samples. Inhibitors compete with activity-based probes for enzyme targets, and this competition is read out by loss of fluorescence (for fluorophore-conjugated probes) or MS (for biotinylated probes) signals (not shown). m/z, mass to charge ratio.
Table 1.
Representative activity-based probes and their application to cancer research
| Structure* | Enzyme class | Applications in cancer |
|---|---|---|
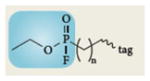 |
Serine hydrolases | Identified increased KIAA1363 (REFS 49,52,53) and MAGL11 activities in aggressive human cancer lines11,52 and primary tumours11,49. Identified increased uPA and tPA serine protease activities in secreted proteomes of aggressive cancer cells51,52. Identified increased RBBP9 activity in pancreatic carcinomas54 |
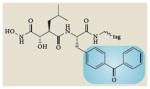 |
Metalloproteinases | Identified increased neprilysin activity in aggressive melanoma cell lines31 |
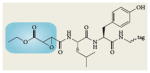 |
Cysteine proteases | Identified increased cathepsin cysteine protease activities in pancreatic islet tumours57. Used for in vivo imaging of tumour cathepsin activity57,60 |
 |
Kinases | Inhibitor selectivity profiling of kinase inhibitors35,71 |
 |
Caspases | In vivo and ex vivo visualization of apoptosis in colon tumour-bearing mice treated with Apomab62 |
 |
Deubiquitylases | Identified increased carboxy-terminal hydrolase UCHL3 and UCH37 activity in HPV cervical carcinomas58 |
 |
Cytochrome P450s | Identified the aromatase inhibitor anastrazole as an inducer of CYP1A2 activity68 |
HPV, human papilloma virus; tPA, tissue plasminogen activator; uPA, urinary plasminogen activator.
Blue boxes around structures represent the portion of the activity-based probes that react with the active sites of enzymes.
Serine hydrolases are one of the largest and most diverse enzyme classes in mammalian proteomes and include esterases, thioesterases, lipases, amidases and proteases46. Several serine hydrolases have been implicated in tumorigenesis, including fatty acid synthase10, protein methyl esterase 1 (Ref. 47), and urokinase-type (uPA) and tissue-type (tPA) plasminogen activators48. Fluorophosphonate probes that target the serine hydrolase superfamily20, 27, 28 (Table 1) have been used to discover several deregulated enzymes in cancer49, 50, 51, 52. Using ABPP, we discovered that two serine hydrolases — the uncharacterized enzyme KIAA1363 (Refs 49, 52, 53) and monoacylglycerol (MAG) lipase (MAGL)11 — are highly expressed in aggressive human cancer cells and primary tumours. We also used ABPP to develop selective inhibitors of KIAA1363 and MAGL for the functional characterization of these enzymes in cancer (discussed below; Fig. 2). Using ABPP, Shields and colleagues54 recently determined that the activity of the serine hydrolase retinoblastoma-binding protein 9 (RBBP9) is increased in pancreatic carcinomas, in which it promotes anchorage-independent growth and pancreatic carcinogenesis through overcoming transforming growth factor-β (TGFβ)-mediated antiproliferative signalling by reducing the phosphorylation levels of SMAD2 and SMAD3.
Figure 2. Serine hydrolases KIAA1363 and MAGL regulate lipid metabolic pathways that support cancer pathogenesis.
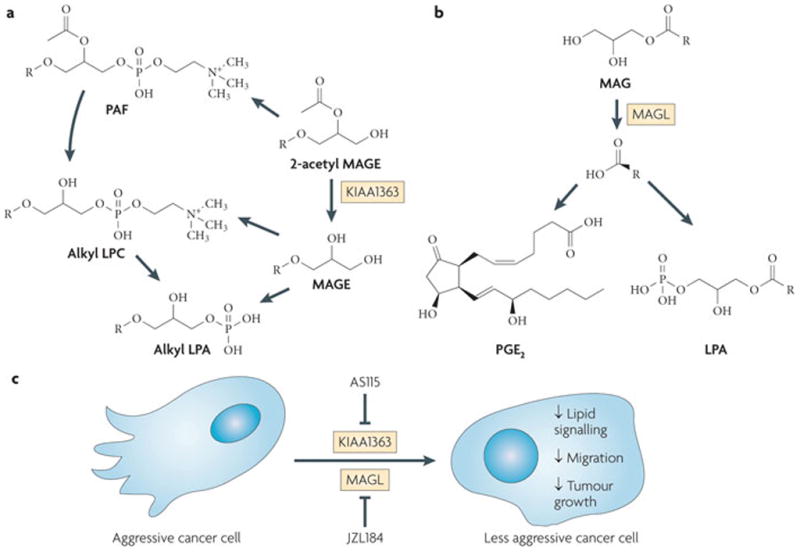
Activity-based protein profiling (ABPP) identified KIAA1363 (part a) and monoacylglycerol (MAG) lipase (MAGL) (part b) as being increased in aggressive human cancer cells from multiple tumour types. Pharmacological and/or RNA interference ablation of KIAA1363 and MAGL coupled with metabolomic analysis revealed specific roles for KIAA1363 and MAGL in cancer metabolism. Disruption of KIAA1363 by the small-molecule inhibitor AS115 lowered monoalkylglycerol ether (MAGE), alkyl lysophosphatidic acid (alkyl LPA) and alkyl lysophosphatidyl choline (alkyl LPC) levels in cancer cells. Disruption of MAGL by the small-molecule inhibitor JZL184 raised MAG levels and reduced free fatty acid, lysophosphatidic acid (LPA) and prostaglandin E2 (PGE2) levels in cancer cells. Disruption of KIAA1363 and MAGL leads to impairments in cancer cell aggressiveness and tumour growth (part c). PAF, platelet-activating factor.
