

Summary
Recent reports demonstrate that the activation and interaction of the protease calpain (CP) and the protein phosphatase calcineurin (CN) are elevated in the late stages of Alzheimer’s disease (AD). However, the extent to which CPs and CN interact during earlier stages of disease progression remains unknown. Here, we investigated CP and CN protein levels in cytosolic, nuclear, and membrane fractions prepared from human postmortem hippocampal tissue from aged non-demented subjects, and subjects diagnosed with mild cognitive impairment (MCI). The results revealed a parallel increase in CP I and the 48 kDa CN-Aα (ΔCN-Aα48) proteolytic fragment in cytosolic fractions during MCI. In primary rat hippocampal cultures, CP-dependent proteolysis and activation of CN was stimulated by application of oligomeric Aβ(1-42) peptides. Deleterious effects of Aβ on neuronal morphology were reduced by blockade of either CP or CN. NMDA-type glutamate receptors, which help regulate cognition and neuronal viability, and are modulated by CPs and CN, were also investigated in human hippocampus. Relative to controls, MCI subjects showed significantly greater proteolytic levels of the NR2B subunit. Within subjects, the extent of NR2B proteolysis was strongly correlated with the generation of ΔCN-Aα48 in the cytosol. A similar proteolytic pattern for NR2B was also observed in primary rat hippocampal cultures treated with oligomeric Aβ and prevented by inhibition of CP or CN. Together, the results demonstrate that the activation and interaction of CPs and CN are increased early in cognitive decline associated with AD and may help drive other pathologic processes during disease progression.
Keywords: Ca2+, Alzheimer’s disease, amyloid, mild cognitive impairment, calcineurin, calpain, NMDA receptors
Introduction
Fundamental to the Ca2+ hypothesis of aging is the presumption that Ca2+ dysregulation increases vulnerability to Alzheimer’s disease (AD), and other age-related neurologic disorders (Toescu et al. 2004; Thibault et al. 2007; Toescu & Verkhratsky 2007). While cumulative evidence from numerous studies on amyloid-bearing transgenic mice and/or amyloid-treated nervous tissue has built a compelling case for the Ca2+ hypothesis (Canzoniero & Snider 2005; Bezprozvanny & Mattson 2008; Green & LaFerla 2008; Small 2009), the link between Ca2+ and disease progression in human studies has been somewhat weaker, as comparisons are generally made between subjects at polar ends of the disease spectrum (i.e. between non-demented pathologically-normal subjects and subjects with pathologically-confirmed AD). Based on these comparisons, it remains unclear as to whether Ca2+ dysregulation is an antecedent or a consequence (or both), of AD pathology. If changes in Ca2+ regulation help drive pathology, as set forth by the Ca2+ hypothesis, then alterations in Ca2+ signaling mechanisms should emerge in the early stages of the disease, when signs of mild cognitive impairment (MCI) are first diagnosed. It is notable that clinical MCI subjects show several anatomical and biochemical anomalies in the hippocampus consistent with Ca2+ dysregulation, including loss of synapses and/or synaptic proteins (Scheff et al. 2006; Scheff et al. 2007; Sultana et al. 2010) and neural atrophy (Apostolova et al. 2010). However, few studies have directly investigated Ca2+ signaling mechanisms in human MCI brain tissue.
The Ca2+-dependent protease calpain (CP) and the Ca2+/calmodulin-dependent protein phosphatase calcineurin (CN) play critical roles in regulating neuronal structure and function, and exhibit elevated activity levels in AD brain tissue and/or in experimental models of AD (Saito et al. 1993; Kuwako et al. 2002; Liu et al. 2005; Norris et al. 2005; Dineley et al. 2007; Vaisid et al. 2007; Kuchibhotla et al. 2008; Rao et al. 2008; Reese et al. 2008; Abdul et al. 2009; Wu et al. 2010). In the last 10 years, it has become increasingly clear that extensive interactions between CP and CN lead to diminished neuronal function and viability (See & Loeffler 2001; Wu et al. 2004; Shioda et al. 2006; Wu et al. 2007; Huang et al. 2010). During severe Ca2+ dysregulation, for instance, CP binds to and proteolyzes the 60 kDa CN catalytic subunit (CN-A), converting it to several high-activity fragments (Wu et al. 2004). The most commonly studied fragment has a molecular weight of 45–48 kDa (ΔCN-A48) and completely lacks a critical C-terminal autoinhibitory domain (AID) responsible for limiting CN activity when Ca2+/calmodulin levels are low (Tallant et al. 1988; Wang et al. 1989). Without the AID, ΔCN-A 48 is active even after intracellular Ca2+ recovers to basal levels. A larger, 57 kDa CP-generated fragment (ΔCN-A57) has also been described. Although the AID is largely intact in the ΔCN-A57 species, this fragment, like ΔCN-A 48, shows greater basal activity compared to full-length CN-A (Liu et al. 2005).
