

Abstract
Purpose
To identify the underlying genetic defect in four generations of a Chinese family affected with bilateral congenital posterior subcapsular cataracts.
Methods
Clinical data from patients in the family were recorded by slit-lamp photography. Genomic DNA samples were extracted from peripheral blood of the pedigree members. Mutation screening was performed in the candidate gene by bidirectional sequencing of the amplified products. The mutation was verified by restriction fragment length polymorphism (RFLP) analysis.
Results
The congenital cataract phenotype of the pedigree was identified as posterior subcapsular by slit-lamp photography. Sequencing of the candidate genes detected a heterozygous c.5C→T change in the coding region of the βB2-crystallin gene (CRYBB2), resulting in the substitution of a highly conserved alanine to valine (p. A2V). All nine family members affected with cataracts were positive for this change, but it was not observed in any of the unaffected members of the family. The transition resulted in the loss of a HaeIII restriction site in the affected members of the pedigree, which was present in the unaffected family members and in all of the 100 unrelated individuals tested.
Conclusions
This study has identified a novel CRYBB2 gene mutation, resulting in the amino substitution p. A2V in a Chinese family with posterior subcapsular congenital cataracts. This mutation is probably the causative lesion for the observed phenotype in this family.
Introduction
Congenital cataracts, the loss of eye lens transparency, are a leading cause of visual impairment or blindness in childhood. Depending on regional socioeconomic development, its prevalence varies from 1−6 cases per 10,000 live births in industrialized countries [1,2] to 5−15 per 10,000 in the poorest areas of the world [3,4]. Globally, congenital cataracts account for approximately one-tenth of childhood blindness due to different causes including infections during embryogenesis, metabolic disorders (galactosemia), and gene defects [5]. As inherited cataracts correspond to 8−25% of congenital cataracts [6], genetic mutation is likely the most common cause. Although autosomal recessive and X-linked forms of inheritance also exist, autosomal dominance is the major form of inheritance of congenital cataracts [7]. Autosomal dominant congenital cataracts (ADCC) have been reported to be caused by mutations in 24 different genes to date: approximately half of the mutations are in the crystallin genes and a quarter in connexins genes, with the remainder divided among the genes for heat shock transcription factor-4 (HSF4), aquaporin-0 (AQP0, MIP), paired-like homeodomain 3 (PITX3), v-maf musculoaponeurotic fibrosarcoma oncogene homolog (MAF), chromatin modifying protein (CHMP4B), lens intrinsic membrane protein 2 (LIM2), beaded filament structural protein-2 (BFSP2) [5,8], the forkhead box protein E3 (FOXE3) [9], Drosophila eyes absent gene 1 (EYA1) [10], intermediate filament protein vimentin (VIM) [11], and carboxylic acid transporter family SLC16A12 [12].
According to the location of the lens opacities, and a detailed description of the shape and appearance, a comprehensive approach is to classify the complex spectrum of morphological variations of congenital cataracts. The cataract phenotype can be divided into the following categories: total, nuclear, cortical, anterior subcapsular, posterior subcapsular, lamellar, cerulean, pulverulent, sutural, coralliform, wedge-shaped, and polymorphic cataracts [5,13]. Mutations in different genes may be associated with similar phenotypes due to genetic heterogeneity [14]. To date, congenital posterior subcapsular cataracts have been related to these genes: CRYAB [15,16], CRYBA1 [17], GJA8, receptor tyrosine kinase gene (EPHA2) [18], PITX3 [19], and CHMP4B [20].
In this paper, a four-generation family affected with congenital posterior subcapsular cataracts was investigated in an attempt to identify the genetic defect associated with their cataract phenotype.
Methods
Clinical evaluation and DNA specimens
The four generation of the family suffering with ADCC were recruited from the Eye Center of Affiliated Second Hospital, College of Medicine, Zhejiang University, Hangzhou, China. Informed consent in accordance with the Zhejiang Institutional Review Board was obtained from all participants and the study protocol adhered to the tenets of the Declaration of Helsinki. In total, 27 individuals participated: nine affected and 18 unaffected. Of the 18, nine were spouses (Figure 1A). Also, 100 unrelated control subjects were recruited. Detailed medical histories were obtained by interviewing all individuals. All participants underwent ophthalmologic examinations, including visual acuity and slit-lamp examination with dilated pupils. Five of the affected members had undergone cataract extraction surgery. Phenotypes were documented by slit-lamp photography (Figure 1B-D). Blood samples were obtained by venipuncture, collected in Vacutainer tubes (Becton-Dickinson, Franklin Lakes, NJ) containing EDTA. Leukocyte genomic DNA was extracted using the QIAmp Blood kit (Qiagen, Duesseldorf, Germany).
Figure 1.