ABPP has also contributed to our knowledge of serine protease activities in cancer. In the course of characterizing an in vivo-derived variant of the human breast cancer line MDA-MB-231, termed 231MFP cells, we determined that these cells possess increased uPA and tPA activity in their secreted proteome51. These cells also show increased tumour growthin vivo, suggesting that deregulated proteolytic activity could contribute to their increased pathogenicity. Madsen and colleagues55 compared serine hydrolase activities in high and low intravasating variants of the human fibrosarcoma HT-1080 cell line and found increased uPA activity in the high-intravasating variant, in which the protease controlled tumour cell intravasation. Interestingly, in these examples, alterations in protease activity occurred without significant changes in mRNA51 or protein55 expression.
Interrogating the activities of other protease families has also provided new insights into deregulated proteolytic processes in cancer. Using epoxide-electrophile probes for cysteine proteases56 (Table 1), Joyce and colleagues57 found that cathepsin activity is higher in angiogenic vasculature and at the invasive fronts of carcinomas, and that pharmacological ablation of a wide range of cathepsins impaired the angiogenic switch, tumour growth, vascularity and invasiveness. They also found that cysteine cathepsins are increased in human papilloma virus (HPV)-induced cervical carcinomas57. Profiling metalloproteinase activities with photoreactive, hydroxamate activity-based probes (Table 1) has uncovered neprilysin as a membrane-associated glycoprotein that has increased activity in aggressive human melanoma lines compared with less-aggressive counterparts31. Comparison of ubiquitin-specific protease activities using the haemagglutinin-tagged ubiquitin-vinyl methyl ester probe (Table 1) revealed that the deubiquitylases ubiquitin-carboxyl esterase-L3 (UCHL3) and UCH37 were upregulated in HPV-positive tumours compared with adjacent normal cervical tissue58. Comparison of a heptaoma cell line that expressed a stably replicating hepatitis C virus subgenomic replicon RNA with the parental cell line using ABPP probes composed of an N-acetylated amino acid that mimicked the P1 position in the peptide substrates of the protease, identified several differentially regulated enzyme activities, some of which were decreased or increased during HCV replication59.
ABPP has also been used for imaging enzyme activities. Bogyo and colleagues60 have introduced quenched near-infrared fluorescent activity-based probes (qNIRF-ABPs) to image cysteine protease activities in tumour xenografts in vivo and ex vivo. These probes emit a fluorescent signal only after covalently modifying a specific protease target, and they can also be used to monitor small-molecule inhibition of protease targets both biochemically and by direct imaging methods. The same researchers have implemented a similar approach using a highly selective aza-peptidyl asparadinyl epoxide qNIRF-ABP61 to target a specific lysosomal cysteine protease, legumain, the expression of which is increased in many human cancers. In another study, mice were treated with dexamethasone, which induced apoptosis and caspase activation in the thymus, both of which were visualized in vivo using a caspase-directed activity-based probe62 (Table 1). This probe could also detect apoptosis that was induced by the monoclonal antibody Apomab in mice bearing xenografted human colorectal tumours62. Further-more, Blum and colleagues63 demonstrated that among contrast agents for protease activities (small peptides, large polymer-based quenched fluorescent substrates and fluorescently labelled ABPP probes), fluorescent ABPP probes showed more rapid and selective uptake into tumours and overall brighter signal compared with substrate-based probes. These approaches can potentially be used in the clinic to define tumour margins, diagnose tumour grade and assess drug-target occupancy in vivo.
ABPP for inhibitor discovery in cancer
Because activity-based probes label the active sites of their enzyme targets, they can form the basis for a competitive screen for enzyme inhibitors27, 64, 65. This competitive ABPP platform has several advantages compared with conventional substrate assays, as inhibitor screens can be conducted directly in complex proteomes and allow concurrent optimization of potency and selectivity against many enzymes in parallel (Fig. 1). Inhibitors can also be developed for uncharacterized enzymes that lack known substrates53, 66. Beyond its important role in inhibitor discovery, competitive ABPP has been used to identify drug targets and off-targets in preclinical or clinical development to gauge mechanism of action and safety31, 37, 67, 68.
