

Sir, Garden et al. (2009) reported an important experimental study highlighting a potential mechanism for neuronal dysfunction distant from the site of damage, specifically a loss of synaptic plasticity in the retrosplenial/posterior cingulate cortex (PCC) after anterior thalamic lesion in the rat. In the discussion section of their article, they make the assumption that this phenomenon plays a role in the early episodic memory impairment characterizing Alzheimer’s disease: the PCC would be disconnected from the anterior thalamic nucleus - affected by early neuronal/synaptic loss - through disruption of the cingulum bundle. This would in turn lead to PCC hypometabolism, which occurs very early in Alzheimer’s disease and already at the stage of amnestic mild cognitive impairment (Minoshima et al., 1997, Chételat et al., 2003a, b). The study by Garden et al. (2009) is therefore important for the understanding of the pathophysiology of the memory impairment that characterizes early Alzheimer’s disease as the proposed underlying synaptic mechanism could be amenable to specific pharmacological modulation.
Garden et al. (2009) also allude to the current debate about the relative importance of disconnection versus local atrophy/direct neuronal damage in the PCC hypometabolism observed in amnestic mild cognitive impairment and early Alzheimer’s disease. They write: “The notion that the retrosplenial/posterior cingulate hypometabolism in Alzheimer’s disease is solely due to a disconnection from sites thought to atrophy prior to the retrosplenial cortex, such as the hippocampus or anterior thalamus (Chételat et al., 2008; Villain et al., 2008) is, however, probably incorrect.” We thank them for citing our work, but feel they have somewhat misquoted us by associating us to the term “solely”. As a matter of fact, we fully agree with their statement that disconnection is not the only mechanism for PCC hypometabolism in Alzheimer’s disease, and have consistently stated so in our publications (Chételat et al., 2008; Villain et al., 2008). First of all, early studies that assessed the effect of correction of the 2-fluoro-2-deoxy-D-glucose positron emission tomography (18)fluoro-deoxyglucose positron emission tomography (FDG-PET) data for partial volume effects demonstrated that atrophy significantly contributes to the PCC hypometabolism observed in raw FDG-PET images in Alzheimer’s disease. For instance, Ibanez et al. (1998) found that following partial volume effect correction, PCC hypometabolism, though still present, had notably reduced. Using voxel-based morphometry, we were among the first to provide in vivo evidence of significant grey matter volume loss in the PCC in both Alzheimer’s disease (Baron et al., 2001) and amnestic mild cognitive (Chételat et al., 2002), providing a straightforward explanation for the above findings. Thus, there can be no doubt that PCC hypometabolism in raw FDG-PET images partly reflects grey matter volume loss. However, we also showed, by directly comparing the degree of grey matter atrophy and partial volume effect-corrected hypometabolism in early Alzheimer’s disease, that a significant fraction of PCC hypometabolism is due to mechanisms other than local atrophy, and speculated that this could represent, at least in part, disconnection effects from the hippocampal region via the cingulum bundle (Chételat et al., 2008). In a subsequent study, we then provided strong support to this hypothesis by combining voxel-based morphometry and FDG-PET, demonstrating a significant correlation between cingulum bundle disruption and PCC hypometabolism (Villain et al., 2008). Thus, disconnection also plays a part in the observed PCC hypometabolism.
Additional intriguing issues, somewhat tricky to address, are (i) does PCC hypometabolism remain related to local atrophy even after correcting for partial volume effects, as a reflection of local burden of neuronal damage? and (ii) do additional mechanisms also contribute to PCC hypometabolism above and beyond actual volume loss and disconnection effects? To address these, we have reanalysed our data from Villain et al. (2008) aiming to assess the independent contribution of local atrophy and disconnection to the residual PCC hypometabolism after correction for partial volume effects, and the remaining part of PCC hypometabolism not explained by these two factors. Using the mean individual metabolic and grey matter Z-scores in the clusters of interest, we found partial volume effect-corrected PCC hypometabolism to be significantly related to local atrophy (Pearson r2=0.42; P=0.003) as well as cingulum bundle volume (Pearson r2=0.7; P=10−5; Fig. 1, blue slopes). We then included both the cingulum bundle white matter Z-scores and the PCC grey matter Z-scores as independent variables in a multiple regression analysis with PCC hypometabolism as the dependent variable. The model was found to be highly significant as a whole (multiple r2=0.70; P=10−5); the cingulum bundle alone was found to provide a significant independent contribution to this model (semi-partial r2=0.28; P=10−3), but PCC atrophy alone did not independently contribute to the model (semi-partial r2<0.01; p=0.68; see Fig. 1, orange slopes). Thus, overall these data suggest that local atrophy does not independently contribute to PCC hypometabolism after partial volume effect correction, i.e. FDG uptake per gram of brain tissue is not influenced by factors conducting to, associated with or downstream of, local atrophy, while disconnection explains a large fraction (here 70%) of this measurement. Beyond methodological factors, the remaining unexplained 30% of total variance probably reflects other non-atrophy-related functional alterations, e.g. amyloid deposition, and/or disconnection from other bundles. We are currently analysing the data from a longitudinal study which will hopefully help in elucidating the temporal sequence of events and the causal relationships between these.