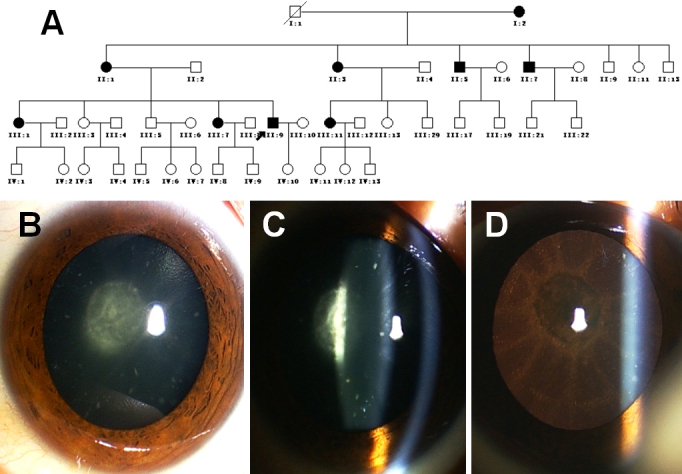
Clinical features of the family. A: Pedigree of the autosomal dominant congenital cataract. The proband is marked with an arrow. Squares and circles indicate males and females respectively. Black and white symbols represent affected and unaffected individuals, respectively. B, C, and D: Slit-lamp photograph of the proband (III:9) shows a posterior subcapsular cataract.
Mutation analysis
Genomic DNA samples from affected and unaffected members of the family were screened for mutations in CRYAA, CRYAB, CRYBA1, CRYBB2, CRYGC, CRYGD, GJA3, GJA8, PITX3, and CHMP4B by a combination of direct sequencing and RFLP analysis. The coding regions of candidate genes were amplified by polymerase chain reaction (PCR) using previously published primer sequences (Table 1) [9,20-26]. The cycling conditions for PCR were 95 °C preactivation for 5 min, 10 cycles of touchdown PCR with 0.5 °C down per cycle from 60 °C to 55 °C, followed by 30 cycles with denaturation at 95 °C for 25 s, annealing at 55 °C for 25 s, and extension at 72 °C for 40 s. PCR products were isolated by electrophoresis on 3% agarose gels and sequenced using the BigDye Terminator Cycle sequencing kit V 3.1 (ABI Applied Biosystems; Sangon Co, China) on an ABI PRISM 3730 Sequence Analyzer (ABI), according to the manufacturer’s instructions. Sequencing results were analyzed using Chromas 1.62 and compared with sequences from NCBI GenBank (CRYAA: 21q22.3; NM_000394, CRYAB: 11q22; NG_009824, CRYBA1: 17q11-q12; NM_005208, CRYBB2: 22q11.2; NM_000496, CRYGC: 2q33-q35; NM_020989, CRYGD: 2q33-q35; NM_006891.3, GJA3: 13q11-q13; NM_021954, GJA8: 1q21-q25; NM_005267, PITX3: 10q25; NM_005029, and CHMP4B: 20q11.22; NM_176812).
Table 1. Polymerase chain reaction primers and product sizes of human CRYBB2 genes.
| Name | Primer sequence (5′-3′) | Tm (°C) | Product size (bp) |
|---|---|---|---|
| CRYBB22F |
5′-CCAGGTCCTCACTGCTGCTTC-3′ |
60.34 |
205 |
| CRYBB22R |
5′-CCCATTTTACAGAAGGGCAAC-3′ |
58.01 |
|
| CRYBB23F |
5′-ACCCTTCAGCATCCTTTGG-3′ |
57.56 |
314 |
| CRYBB23R |
5′-GCAGACAGGAGCAAGGGTAG-3′ |
61.9 |
|
| CRYBB24F |
5′-GCTTGGAGTGGAACTGACCTG-3′ |
61.92 |
244 |
| CRYBB24R |
5′-GGCAGAGAGAAAGTAGGATGATG-3′ |
61.98 |
|
| CRYBB25F |
5′-GCCCCCTCACCCATACTC-3′ |
61.86 |
242 |
| CRYBB25R |
5′-CCCCAGAGTCTCAGTTTCCTG-3′ |
61.92 |
|
| CRYBB26F |
5′CCTAGTGGCTTATGGATGCTC-3′ |
59.97 |
347 |
| CRYBB26R | 5′-TCTTCACTTGGAGGTCTGGAG-3′ | 59.97 |
Restriction fragment length polymorphism analysis
Genetic variations were confirmed by the absence of cleavage sites for the restriction enzyme HaeIII in affected family members. For controls, we analyzed 100 DNA samples from ophthalmologically normal individuals of the same ethnic background as the family members. PCR products of exon 2 of the CRYBB2 gene were digested for 10 h at 37 °C with HaeIII (TAKARA, Dalian, China), then electrophoresized in 5% polyacrylamide gels and analyzed under ultraviolet (UV) light.
Computational algorithms
Computational methods have been shown to be effective in predicting whether a specific amino acid substitution of a protein sequence is deleterious or neutral to the function of the protein. Three methods were used: Sorting Intolerant from Tolerant amino acid substitutions (SIFT), Polymorphism Phenotyping (PolyPhen), and Grantham score difference (Align-GVGD).