Competitive ABPP has served as a principal assay for screening directed libraries of inhibitors and for optimizing their selectivity against serine hydrolases that are expressed in cancer cell and tissue proteomes. This effort has led to the identification of two carbamate agents, AS115 (Ref. 53) and JZL184 (Ref. 69), which are potent and selective inhibitors of KIAA1363 and MAGL, respectively. Competitive ABPP has also been used to explore the full target profile for anti-cancer drugs. Profiling cytochrome P450 enzymes with clickable aryl-alkyne probes (Table 1) showed that the aromatase inhibitor anastrazole, which is approved for breast cancer therapy, significantly increases the activity (as determined by probe labelling) of CYP1A2 and decreases the activity of CYP2C19 (Ref. 68). These results indicate that anastrazole interacts with multiple P450 enzymes and, in at least one case (CYP1A2), might cooperatively enhance the binding of other drugs. Characterization of the matrix metalloproteinase (MMP)-directed inhibitor GM6001 (ilomastat) by competitive ABPP revealed that this agent also inhibits several metalloproteinases outside of the MMP family31, which could explain some of the toxicity issues that broad-spectrum MMP inhibitors have confronted in clinical development70. Kinase-directed activity-based probes (Table 1) have been used to identify 39 kinase targets of the broad-spectrum inhibitor staurosporine in cancer cell lines35. Activity-based probes based on the structure of the PI3K inhibitor wortmannin37, 38, revealed that this natural product also targets members of the polo-like clan of kinases37, 71. The integration of competitive ABPP platforms into the preclinical and clinical development of cancer therapeutics has the potential to clarify the mechanism of action and reduce off-target toxicity for future drug candidates.
Finally, we should note that competitive ABPP experiments have historically been analysed using one-dimensional (1D) SDS–PAGE or MS, which are limited in throughput to compound libraries of a modest size (200–300 compounds). This limitation has recently been addressed, at least in part, by coupling competitive ABPP with fluorescence polarization (fluopol-ABPP)72, which provides a homogeneous assay that is compatible with high-throughput screening and which can be adapted to different classes of enzymes and activity-based probes. Fluopol-ABPP has been successfully used to discover selective inhibitors for two cancer-related enzyme targets, the hydrolytic enzyme RBBP9 and the thioltransferase glutathione S-transferase omega 1 (GSTO1)72, 73.
Integration of ABPP with other profiling methods
Integration of ABPP with metabolomics
Metabolomics has emerged as a powerful method for broadly assessing the biochemical functions of enzymatic pathways in normal physiology and disease23, 24 (Box 1). When complemented with selective inhibitors developed through competitive ABPP, metabolomics can be used to not only identify endogenous substrates and products of enzymes, but also metabolites that are upstream or downstream of these immediate substrates and products, allowing the integration of individual enzymatic reactions into the larger metabolic networks of cancer cells. Two examples of how coupling of ABPP with metabolomics has helped in defining contributions made by enzymes in cancer are discussed below.
Box 1. Metabolomics.
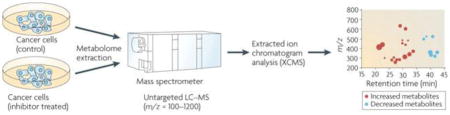
Metabolomics has emerged as a powerful approach for investigating enzyme function in living systems23, 24. Metabolomic experiments in the context of enzyme studies typically start with the extraction of metabolites from control and enzyme-disrupted biological systems, followed by metabolite detection and comparative data analysis. For example, lipophilic metabolites can be enriched from cells or tissues by organic extraction. Mass spectrometry (MS) has become a primary analytical method for surveying metabolites in complex biological samples, with upfront separation accomplished by liquid chromatography (LC MS) or gas chromatography (GC MS). MS experiments can be carried out using targeted92 or untargeted93, 94 approaches, depending on whether the objective is to profile and quantitate known metabolites or to broadly scan for metabolites across a large mass range, respectively. As metabolomic experiments generate a large amount of data, powerful software tools are needed for identification and quantitation of ions in LC MS data sets (see the figure; the mass to charge ratio (m/z) is indicated). One such program is XCMS95, which aligns, quantifies and statistically ranks ions that are altered between two sets of metabolomic data. This program can be used to rapidly identify metabolomic signatures of various disease states or to assess metabolic networks that are regulated by an enzyme using pharmacological or genetic tools that modulate enzyme function. Additional databases assist in metabolite structural characterization, such as HMDB96, 97, METLIN98, 99 and LIPID MAPS100.
A role for KIAA1363 in regulating pro-tumorigenic ether lipids
As mentioned above, ABPP studies identified increased KIAA1363 activity in both aggressive human cancer cell lines53 and primary tumours49, and have identified AS115 as a potent and selective inhibitor of this enzyme53. Untargeted liquid chromatograpy (LC)–MS analysis of lipophilic metabolites from AS115-treated cancer cells revealed that KIA1363 regulates an unusual class of lipids — the monoalkylglycerol ethers (MAGEs)11. Previous studies had shown that tumours contained increased levels of MAGE and other ether lipids and identified positive correlations between ether lipid content and tumorigenicity in cancer cells74, 75. However, the enzymes that regulate ether lipid metabolism in cancer have remained enigmatic. Additional studies showed that KIAA1363 is the principal 2-acetyl MAGE hydrolase in cancer cells, providing one potential pathway by which this enzyme could influence ether lipid content. Importantly, stable knock down of KIAA1363 by RNA interference (RNAi) also led to a reduction in MAGE levels, and this effect was found to further perturb other pro-tumorigenic lipids, including alkyl lysophosphatidic acid (alkyl-LPA)53. These metabolic changes in cancer cells correlated with reductions in migration and in vivo tumour growth, thus pointing to an important role for the KIAA1363–ether lipid pathway in supporting cancer pathogenesis53.