Recent studies have reported increased temporal cortex levels for both ΔCN-Aα 48 and ΔCN-Aα 57 in patients with confirmed AD (Liu et al. 2005; Wu et al. 2010). The purpose of the present study was to investigate whether these changes are apparent when clinical memory deficits and neuroanatomical anomalies first appear (i.e. during MCI). We show for the first time that CP and ΔCN-Aα 48 levels are significantly greater in human hippocampal cytosolic fractions prepared from MCI relative to age-matched control subjects. Importantly, CP and ΔCN-Aα 48 levels were positively correlated to one another with-in subjects and linked to altered proteolytic processing of the NR2B subunit of the NMDA receptor. Using rat primary hippocampal cultures, we also offer the first evidence that CP-mediated proteolysis and activation of CN is+ a critical mechanism through which oligomeric Aβ(1-42) peptides cause neurodegeneration. The results provide important support for the Ca2+ hypothesis of aging and AD.
Results
Increase in “activated” calpain I levels with MCI
Earlier studies by other investigators suggest that, with Ca2+ dysregulation, CP I is converted from an inactive 80 kDa protein to an activated 76 kDa fragment through autoproteolysis. Presently, however, little information is available regarding the levels of activated CP I in human subjects during putative early stages of AD, such as MCI. To address this issue, we used Western blot to detect and quantify CP I protein levels in cytosolic and nuclear fractions from post mortem human hippocampal tissue of 10 MCI and 10 age-matched control subjects (see Table 1 for subject information). As shown in Figure 1A, CP I commonly exhibited two major bands (80 and 76 kDa) in both subject groups. A few samples in each group also showed another minor band at 78 kDa. Similar banding patterns for CP I in human brain tissue have been shown in several earlier studies (Saito et al. 1993; Veeranna et al. 2004; Liu et al. 2005; Marcum et al. 2005). As shown in Figure 1B, the 76 kDa-to-80 kDa ratio for CP I was more than two fold greater in the MCI subjects (p < 0.01; Fig. 1B), suggesting that CP I activity is increased during early stages of clinical cognitive decline. MCI subjects also showed slightly greater nuclear CP I levels, however, this difference was not significant.
Table 1.
Subject information
| Groups | n | Male/Female | Age mean ± SEM | Postmortem Autopsy Interval mean ± SEM | MMSE* mean ± SEM |
|---|---|---|---|---|---|
| Control | 10 | 3/7 | 89.75 ± 1.87 | 3.06 ± 0.25 | 28.75 ± 0.43 |
| MCI | 10 | 6/4 | 89.2 ± 1.65 | 3.03 ± 0.28 | 24.10 ± 1.21 |
MMSE = mini mental state examination score
Fig. 1. Increase in activated form of calpain I with MCI.

(A) Representative western blot for CP I in cytosolic (C) and nuclear (N) fractions prepared from hippocampal tissue samples from two control and two MCI patients. Histone-3 (His-3) levels were also probed to confirm the purity of nuclear fractions, and GAPDH was used as a loading control. (B) Mean ± SD CP I 76/80 kDa ratios measured across all subjects. *p<0.01
Selective changes in CN proteolysis with MCI
CN is among the many targets of CP-mediated proteolysis in central nervous system. Proteolysis of CN by CP can result in the removal of the AID from the CN-A subunit, yielding a 45–48 kDa proteolytic fragment with high catalytic activity (Tallant et al. 1988; Wang et al. 1989). A similar 48 kDa fragment was recently found at elevated levels in nuclear tissue fractions harvested from subjects with severe AD (Wu et al. 2010). And in an earlier study, Liu et al 2005 revealed an increase in the proteolysis of CN to a highly active 57 kDa fragment in late stage AD brain samples. Last year, we reported that CN signaling in human hippocampus was elevated early in cognitive decline and continued to rise as dementia worsened during the later stages of AD (Abdul et al., 2009). Together, these results, along with evidence for increased CP I activation levels in MCI (Fig. 1), suggest that elevated CN signaling in early AD may be due to CP-mediated proteolysis. We therefore investigated this possibility in the same tissue fractions shown above (Fig. 1) using anti-CN-A antibodies that recognize the full length and truncated fragments of the CN-Aα and CN-Aβ isoforms.