SIFT uses sequence homology to predict whether an amino acid substitution will affect protein function and, thus, potentially contribute to a disease. It is based on the approach that important amino acids tend to be highly conserved across species. SIFT, which assigns scores from 0 to 1, predicts substitutions with scores less than 0.05 as deleterious, whereas those greater than or equal to 0.05 are considered to be tolerated [27]. PolyPhen takes into account the evolutionary conservation of the amino acid subjected to the mutation and the physicochemical characteristics of the wild-type and mutated amino acid residue and the consequence of the amino acid change for the structural properties of the protein [6]. Grantham Variation (GV) measures the degree of biochemical variation among amino acids found at a given position in the multiple sequence alignment: Grantham Deviation (GD) reflects the 'biochemical distance' of the mutant amino acid from the observed amino acid at a particular position (given by GV). Align-GVGD can be used to predict the transactivation activity of each missense substitution [28]. A value of GV=0 corresponds to a residue that is invariant in the alignment, a value of GV of 60–65 is the upper limit of conservative variation across species, and a value of GV>100 is indicative of positions that are under little functional constraint. A value of GD=0 corresponds to a missense substitution that is within the cross-species range of variation at its position in the protein; at invariant positions (GV=0); GD=60–65 is the upper limit of a conservative missense substitution [29]. Additionally, for hydropathy analysis we used Kyte-Doolittle hydropathy plots. All hydropathies for both wild-type and mutants were calculated in a default window size of 7.
Results
Clinical evaluation
The cataract exhibited an autosomal dominant inheritance pattern in the family under investigation (Figure 1A). Opacification of the lens was bilateral in all affected individuals. There was no family history of other ocular or systemic abnormalities. Five of the nine patients had undergone lens surgery. Slit-lamp examination of the eyes in the remaining patients without cataract removal showed opacity in the posterior subcapsular region with additional dot-like opacification in its cortex of bilateral lenses (Figure 1B-D). Visual acuity in the unoperated eyes of those affected individuals ranged from 0.6 to 0.02. Most affected individuals noticed their visual impairments in their twenties, and then their visual acuity decreased gradually until surgery was required to improve their visual function after the age of 40.
Mutation screening
Bidirectional sequencing of the coding regions of the candidate genes showed a heterozygous change, C>T (Figure 2), at position 5 (c.5C>T) of the CRYBB2 gene in all nine affected individuals, leading to the replacement of a highly conserved alanine with valine at the second amino acid position (p. A2V). This substitution was not seen in the unaffected individuals or in the 100 unrelated control subjects (200 chromosomes) from the same Chinese population (data not shown).
Figure 2.
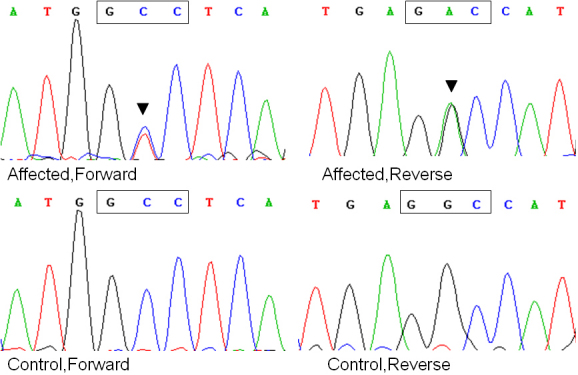
Forward and reverse sequence analysis of the affected and unaffected individuals in the ADCC Chinese family, showing a heterozygous mutation (c.5C>T) in exon 2 of CRYBB2 (black triangles).
Restriction fragment length polymorphism analysis
The mutation was confirmed by a HaeIII digest of the PCR-amplified exon 2 of the CRYBB2 gene. This mutation resulted in the loss a HaeIII restriction site in all the affected members of the Chinese family under investigation, but was not detected in the 100 unrelated normal controls or unaffected pedigree members (Figure 3A).
Figure 3.
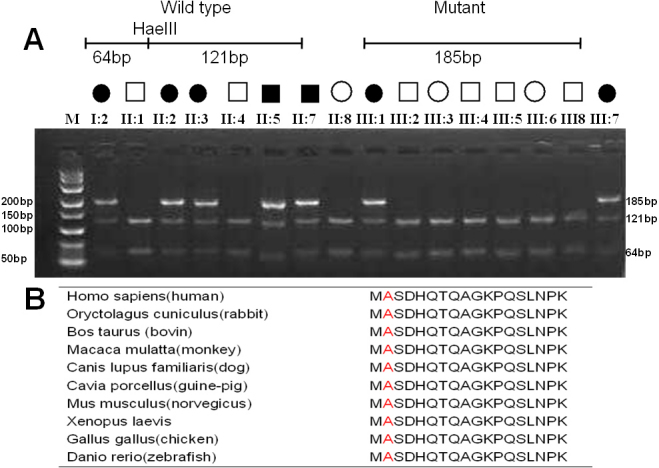
The CRYBB2 mutation cosegregates with the disease in the family. A: Restriction fragment length analysis (RFLP) showing the loss of the HaeIII restriction site in heterozygous individuals with the A2V mutation (185 bp) but it was present in unaffected individuals (121 bp and 64 bp). B: Multiple-sequence alignment in CRYBB2 from different species reveals that codon 2, where the mutation (p. A2V) occurred, is highly conserved (highlighted in red).