A role for MAGL in regulating fatty acid pathways in cancer cells
Competitive ABPP identified a potent and selective MAGL inhibitor, JZL184 (Ref. 69), which increases MAG levels in multiple tissues in mice without concurrent changes in global free fatty acid levels76. In contrast to this metabolic profile, MAGL inhibition in aggressive melanoma cells, and ovarian and breast cancer cells, not only raised MAGs but also lowered free fatty acid levels — results that were confirmed by RNAi knock down of MAGL11. MAGL reductions also impaired cancer cell migration, invasion and serum-free cell survival in vitro, as well as tumour xenograft growth in vivo11. These effects were rescued by the addition of free fatty acids in vitro or treatment with a high-fat diet in vivo, which supports a pro-tumorigenic role for MAGL-generated fatty acids. Also consistent with this premise, overexpression of wild-type, but not catalytically dead, MAGL was sufficient to increase free fatty acids and confer malignant properties on non-aggressive cancer cells11. Metabolomic profiles revealed that the MAGL fatty acid pathway feeds into a larger lipid network that includes the pro-tumorigenic signalling molecules LPA and prostaglandin E2 (PGE2)11. These results suggest that as tumour development progresses, cancer cells with increased MAGL activity produce more cellular fatty acids, which can serve as building blocks for lipid transmitters that further drive cancer malignancy.
Additional metabolomic studies in cancer
Metabolomics has also been a useful technology for characterizing enzymes that are genetically mutated in cancer. An integrated genomic analysis, consisting of sequencing 22,661 protein-coding genes coupled with high-density oligonucleotide array analysis of 22 human glioblastoma (World Health Organization (WHO) grade IV) samples, identified mutations in the enzyme cytosolic isocitrate dehydrogenase 1 (IDH1) as a common feature of a major subset of primary human brain cancers7. These mutations produce a single amino acid change in the IDH1 active site, resulting in the loss of the ability of the enzyme to convert isocitrate to α-ketoglutarate. Metabolomics revealed that cancer-associated mutations also result in a new catalytic activity — the NADPH-dependent reduction of α-ketoglutarate to R(2)-2-hydroxyglutarate (2-HG)7. Human malignant gliomas with an IDH1 mutation exhibit markedly increased levels of 2HG compared with gliomas without this mutation7. These intriguing findings designate 2-HG as a potential onco-metabolite. Consistent with this hypothesis, an excess accumulation of 2-HG has been shown to lead to an increased risk of brain tumour development in patients with inborn errors of 2-HG metabolism77.
ABPP and proteases
Proteases have long been implicated as drivers of tumorigenesis and are important for early tumour progression, as well as invasion and metastasis78,79, 80, 81. Increased protease activity is also useful as a diagnostic marker for many cancers78, 79, 80, 81. Although proteases catalyse one of the most pervasive post-translational modifications in living systems, most of these enzymes remain incompletely characterized with respect to their endogenous substrates. Whereas technologies such as ABPP are useful for identifying deregulated proteolytic activities, additional methods are required to identify the substrates of proteases. To address this issue, several proteomic approaches have been developed that globally profile protease substrate relationships in native biological systems82, 83, 84, 85, 86 (Fig. 3). One group of proteomic methods, N-terminal labelling techniques, relies on chemically tagging neo-N termini that are produced through proteolytic cleavage events. The chemically tagged N-terminal peptides can then be enriched and analysed using MS methods. Many variations of this strategy have been developed and have been instrumental in identifying exact sites of proteolytic cleavage for various proteases, including caspases84 and MMPs85. Wells and colleagues84 have introduced an engineered enzyme (subtiligase) to tag and enrich nascent N termini in complex protein mixtures. Application of this technique to study apoptosis led to the identification of 292 caspase-cleaved proteins84. Overall and colleagues85 have recently developed the terminal amine isotopic labelling of substrates (TAILS) technique, which uses an amine-reactive polymer to capture internal peptides leaving only labelled neo-N termini and mature blocked N termini for MS analysis. They used this technique to extensively study the substrate profile of MMP2 in mouse fibroblast secretomes and identified 288 potential substrates for this protease85. Van Damme et al.82 developed an N-terminal enrichment strategy, which relies on combined fractional diagonal chromatography (COFRADIC), and so identified 93 caspase-mediated cleavage events during CD95-induced apoptosis. Also of note is the N-CLAP method developed by Jaffrey and colleagues83, which uses a chemical treatment strategy to selectively label N-terminal amines on proteins.
Figure 3. Proteomic strategies for mapping protease substrates.