As shown in Figure 2A, both CN-A isoforms (α and β) in human hippocampus were detected as four primary bands with molecular weights of ~60, ~57, ~48, and ~37 kDa. The 60 kDa band represents full-length CN-A, and the 57 and 48 kDa bands could be strongly induced in human hippocampal whole tissue homogenates treated for 15 min with 1 mM Ca2+ and purified CP I (Fig. 2A), consistent with CP I-mediated proteolysis as shown in other reports (Wu et al. 2004). The full length 60 kDa band is referred to from here out as FLCN-A, while the 57 and 48 kDa fragments are referred to as ΔCN-A57 and ΔCN-A48. Note that the 37 kDa band was not dependent on CP I activity (Fig. 2A), suggesting that this fragment is generated by another protease or perhaps represents an alternative splice variant of CN-A.
Fig. 2. Increased calpain-mediated truncation/activation of CN-A α with MCI.

(A) Western blots of CN-Aα and CN-Aβ in human hippocampal brain tissue extracts incubated at 37 °C for 15 min in the presence (lanes 2 and 4) or absence (lanes 1 and 3) of 2 mM DTT, 1 unit of recombinant pig-calpain1 (CP I) and 2mM CaCl2 (Ca2+). (B) Representative Western blots for CN-Aα and CN-Aβ in cytosolic (C) and nuclear (N) fractions prepared from hippocampal tissue samples from two control and two MCI patients. Histone-3 (His-3) levels were also probed to confirm the purity of nuclear fractions, while GAPDH was used as a loading control. (C) Cytosolic and nuclear accumulation (mean ± SD) of full-length (FL), and Δ57 kDa and Δ48 kDa proteolyzed fragments of CN-Aα measured across all subjects. *p<0.05, #p<0.005, +p<0.001
When analyzed in different subcellular fractions across subject groups, both CN-Aα and CN-Aβ occurred predominantly as full-length proteins and no group differences were observed for FLCN-Aα or FLCN-Aβ in any fraction (Fig. 2B and see Supplementary Figure 1A and B). In contrast, changes in CN proteolysis were apparent, but depended strongly on the isoform and subcellular fraction examined. For the CN-Aα isoform, overall ΔCN-Aα48 levels were significantly higher (p < 0.05) in MCI subjects (Fig. 2E), due primarily to a large significant increase in the cytosolic fraction (p < 0.01). In fact, approximately 25% of all available cytosolic CN-Aα in the MCI group was of the ΔCN-Aα48 variety. Moreover, within subject comparisons revealed a strong positive correlation (r = 0.57, p<0.05) between ΔCN-Aα48 and CP I in the cytosol, consistent with elevated CP-mediated proteolysis during MCI (Fig. 3C). Interestingly,ΔCN-Aα48 was significantly reduced (p < 0.001) in the nucleus of MCI subjects (Fig. 2E). However, the absolute amounts of nuclear ΔCN-Aα48 were very low for both groups (< 10% of all CN Aα in the nucleus). Although ΔCN-Aα57 was the most abundant proteolytic fragment across cellular fractions, no differences in its levels were observed between control and MCI subjects (Fig. 2D), nor was a significant correlation observed between ΔCN-Aα57 and CP I in any cellular fraction (Fig. 3B).
Fig. 3. Interrelationships between cytosolic-associated CP and CN truncation.

Scatterplots showing ranked CP I levels as a function of either FLCN-Aα (A), ΔCN-Aα57 (B), or ΔCN-Aα48 (C) in hippocampal cytosolic fractions from all subjects.
Unlike the CN-Aα isoform, total levels for CN-Aβ proteolytic fragments (ΔCN-Aβ57,ΔCN-Aβ48 and ΔCN-Aβ37 kDa) exhibited high intra-group variability with no significant differences across subject groups (Fig. 2B, average data not shown). Moreover, even though CP I is capable of cleaving CN-Aβ in vitro (Fig. 2A), we observed no significant correlation between CP I and any CN-Aβ proteolytic fragment (data not shown).
Finally, because a sizable pool of CN is associated with the plasma membrane, where it is tethered to a variety of binding proteins and juxtaposed to neurotransmitter receptors and ion channels, we also investigated CN levels in membrane fractions from control and MCI subjects (supplementary Fig. 2A and 2B). Western blots revealed one primary band for each CN-A isoform, which appeared at similar levels in both subject groups. When analyzed side-by-side with tissue fractions that contain both full-length and proteolyzed CN (i.e. human hippocampal cytosolic and nuclear extracts, and primary rat hippocampal culture homogenates), membrane-associated CN migrated more closely with the 60 kDa product (supplementary Fig. 2C). This result suggests that membrane-associated CN exists largely as a full-length protein.