Multiple-sequence alignment and mutation analysis
Using the NCBI websites, a multiple sequence alignment showed that the alanine at position 2 of human the CRYBB2 protein (Homo sapiens, NP_000487.1) is highly conserved in various species including Oryctolagus cuniculus (NP_001082786.1), Bos taurus (NP_777232.1), Macaca mulatta (NP_001116366.1), Canis lupus familiaris (NP_001041578.1), Cavia porcellus (NP_001166542.1), Mus musculus (NP_031799.1), Xenopus laevis (NP_001086491.1), Gallus gallus (NP_990506.2), and Danio rerio (NP_001018138.1; Figure 3B).
Computational analysis
Computational protein analysis of A2V CRYBB2 revealed the following results: the SIFT method revealed a score of 0.00, meaning the substitution is intolerant. PolyPhen analysis produced a score of 1.603, which is predicted to be “probably damaging.” Finally, Align-GVGD showed a score of GV0.00, GD65.28, which belongs to class C65 and means “most likely to interfere with function.” All of these results indicated the A2V substitution was likely deleterious and possibly contributed to the disease. The Kyte-Doolittle algorithm for hydrophobicity analysis showed the local hydrophobicity at and near the altered amino acid was increased (Figure 4).
Figure 4.
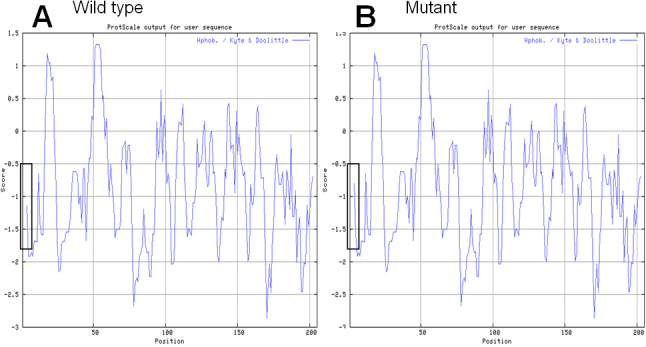
Hydropathy analysis of the mutant protein. Kyte-Doolittle hydropathy plot of CRYBB2WT (A) and CRYBB2A2V (B). The x-axis represents position of amino acids. The y-axis represents the hydropathy value in a default window size of 7. The region of interest is marked by white boxes. The increase in hydrophobicity in the mutant form is evident.
Discussion
Crystallins are known to constitute about 90% of the water-soluble proteins of the lens and contribute to transparency and refractive properties, due to a uniform concentration gradient in the lens. The vertebrate crystallins are divided into two families: α-crystallins and the β- and γ-crystallin families [30,31]. The β- and γ-crystallins share a common feature of anti-parallel β sheets in the proteins, referred to as the “Greek key motif.” The Greek key motif are agreed to be among the most stable of structures in proteins. Computer-based analyses suggest that they form an interdomain association: intramolecular in the γ-crystallins, and intermolecular in the β-crystallins. The detailed analysis of the mutations in the β- and γ-crystallin encoding genes might help to identify those amino acids which are the important corner stones for this function [32]. A second functional aspect of the Greek key motif is its Ca2+ binding properties. A human lens model of cortical cataract had been developed by Sanderson and his coworkers to study the role of Ca2+ in cataractogenesis [33].Recently, Fischer et al. [34] reported on the effects of β- and γ-crystallins in axon regeneration of retinal ganglion cells.
The β-crystallin gene consists of six exons; the first exon is not translated, the second exon encodes the NH2-terminal extension, and the subsequent four exons are responsible for one Greek key motif each [35]. Mutations in the CRYBB2 gene in humans and mice have been reported to induce genetic cataracts [8,23,36-49] (Table 2). Previously, nine geographically distinct families have been reported with the same nonsense mutation (Q155X) in exon 6 of the CRYBB2 gene, which is associated with diverse phenotypes. This mutation creates a premature stop codon (Q155X) and results in an in-frame stop codon at nucleotide 475 of exon 6 that may cause a truncation of 50 amino acids from the COOH-terminus of βB2-crystallin, which destabilizes the domain structure of betaB2-crystallin. Using a mammalian two-hybrid system assay, spectroscopy (circular dichroism and fluorescence) and fast performance liquid chromatography (FPLC), the Q155X mutant shows not only decreased ordered structure and stability, but also a decrease in protein–protein interactions with βB2-crystallin mutant, which might contribute to cataract formation [50]. This mutation is explained by a gene conversion mechanism between the CRYBB2 gene and its pseudogene CRYBB2P1. The diversity of the phenotypes may be caused by variations in the promoter region, possibly influencing the expression of the CRYBB2 protein in the lens or other crystallin genes as modifiers from surrounding loci [37,39]. The other mutations were found in exons 3, 5, and 6. A2V in the CRYBB2 gene characterized here is the first reported mutation in exon 2 (NH2-terminal extension) of the CRYBB2 gene.