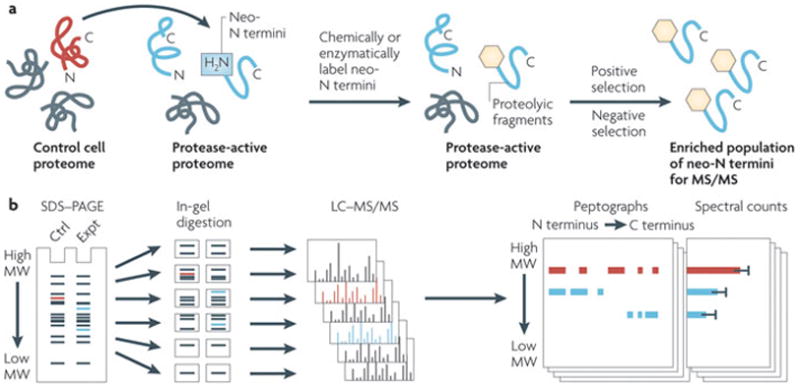
a) Amino terminal labelling techniques use chemical or enzymatic methods to selectively label neo-N termini that are created on protease treatment. The labelled N-terminal peptides can then be enriched through positive selection methods (such as the subtiligase method84) or, alternatively, the internal unmodified peptides can be removed through negative selection methods (such as terminal amine isotopic labelling of substrates (TAILS) methods85). The remaining pool of enriched labelled neo-N termini can then be analysed using tandem mass spectrometry (MS) and the exact sites of proteolytic cleavage can be assigned. b) An alternative proteomic method for protease substrate identification, Protein Topography and Migration Analysis Platform (PROTOMAP86) combines one-dimensional (1D) SDS–PAGE fractionation with liquid chromatograpy (LC)–MS analysis. In a typical PROTOMAP experiment, proteomes from control (Ctrl; red) and experimental (protease-active) (Expt; blue) systems are separated by 1D SDS–PAGE. The lanes are cut into bands at fixed intervals, digested with trypsin and analysed by LC–MS/MS to generate data that are integrated into peptographs, which plot sequence coverage for a given protein in the horizontal dimension (N to C terminus; left to right) versus gel migration in the vertical dimension. Spectral count values for each protein in each gel band provide quantitation. Cleaved proteins are identified by shifts in migration from higher to lower molecular weight (MW) in Ctrl versus Expt systems.
Although N-terminal labelling methods have provided valuable insights into biological systems that are controlled by proteases, they possess some drawbacks. Perhaps most notably, the identification of a cleavage event relies on the detection of a single peptide from the C-terminal portion of a cleaved protein, and therefore does not provide information on the size or stability of the remaining protein fragments. With this consideration in mind, we developed a platform for protease substrate discovery that aims to provide a more complete topological map of proteolytic events occurring in biological systems. This approach, termed the Protein Topography and Migration Analysis Platform (PROTOMAP), consists of the fractionation of active and inactive protease samples by 1D SDS PAGE, followed by LC–MS analysis of tryptically digested proteins from individual gel bands86, 87 (Fig. 3). Identified proteins are subsequently assembled into two-dimensional peptographs that combine sequence coverage with gel migration information, and when coupled with spectral counting data this yields a semi-quantitative topographical map for all detectable proteins (and their cleavage products) in a sample. PROTOMAP has been used to study the intrinsic apoptotic pathway in Jurkat T cells, resulting in the discovery of more than 250 protein cleavage events in apoptotic cells, most of which had not previously been reported in the literature86, 87. Interestingly, a meta-analysis has revealed substantial congruency in the proteolytic events identified by the subtiligase and PROTOMAP methods in apoptotic cells73, indicating that these approaches provide complementary ways to characterize proteolytic pathways in biological systems. We envision that coupling protease–substrate discovery platforms with ABPP should offer a versatile and a potentially routine way to map deregulated proteolytic pathways in cancer and other pathophysiological processes. The information acquired by protease–substrate discovery platforms should also guide the development of new substrate-derived ABPP probes for specific proteases.
Conclusions
Chemical labelling methods have become centrepieces for a wide range of proteomic investigations, whether for measuring the expression88 and the post-translational modification89, 90, 91 of proteins or, as we discuss above, the activity state of proteins. We have attempted to highlight how one such chemoproteomic technology, ABPP, has enabled the discovery of new enzyme activities deregulated in human cancer. Importantly, ABPP also provides a built-in assay that can be used to develop inhibitors to assess the functional role of enzymes in cancer. Activity-based probes have found additional uses as, for example, imaging agents to visualize enzyme activities in tumours. An extension of these studies into a clinical setting could enable rapid imaging, staging and diagnosis of various types of cancers both in biopsy samples and in vivo. Looking forwards, we also anticipate that ABPP might have an important role in demystifying the process of target identification for chemical genomic screens in cancer cells. Lead compounds emerging from phenotypic screens could be transformed into ABPP probes by introducing, for example, photoreactive groups and clickable affinity handles to facilitate the identification of protein targets and sites of probe labelling.
With the advent of fluopol-ABPP, inhibitor discovery and optimization can now be carried out in a high-throughput manner, allowing cancer-relevant enzyme targets to be screened against extensive small-molecule libraries, such as those available as part of the US National Institutes of Health-supported Molecular Libraries Screening initiative. By coupling ABPP with other large-scale profiling methods, such as metabolomics and proteomics, important insights can be gained into how certain enzymes are used or hijacked to carry out biochemical tasks that fuel tumorigenesis. Such enzymes could be important targets for the next generation of cancer therapeutics.