Figure 1. Contribution of local atrophy and cingulum bundle disruption to PVE-corrected PCC hypometabolism.
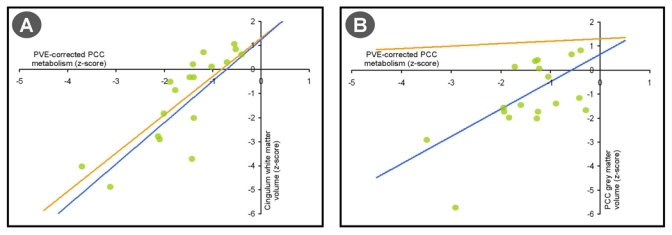
PCC hypometabolism is significantly correlated to both Cingulum white matter atrophy (A) and PCC Grey Matter atrophy (B) when assessed separately (blue slopes). However, only Cingulum bundle disruption remained significantly related to PVE-corrected PCC hypometabolism when controlling for PCC atrophy (A, orange slopes), whereas PCC atrophy did not independently contribute to PCC hypometabolism when Cingulum white matter atrophy was controlled (B, orange).
References
- Baron JC, Chételat G, Desgranges B, Perchey G, Landeau B, de la Sayette V, et al. In vivo mapping of gray matter loss with voxel-based morphometry in mild Alzheimer’s Disease. NeuroImage. 2001;14:298–308. doi: 10.1006/nimg.2001.0848. [DOI] [PubMed] [Google Scholar]
- Chételat G, Desgranges B, de la Sayette V, Viader F, Eustache F, Baron JC. Mapping gray matter loss with voxel-based morphometry in mild cognitive impairment. NeuroReport. 2002;13:1939–43. doi: 10.1097/00001756-200210280-00022. [DOI] [PubMed] [Google Scholar]
- Chételat G, Desgranges B, de la Sayette V, Viader F, Berkouk K, Landeau B, et al. Dissociating atrophy and hypometabolism impact on memory in mild cognitive impairment. Brain. 2003a;126:1955–67. doi: 10.1093/brain/awg196. [DOI] [PubMed] [Google Scholar]
- Chételat G, Desgranges B, de la Sayette V, Viader F, Eustache F, Baron JC. Mild cognitive impairment: FDG-PET predicts who is to rapidly convert to Alzheimer's disease. Neurology. 2003b;60:1374–77. doi: 10.1212/01.wnl.0000055847.17752.e6. [DOI] [PubMed] [Google Scholar]
- Chételat G, Desgranges B, Landeau B, Mézenge F, Poline JB, de la Sayette V, et al. Direct voxel-based comparison between grey matter hypometabolism and atrophy in Alzheimer’s disease. Brain. 2008;131:60–71. doi: 10.1093/brain/awm288. [DOI] [PubMed] [Google Scholar]
- Garden DL, Massey PV, Caruana DA, Johnson B, Warburton EC, Aggleton JP, et al. Anterior thalamic lesions stop synaptic plasticity in retrosplenial cortex slices: expanding the pathology of diencephalic amnesia. Brain. 2009;132:1847–57. doi: 10.1093/brain/awp090. [DOI] [PubMed] [Google Scholar]
- Ibáñez V, Pietrini P, Alexander GE, Furey ML, Teichberg D, Rajapakse JC, et al. Regional glucose metabolic abnormalities are not the result of atrophy in Alzheimer's disease. Neurology. 1998;50:1585–93. doi: 10.1212/wnl.50.6.1585. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol. 1997;42:85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- Villain N, Desgranges B, Viader F, de la Sayette V, Mézenge F, Landeau B, et al. Relationships between hippocampal atrophy, white matter disruption and gray matter hypometabolism in Alzheimer's disease. J Neurosci. 2008;28:6174–81. doi: 10.1523/JNEUROSCI.1392-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]