Table 2. Previous CRYBB2 gene mutations associated with congenital cataracts.
| Bp exchange | Aa exchange | Biologic consequence | Origin of family | Reference |
|---|---|---|---|---|
|
Human mutations | ||||
| c. G607A |
p. V187M |
Nuclear cataract |
Basotho |
[36] |
| c. C475T |
p. Q155X |
Sutural opacity and fish tail-like branches |
American |
[37] |
| c. C475T |
p. Q155X |
Cerulean |
American |
[8] |
| c. C475T |
p. Q155X |
ADCC |
Canadian |
[38] |
| c. C475T |
p. Q155X |
ADCC |
Chilean |
[39] |
| c. C475T |
p. Q155X |
Progressive polymorphic coronary ADCC |
Indian |
[40] |
| c. C92T |
p. S31W |
Coronary cataract |
Chinese |
[41] |
| c. C475T |
p. Q155X |
Cerulean ADCC |
Chinese |
[42] |
| c. C475T |
p. Q155X |
Progressive polymorphic |
Chinese |
[43] |
| c. G465T |
p. W151C |
Central nuclear |
Indian |
[44] |
| c. A383T |
p. D128V |
Dominant ring-shaped cortical cataract |
Germany |
[45] |
| c.C489A |
p. Y159X |
ADCC |
Danish |
[46] |
| c.C433T |
p. R145W |
ADCC |
Danish |
[46] |
| c.A440G |
p. Q147R |
ADCC |
Danish |
[46] |
| c.C449T |
p. T150M |
ADCC |
Danish |
[46] |
|
Mouse mutations | ||||
| Intron 5:_57 A->T |
Splicing: 19 new amino acids in front of exon 6 |
Progressive cataract |
[47] |
|
| c. T560A |
p. V187Q |
Progressive cataract |
[35] |
|
| 585–587 Deletion | 195–198 ΔQSVR | Progressive cataract | [48,49] | |
βB2-crystallin, the major component of β-crystallin, is a homodimer at low concentrations, and can form a heterodimer with other beta-crystallins under physiologic conditions [51]. Based on the structure from X-ray refraction studies for homodimer βB2-crystallin, each subunit includes 16 β-strands, eight in the NH2-terminal domain and eight in the COOH-terminal domain [51]. The results of NMR spectroscopic studies indicate that the terminal extensions of beta B2-crystallin appear to be of little ordered conformation, are accessible to solvent and flex freely from the main body of the protein [52].Earlier studies indicated that the NH2-terminal extension of β-crystallin played an important role in oligomerization [53].Recent experimental data suggest that the NH2- and COOH-terminal arms appear to be involved in preventing the formation of higher homo- and hetero oligomers [54].The long and flexible NH2-terminal extension of the CRYBB2 (PDB structure 1YTQ) might be critical for mediating protein interactions. Thus, the A2V substitution may influence homo- and heteromolecular interactions, which would contribute to cataract formation. In addition, lens crystallins are known to be susceptible to a wide variety of post-translational modifications such as acetylation, deamidation, methylation, oxidation, phosphorylation, and truncation of terminal extensions by thiol proteases. Age-related proteolytic processing of human lens β-crystallins occurs mainly at the NH2-terminal extensions [55]. This single base substitution may play a role in post-translational modifications of crystallins, which would lead to protein structure and function alteration.
By the SIFT, PolyPhen, and Align-GVGD programs, we evaluated the possible effect of this amino acid substitution (p. A2V) on βB2-crystallin protein function. The programs were used to determine whether a specific amino acid substitution would lead to an altered protein structure and function, based on sequence homology and structural information. As the isolated predictive value of these programs can be increased by their combination [6,56], it is believed that the A2V mutation alters the protein structure of the βB2-crystallin protein to such an extent it may contribute to the disease.
Also, hydropathy analysis revealed a variation in the physicochemical properties of the critical region in the A2V mutant (Figure 4B) compared with wild-type βB2-crystallin (Figure 4A). The environment surrounding the amino acid “V” in the mutant protein is more hydrophobic. Thus, the increase in hydrophobicity in the mutant form might affect the solubility of the mutant protein and hence contribute to cataract formation.
Conclusions
A novel A2V mutation of the CRYBB2 protein was identified and characterized in a Chinese family presenting with the posterior subcapsular type of congenital cataract. Further experiments on this cataract-related genetic defect and the factors that modify their variable phenotypes will improve our understanding of the mechanism of cataract formation and illuminate the developmental biology and biochemistry of the lens.
Acknowledgments
We thank the family for participating in this study. This research is supported by grant from Key Projects in the National Science & Technology Pillar Program in the Eleventh Five Year Plan Period(2006BAI02B04) and the Major Innovative Research Team Program of Zhejiang provice (2009R50039).