Acknowledgments
This work was supported by the US National Institutes of Health (CA087660 and CA132630), the American Cancer Society (D.K.N.), the California Breast Cancer Research Foundation (M.M.D.), the ARCS Foundation (M.M.D.) and the Skaggs Institute for Chemical Biology.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Deberardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 4.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. A seminal paper describing how cancer cells primarily depend on glycolysis for energy, showing that cancer cells have a fundamentally altered metabolism compared with normal cells. [DOI] [PubMed] [Google Scholar]
- 5.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. This paper shows that the M2 isoform of pyruvate kinase is responsible for the Warburg effect. [DOI] [PubMed] [Google Scholar]
- 6.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 7.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wise DR, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeBerardinis RJ, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. A comprehensive review of the roles of enhanced fatty acid synthase andde novo lipogenesis in cancer. [DOI] [PubMed] [Google Scholar]
- 11.Nomura DK, et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. This paper used ABPP in combination with metabolomics to show that the lipolytic enzyme MAGL regulates pro-tumorigenic fatty acid products in cancer cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nature Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 13.Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nature Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- 14.Golub TR, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 15.Patterson SD, Aebersold RH. Proteomics: the first decade and beyond. Nature Genet. 2003;33:311–323. doi: 10.1038/ng1106. [DOI] [PubMed] [Google Scholar]
- 16.Yates JR. Mass spectral analysis in proteomics. Annu Rev Biophys Biomol Struct. 2004;33:297–316. doi: 10.1146/annurev.biophys.33.111502.082538. [DOI] [PubMed] [Google Scholar]
- 17.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 18.Cravatt BF, Simon GM, Yates JR., 3rd The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 19.Kobe B, Kemp BE. Active site-directed protein regulation. Nature. 1999;402:373–376. doi: 10.1038/46478. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. This paper describes the principles of ABPP and development of probes for the serine hydrolase superfamily. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 22.Fonovic M, Bogyo M. Activity based probes for proteases: applications to biomarker discovery, molecular imaging and drug screening. Curr Pharm Des. 2007;13:253–261. doi: 10.2174/138161207779313623. [DOI] [PubMed] [Google Scholar]
- 23.Vinayavekhin N, Homan EA, Saghatelian A. Exploring disease through metabolomics. ACS Chem Biol. 2010;5:91–103. doi: 10.1021/cb900271r. [DOI] [PubMed] [Google Scholar]
- 24.Saghatelian A, et al. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry. 2004;43:14332–14339. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 25.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Amer Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 26.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 27.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 28.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Direct visualization of serine hydrolase activities in complex proteome using fluorescent active site-directed probes. Proteomics. 2001;1:1067–1071. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 29.Greenbaum D, et al. Chemical approaches for functionally probing the proteome. Mol Cell Proteomics. 2002;1:60–68. doi: 10.1074/mcp.t100003-mcp200. [DOI] [PubMed] [Google Scholar]
- 30.Kato D, et al. Activity-based probes that target diverse cysteine protease families. Nature Chem Biol. 2005;1:33–38. doi: 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- 31.Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Activity-based probes for the proteomic profiling of metalloproteases. Proc Natl Acad Sci USA. 2004;101:10000–10005. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sieber SA, Niessen S, Hoover HS, Cravatt BF. Proteomic profiling of metalloprotease activities with cocktails of active-site probes. Nature Chem Biol. 2006;2:274–281. doi: 10.1038/nchembio781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan EW, Chattopadhaya S, Panicker RC, Huang X, Yao SQ. Developing photoactive affinity probes for proteomic profiling: hydroxamate-based probes for metalloproteases. J Am Chem Soc. 2004;126:14435–14446. doi: 10.1021/ja047044i. [DOI] [PubMed] [Google Scholar]
- 34.Li YM, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 35.Patricelli MP, et al. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry. 2007;46:350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- 36.Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nature Chem Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, et al. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem Biol. 2005;12:99–107. doi: 10.1016/j.chembiol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Yee MC, Fas SC, Stohlmeyer MM, Wandless TJ, Cimprich KA. A cell-permeable activity-based probe for protein and lipid kinases. J Biol Chem. 2005;280:29053–29059. doi: 10.1074/jbc.M504730200. [DOI] [PubMed] [Google Scholar]
- 39.Kumar S, et al. Activity-based probes for protein tyrosine phosphatases. Proc Natl Acad Sci USA. 2004;101:7943–7948. doi: 10.1073/pnas.0402323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salisbury CM, Cravatt BF. Activity-based probes for proteomic profiling of histone deacetylase complexes. Proc Natl Acad Sci USA. 2007;104:1171–1176. doi: 10.1073/pnas.0608659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salisbury CM, Cravatt BF. Optimization of activity-based probes for proteomic profiling of histone deacetylase complexes. J Am Chem Soc. 2008;130:218–2194. doi: 10.1021/ja074138u. [DOI] [PubMed] [Google Scholar]
- 42.Hekmat O, Kim YW, Williams SJ, He S, Withers SG. Active-site peptide —fingerprinting of glycosidases in complex mixtures by mass spectrometry Discovery of a novel retaining beta-1,4-glycanase in Cellulomonas fimi. J Biol Chem. 2005;280:35126–35135. doi: 10.1074/jbc.M508434200. [DOI] [PubMed] [Google Scholar]
- 43.Vocadlo DJ, Bertozzi CR. A strategy for functional proteomic analysis of glycosidase activity from cell lysates. Angew Chem Int Ed Engl. 2004;43:5338–5342. doi: 10.1002/anie.200454235. [DOI] [PubMed] [Google Scholar]
- 44.Adam GC, Cravatt BF, Sorensen EJ. Profiling the specific reactivity of the proteome with non-directed activity-based probes. Chem Biol. 2001;8:81–95. doi: 10.1016/s1074-5521(00)90060-7. [DOI] [PubMed] [Google Scholar]
- 45.Weerapana E, Simon GM, Cravatt BF. Disparate proteome reactivity profiles of carbon electrophiles. Nature Chem Biol. 2008;4:405–407. doi: 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon GM, Cravatt BF. Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J Biol Chem. 2010;285:11051–11055. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puustinen P, et al. PME-1 protects extracellular signal-regulated kinase pathway activity from protein phosphatase 2A-mediated inactivation in human malignant glioma. Cancer Res. 2009;69:2870–7. doi: 10.1158/0008-5472.CAN-08-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57:25–40. doi: 10.1007/s000180050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jessani N, et al. A streamlined platform for high-content functional proteomics of primary human specimens. Nature Methods. 2005;2:691–697. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- 50.Jessani N, Niessen S, Mueller BM, Cravatt BF. Breast cancer cell lines grown in vivo: what goes in isn’t always the same as what comes out. Cell Cycle. 2005;4:253–255. [PubMed] [Google Scholar]
- 51.Jessani N, et al. Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo. Proc Natl Acad Sci USA. 2004;101:13756–13761. doi: 10.1073/pnas.0404727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jessani N, Liu Y, Humphrey M, Cravatt BF. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc Natl Acad Sci USA. 2002;99:10335–10340. doi: 10.1073/pnas.162187599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiang KP, Niessen S, Saghatelian A, Cravatt BF. An enzyme that regulates ether lipid signaling pathways in cancer annotated by multidimensional profiling. Chem Biol. 2006;13:1041–1050. doi: 10.1016/j.chembiol.2006.08.008. This paper uses ABPP coupled with metabolomics to determine that the uncharacterized enzyme KIAA1363 regulates ether lipid metabolism in aggressive cancer cells. [DOI] [PubMed] [Google Scholar]
- 54.Shields DJ, et al. RBBP9: a tumor-associated serine hydrolase activity required for pancreatic neoplasia. Proc Natl Acad Sci USA. 2010;107:2189–2194. doi: 10.1073/pnas.0911646107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madsen MA, Deryugina EI, Niessen S, Cravatt BF, Quigley JP. Activity-based protein profiling implicates urokinase activation as a key step in human fibrosarcoma intravasation. J Biol Chem. 2006;281:15997–16005. doi: 10.1074/jbc.M601223200. [DOI] [PubMed] [Google Scholar]
- 56.Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem Biol. 2000;7:569–581. doi: 10.1016/s1074-5521(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 57.Joyce JA, et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. This paper uses ABPP to identify the cathepsin class of cysteine proteases as important contributors to cancer pathogenesis. [DOI] [PubMed] [Google Scholar]
- 58.Rolen U, et al. Activity profiling of deubiquitinating enzymes in cervical carcinoma biopsies and cell lines. Mol Carcinog. 2006;45:260–269. doi: 10.1002/mc.20177. [DOI] [PubMed] [Google Scholar]
- 59.Blais DR, et al. Activity-based proteome profiling of hepatoma cells during hepatitis C virus replication using protease substrate probes. J Proteome Res. 2009;9:912–923. doi: 10.1021/pr900788a. [DOI] [PubMed] [Google Scholar]
- 60.Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nature Chem Biol. 2007;3:668–677. doi: 10.1038/nchembio.2007.26. This paper describes the use of ABPP probes for near-infrared imaging of protease activities in tumours in living animals. [DOI] [PubMed] [Google Scholar]
- 61.Lee J, Bogyo M. Development of near-infrared fluorophore (NIRF)-labeled activity-based probes for in vivo imaging of legumain. ACS Chem Biol. 2010;5:233–243. doi: 10.1021/cb900232a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edgington LE, et al. Noninvasive optical imaging of apoptosis by caspase-targeted activity-based probes. Nature Med. 2009;15:967–973. doi: 10.1038/nm.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blum G, Weimer RM, Edgington LE, Adams W, Bogyo M. Comparative assessment of substrates and activity based probes as tools for non-invasive optical imaging of cysteine protease activity. PLoS One. 2009;4:e6374. doi: 10.1371/journal.pone.0006374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greenbaum DC, et al. Small molecule affinity fingerprinting. A tool for enzyme family subclassification, target identification, and inhibitor design. Chem Biol. 2002;9:1085–1094. doi: 10.1016/s1074-5521(02)00238-7. [DOI] [PubMed] [Google Scholar]