References
- 1.Rahi JS, Sripathi S, Gilbert CE, Foster A. Childhood blindness in India: causes in 1318 blind school students in nine states. Eye (Lond) 1995;9:545–50. doi: 10.1038/eye.1995.137. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert C, Foster A. Childhood blindness in the context of VISION 2020–the right to sight. Bull World Health Organ. 2001;79:227–32. [PMC free article] [PubMed] [Google Scholar]
- 3.Apple DJ, Ram J, Foster A, Peng Q. Elimination of cataract blindness: a global perspective entering the new millenium. Surv Ophthalmol. 2000;45:S1–196. [PubMed] [Google Scholar]
- 4.Francis PJ, Berry V, Bhattacharya SS, Moore AT. The genetics of childhood cataract. J Med Genet. 2000;37:481–8. doi: 10.1136/jmg.37.7.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT. Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol. 2004;49:300–15. doi: 10.1016/j.survophthal.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Santana A, Waiswol M, Arcieri ES, Cabral de Vasconcellos JP, Barbosa de Melo M. Mutation analysis of CRYAA, CRYGC, and CRYGD associated with autosomal dominant congenital cataract in Brazilian families. Mol Vis. 2009;15:793–800. [PMC free article] [PubMed] [Google Scholar]
- 7.Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008;19:134–49. doi: 10.1016/j.semcdb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devi RR, Yao W, Vijayalakshmi P, Sergeev YV, Sundaresan P, Hejtmancik JF. Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis. 2008;14:1157–70. [PMC free article] [PubMed] [Google Scholar]
- 9.Brémond-Gignac D, Bitoun P, Reis LM, Copin H, Murray JC, Semina EV. Identification of dominant FOXE3 and PAX6 mutations in patients with congenital cataract and aniridia. Mol Vis. 2010;16:1705–11. [PMC free article] [PubMed] [Google Scholar]
- 10.Azuma N, Hirakiyama A, Inoue T, Asaka A, Yamada M. Mutations of a human homologue of the Drosophila eyes absent gene (EYA1) detected in patients with congenital cataracts and ocular anterior segment anomalies. Hum Mol Genet. 2000;9:363–6. doi: 10.1093/hmg/9.3.363. [DOI] [PubMed] [Google Scholar]
- 11.Müller M, Bhattacharya SS, Moore T, Prescott Q, Wedig T, Herrmann H, Magin T. Dominant cataract formation in association with a vimentin assembly disrupting mutation. Hum Mol Genet. 2009;18:1052–7. doi: 10.1093/hmg/ddn440. [DOI] [PubMed] [Google Scholar]
- 12.Kloeckener-Gruissem B, Vandekerckhove K, Nurnberg G, Neidhardt J, Zeitz C, Nurnberg P, Schipper I, Berger W. Mutation of solute carrier SLC16A12 associates with a syndrome combining juvenile cataract with microcornea and renal glucosuria. Am J Hum Genet. 2008;82:772–9. doi: 10.1016/j.ajhg.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amaya L, Taylor D, Russell-Eggitt I, Nischal KK, Lengyel D. The morphology and natural history of childhood cataracts. Surv Ophthalmol. 2003;48:125–44. doi: 10.1016/s0039-6257(02)00462-9. [DOI] [PubMed] [Google Scholar]
- 14.Lorenz B. Genetic examination in cases of congenital cataract. Ophthalmologe. 2007;104:559–65. doi: 10.1007/s00347-007-1557-2. [DOI] [PubMed] [Google Scholar]
- 15.Liu M, Ke T, Wang Z, Yang Q, Chang W, Jiang F, Tang Z, Li H, Ren X, Wang X, Wang T, Li Q, Yang J, Liu J, Wang QK. Identification of a CRYAB mutation associated with autosomal dominant posterior polar cataract in a Chinese family. Invest Ophthalmol Vis Sci. 2006;47:3461–6. doi: 10.1167/iovs.05-1438. [DOI] [PubMed] [Google Scholar]
- 16.Veromann S. Theoretical considerations regarding the study “Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans”. Am J Hum Genet. 2002;71:684–5. doi: 10.1086/342207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu Z, Ji B, Wan C, He G, Zhang J, Zhang M, Feng G, He L, Gao L. A splice site mutation in CRYBA1/A3 causing autosomal dominant posterior polar cataract in a Chinese pedigree. Mol Vis. 2010;16:154–60. [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang T, Hua R, Xiao W, Burdon KP, Bhattacharya SS, Craig JE, Shang D, Zhao X, Mackey DA, Moore AT, Luo Y, Zhang J, Zhang X. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum Mutat. 2009;30:E603–11. doi: 10.1002/humu.20995. [DOI] [PubMed] [Google Scholar]