- 65.Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nature Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 66.Li W, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J Am Chem Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- 67.Wright AT, Cravatt BF. Chemical proteomic probes for profiling cytochrome p450 activities and drug interactions in vivo. Chem Biol. 2007;14:1043–1051. doi: 10.1016/j.chembiol.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wright AT, Song JD, Cravatt BF. A suite of activity-based probes for human cytochrome P450 enzymes. J Am Chem Soc. 2009;131:10692–10700. doi: 10.1021/ja9037609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Long JZ, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nature Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 71.Liu Y, Jiang N, Wu J, Dai W, Rosenblum JS. Polo-like kinases inhibited by wortmannin. Labeling site and downstream effects. J Biol Chem. 2007;282:2505–2511. doi: 10.1074/jbc.M609603200. [DOI] [PubMed] [Google Scholar]
- 72.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nature Biotechnol. 2009;27:387–394. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bachovchin DA, et al. Oxime esters as selective, covalent inhibitors of the serine hydrolase retinoblastoma-binding protein 9 (RBBP9) Bioorg Med Chem Lett. 2010;20:2254–2258. doi: 10.1016/j.bmcl.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roos DS, Choppin PW. Tumorigenicity of cell lines with altered lipid composition. Proc Natl Acad Sci USA. 1984;81:7622–7626. doi: 10.1073/pnas.81.23.7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Snyder F, Wood R. Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues. Cancer Res. 1969;29:251–257. [PubMed] [Google Scholar]
- 76.Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Schaftingen E, Rzem R, Veiga-da-Cunha M. L -2-hydroxyglutaric aciduria, a disorder of metabolite repair. J Inherit Metab Dis. 2009;32:135–142. doi: 10.1007/s10545-008-1042-3. [DOI] [PubMed] [Google Scholar]
- 78.Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–135. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- 79.Janicke F, et al. Urokinase (uPA) and its inhibitor PAI-1 are strong and independent prognostic factors in node-negative breast cancer. Breast Cancer Res Treat. 1993;24:195–208. doi: 10.1007/BF01833260. [DOI] [PubMed] [Google Scholar]
- 80.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tandon AK, Clark GM, Chamness GC, Chirgwin JM, McGuire WL. Cathepsin D and prognosis in breast cancer. N Engl J Med. 1990;322:297–302. doi: 10.1056/NEJM199002013220504. [DOI] [PubMed] [Google Scholar]
- 82.Van Damme P, et al. Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nature Methods. 2005;2:771–777. doi: 10.1038/nmeth792. [DOI] [PubMed] [Google Scholar]
- 83.Xu G, Shin SB, Jaffrey SR. Global profiling of protease cleavage sites by chemoselective labeling of protein N-termini. Proc Natl Acad Sci USA. 2009;106:19310–19315. doi: 10.1073/pnas.0908958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mahrus S, et al. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell. 2008;134:866–876. doi: 10.1016/j.cell.2008.08.012. This paper describes an advanced N-terminal labelling method to map the precise sites of proteolytic cleavage in complex cellular systems. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleifeld O, et al. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nature Biotechnol. 28:281–288. doi: 10.1038/nbt.1611. [DOI] [PubMed] [Google Scholar]
- 86.Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038. This paper uses an integrated gel and MS platform to map proteolytic cleavage events and their effect on protein levels and structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Simon GM, Dix MM, Cravatt BF. Comparative assessment of large-scale proteomic studies of apoptotic proteolysis. ACS Chem Biol. 2009;4:401–408. doi: 10.1021/cb900082q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 89.Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proc Natl Acad Sci USA. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martin BR, Cravatt BF. Large-scale profiling of protein palmitoylation in mammalian cells. Nature Methods. 2009;6:135–138. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hang HC, et al. Chemical probes for the rapid detection of Fatty-acylated proteins in Mammalian cells. J Am Chem Soc. 2007;129:2744–2745. doi: 10.1021/ja0685001. [DOI] [PubMed] [Google Scholar]
- 92.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saghatelian A, Cravatt BF. Global strategies to integrate the proteome and metabolome. Curr Opin Chem Biol. 2005;9:62–68. doi: 10.1016/j.cbpa.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 94.Vinayavekhin N, Saghatelian A. Untargeted metabolomics. Curr Protoc Mol Biol. 2010;30:1–24. doi: 10.1002/0471142727.mb3001s90. [DOI] [PubMed] [Google Scholar]
- 95.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 96.Wishart DS, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37:D603–D610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wishart DS, et al. HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007;35:D521–D526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sana TR, Roark JC, Li X, Waddell K, Fischer SM. Molecular formula and METLIN Personal Metabolite Database matching applied to the identification of compounds generated by LC/TOF-MS. J Biomol Tech. 2008;19:258–266. [PMC free article] [PubMed] [Google Scholar]
- 99.Smith CA, et al. METLIN: a metabolite mass spectral database. Ther Drug Monit. 2005;27:74–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 100.Schmelzer K, Fahy E, Subramaniam S, Dennis EA. The lipid maps initiative in lipidomics. Methods Enzymol. 2007;432:171–183. doi: 10.1016/S0076-6879(07)32007-7. [DOI] [PubMed] [Google Scholar]