- 19.Burdon KP, McKay JD, Wirth MG, Russell-Eggit IM, Bhatti S, Ruddle JB, Dimasi D, Mackey DA, Craig JE. The PITX3 gene in posterior polar congenital cataract in Australia. Mol Vis. 2006;12:367–71. [PubMed] [Google Scholar]
- 20.Shiels A, Bennett TM, Knopf HL, Yamada K, Yoshiura K, Niikawa N, Shim S, Hanson PI. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet. 2007;81:596–606. doi: 10.1086/519980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanita V, Singh JR, Hejtmancik JF, Nurnberg P, Hennies HC, Singh D, Zhang X. A novel fan-shaped cataract-microcornea syndrome caused by a mutation of CRYAA in an Indian family. Mol Vis. 2006;12:518–22. [PubMed] [Google Scholar]
- 22.Lu S, Zhao C, Jiao H, Kere J, Tang X, Zhao F, Zhang X. Two Chinese families with pulverulent congenital cataracts and Delta G91 CRYBA1 mutations. Mol Vis. 2007;13:1154–60. [PubMed] [Google Scholar]
- 23.Litt M, Carrero-Valenzuela R, LaMorticella DM, Schultz DW, Mitchell TN, Kramer P, Maumenee IH. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum Mol Genet. 1997;6:665–8. doi: 10.1093/hmg/6.5.665. [DOI] [PubMed] [Google Scholar]
- 24.Zhang LY, Yam GHF, Fan DSP, Tam POS, Lam DSC, Pang CP. A novel deletion variant of gamma D-crystallin responsible for congenital nuclear cataract. Mol Vis. 2007;13:2096–104. [PubMed] [Google Scholar]
- 25.Hansen L, Yao WL, Eiberg H, Funding M, Riise R, Kjaer K, Hejtmancik JF, Rosenberg T. The congenital “ant-egg” cataract phenotype is caused by a missense mutation in connexin46. Mol Vis. 2006;12:1033–9. [PubMed] [Google Scholar]
- 26.Schmidt W, Klopp N, Illig T, Graw J. A novel GJA8 mutation causing a recessive triangular cataract. Mol Vis. 2008;14:851–6. [PMC free article] [PubMed] [Google Scholar]
- 27.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–25. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29:1342–54. doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graw J. Cataract mutations and lens development. Prog Retin Eye Res. 1999;18:235–67. doi: 10.1016/s1350-9462(98)00018-4. [DOI] [PubMed] [Google Scholar]
- 31.Andley UP. Crystallins in the eye: Function and pathology. Prog Retin Eye Res. 2007;26:78–98. doi: 10.1016/j.preteyeres.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Graw J. Genetics of crystallins: cataract and beyond. Exp Eye Res. 2009;88:173–89. doi: 10.1016/j.exer.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Sanderson J, Marcantonio JM, Duncan G. A human lens model of cortical cataract: Ca2+-induced protein loss, vimentin cleavage and opacification. Invest Ophthalmol Vis Sci. 2000;41:2255–61. [PubMed] [Google Scholar]
- 34.Fischer D, Hauk TG, Muller A, Thanos S. Crystallins of the beta/gamma-superfamily mimic the effects of lens injury and promote axon regeneration. Mol Cell Neurosci. 2008;37:471–9. doi: 10.1016/j.mcn.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Graw J, Loster J, Soewarto D, Fuchs H, Reis A, Wolf E, Balling R, Hrabé de Angelis M. Aey2, a new mutation in the betaB2-crystallin-encoding gene of the mouse. Invest Ophthalmol Vis Sci. 2001;42:1574–80. [PubMed] [Google Scholar]
- 36.Mothobi ME, Guo SR, Liu YY, Chen Q, Yussuf AS, Zhu XL, Fang Z. Mutation analysis of congenital cataract in a Basotho family identified a new missense allele in CRYBB2. Mol Vis. 2009;15:1470–5. [PMC free article] [PubMed] [Google Scholar]
- 37.Vanita SV. Reis A, Jung M, Singh D, Sperling K, Singh JR, Bürger J. A unique form of autosomal dominant cataract explained by gene conversion between beta-crystallin B2 and its pseudogene. J Med Genet. 2001;38:392–6. doi: 10.1136/jmg.38.6.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gill D, Klose R, Munier FL, McFadden M, Priston M, Billingsley G, Ducrey N, Schorderet DF, Héon E. Genetic heterogeneity of the Coppock-like cataract: A mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci. 2000;41:159–65. [PubMed] [Google Scholar]
- 39.Bateman JB, von-Bischhoffshaunsen FRB, Richter L, Flodman P, Burch D, Spence MA. Gene conversion mutation in crystallin, beta-B2 (CRYBB2) in a Chilean family with autosomal dominant cataract. Ophthalmology. 2007;114:425–32. doi: 10.1016/j.ophtha.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 40.Li FF, Zhu SQ, Wang SZ, Gao C, Huang SZ, Zhang M, Ma X. Nonsense mutation in the CRYBB2 gene causing autosomal dominant progressive polymorphic congenital coronary cataracts. Mol Vis. 2008;14:750–5. [PMC free article] [PubMed] [Google Scholar]
- 41.Lou D, Tong JP, Zhang LY, Chiang SW, Lam DS, Pang CP. A novel mutation in CRYBB2 responsible for inherited coronary cataract. Eye (Lond) 2009;23:1213–20. doi: 10.1038/eye.2008.222. [DOI] [PubMed] [Google Scholar]
- 42.Wang L, Lin H, Gu JZ, Su H, Huang SZ, Qi Y. Autosomal-Dominant Cerulean Cataract in a Chinese Family Associated with Gene Conversion Mutation in Beta-B2-Crystallin. Ophthalmic Res. 2009;41:148–53. doi: 10.1159/000209668. [DOI] [PubMed] [Google Scholar]
- 43.Yao K, Tang X, Shentu X, Wang K, Rao H, Xia K. Progressive polymorphic congenital cataract caused by a CRYBB2 mutation in a Chinese family. Mol Vis. 2005;11:758–63. [PubMed] [Google Scholar]
- 44.Santhiya ST, Manisastry SM, Rawlley D, Malathi R, Anishetty S, Gopinath PM, Vijayalakshmi P, Namperumalsamy P, Adamski J, Graw J. Mutation analysis of congenital cataracts in Indian families: Identification of SNPs and a new causative allele in CRYBB2 gene. Invest Ophthalmol Vis Sci. 2004;45:3599–607. doi: 10.1167/iovs.04-0207. [DOI] [PubMed] [Google Scholar]
- 45.Pauli S, Soker T, Klopp N, Illig T, Engel W, Graw J. Mutation analysis in a German family identified a new cataract-causing allele in the CRYBB2 gene. Mol Vis. 2007;13:962–7. [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen L, Mikkelsen A, Nurnberg P, Nurnberg G, Anjum I, Eiberg H, Rosenberg T. Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci. 2009;50:3291–303. doi: 10.1167/iovs.08-3149. [DOI] [PubMed] [Google Scholar]
- 47.Ganguly K, Favor J, Neuhauser-Klaus A, Sandulache R, Puk O, Beckers J, Horsch M, Schädler S, Vogt Weisenhorn D, Wurst W, Graw J. Novel allele of crybb2 in the mouse and its expression in the brain. Invest Ophthalmol Vis Sci. 2008;49:1533–41. doi: 10.1167/iovs.07-0788. [DOI] [PubMed] [Google Scholar]
- 48.Duprey KM, Robinson KM, Wang Y, Taube JR, Duncan MK. Subfertility in mice harboring a mutation in betaB2-crystallin. Mol Vis. 2007;13:366–73. [PMC free article] [PubMed] [Google Scholar]
- 49.Chambers C, Russell P. Deletion mutation in an eye lens beta-crystallin. An animal model for inherited cataracts. J Biol Chem. 1991;266:6742–6. [PubMed] [Google Scholar]
- 50.Liu BF, Liang J. Interaction and biophysical properties of human lens Q155*beta B2-crystallin mutant. Mol Vis. 2005;11:321–7. [PubMed] [Google Scholar]
- 51.Liu BF, Liang JJ. Domain interaction sites of human lens betaB2-crystallin. J Biol Chem. 2006;281:2624–30. doi: 10.1074/jbc.M509017200. [DOI] [PubMed] [Google Scholar]
- 52.Carver JA, Cooper PG, Truscott RJ. 1H-NMR spectroscopy of beta B2-crystallin from bovine eye lens. Conformation of the N- and C-terminal extensions. Eur J Biochem. 1993;213:313–20. doi: 10.1111/j.1432-1033.1993.tb17764.x. [DOI] [PubMed] [Google Scholar]
- 53.Berbers GA, Hoekman WA, Bloemendal H, de Jong WW, Kleinschmidt T, Braunitzer G. Proline- and alanine-rich N-terminal extension of the basic bovine beta-crystallin B1 chains. FEBS Lett. 1983;161:225–9. doi: 10.1016/0014-5793(83)81013-8. [DOI] [PubMed] [Google Scholar]
- 54.Trinkl S, Glockshuber R, Jaenicke R. Dimerization of beta B2-crystallin: the role of the linker peptide and the N- and C-terminal extensions. Protein Sci. 1994;3:1392–400. doi: 10.1002/pro.5560030905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dolinska MB, Sergeev YV, Chan MP, Palmer I, Wingfield PT. N-terminal extension of beta B1-crystallin: identification of a critical region that modulates protein interaction with beta A3-crystallin. Biochemistry. 2009;48:9684–95. doi: 10.1021/bi9013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan PA, Duraisamy S, Miller PJ, Newell JA, McBride C, Bond JP, Raevaara T, Ollila S, Nyström M, Grimm AJ, Christodoulou J, Oetting WS, Greenblatt MS. Interpreting missense variants: comparing computational methods in human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR). Hum Mutat. 2007;28:683–93. doi: 10.1002/humu.20492. [DOI] [PubMed] [Google Scholar]