Oligomeric Aβ stimulates CP-dependent proteolysis and activation of CN
The results on human hippocampus suggest that CN-Aα undergoes significant CP-1-mediated proteolysis during the early phase of cognitive decline associated with AD (Fig. 2 and 3). To further investigate CP-CN interactions in the context of AD, we quantified CN proteolysis in primary rat hippocampal cultures at 3 and 24 h after treatment with pathogenic oligomeric Aβ peptides alone (65 nM), or in the presence of the CP inhibitor calpeptin (10 μM), or the caspase I inhibitor Z-YVAD-FMK (1μM). Cleavage of the well-characterized calpain substrate, α-spectrin, from an ~250 to an ~150 kDa product was used as a positive control for CP-dependent proteolysis. At three hours post-Aβ exposure, very little proteolysis of either CN-Aα or spectrin was observed (Fig. 4A, top panel). However, at the 24 h time point, extensive proteolysis was observed for both of these proteins (Fig. 4B, top panel). This proteolysis was blocked by calpeptin, but not by Z-YVAD-FMK, suggesting a relatively selective role for CP in CN proteolysis.
Fig. 4. Blockade of calpain activity inhibits Aβ-mediated proteolysis and activation of CN in primary hippocampal cultures.

(A–B, top panels) Representative Western blots of spectrin, CN-Aα, and GAPDH in mixed hippocampal cultures treated 3 (A) or 24 h (B) prior with oligomeric Aβ(1-42) in the presence or absence of the potent CP inhibitor calpeptin (CLPT) or the caspase I inhibitor Z-YVAD-FMK (Z-YVAD). (A–B, bottom panels) Mean ± SEM NFAT-luciferase activity (% control) in separate primary hippocampal cultures at 3 (A) and 24 h (B) after Aβ(1-42) treatment in the presence or absence of CLPT. (D) Immunofluorescent labeling of MAP2 in mixed hippocampal cultures 24 h after Aβ(1-42) treatment in the presence or absence of CLPT, or FK506. *p<0.05, #p<0.001.
To determine if CN proteolysis resulted in greater CN signaling, we measured the transcriptional activity of the nuclear factor of activated T cells (NFAT, a specific and well-characterized CN substrate) in separate hippocampal cultures following Aβ treatment. As we have shown previously in primary astrocyte cultures (Abdul et al., 2009), oligomeric Aβ strongly stimulated NFAT activation in primary mixed cultures (Fig. 4A, B, bottom panels). And while blockade of CP activity with calpeptin markedly reduced NFAT activation by oligomeric Aβ, this effect was dependent on exposure time. Specifically, calpeptin had little effect within the first three hours of Aβ treatment (Fig. 4A, bottom panel), but reduced NFAT activity by more than 50% (p < 0.05) when assessed after a 24 h exposure (Fig. 4B, bottom panel). These results are consistent with the time-dependent changes observed for CN proteolysis shown in the top panels of Figure 4A and 4B and suggest that elevated CN signaling following Aβ exposure results, in part, from CP I-mediated proteolysis of the CN-Aα isoform.
Aβ peptides have been well-characterized for their deleterious effects on neuronal structure and viability, many of which are mediated through activation of CPs and CN (Jordan et al. 1997; Kuwako et al. 2002; Shankar et al. 2007; Agostinho et al. 2008; Reese et al. 2008; Wei et al. 2008; Tackenberg & Brandt 2009; Lopes et al. 2010; Wu et al. 2010; Zhao et al. 2010). Similar to these reports, we also observed striking degeneration in MAP2-labeled primary neurons exposed for 24 h to oligomeric Aβ peptides, including widespread dendritic blebbing and atrophy (Fig. 4C). Although these effects were greatly ameliorated by blockade of CP activity with calpeptin, signs of degeneration, in particular dendritic blebbing, were still evident. In contrast, cultures treated with Aβ in the presence of the CN inhibitor, FK-506 (2 αM), appeared vibrant and healthy and were visually indistinguishable from control cultures. These results suggest that Aβ-mediated activation of CN, through CP-dependent and independent means, plays a critical role in neurodegeneration.
CP/CN interactions and NR2B proteolysis during MCI
While MCI is associated with elevated Aβ levels (Murphy et al. 2007; Okello et al. 2009), signs of extensive neurodegeneration and cell death are not usually present during this putative stage of AD. Instead, MCI is characterized by a subtle loss of synaptic contacts and/or proteins (Scheff et al. 2006; Murphy et al. 2007; Scheff et al. 2007; Sultana et al. 2010) and mild neural atrophy (Apostolova et al. 2010), which may be early consequences of amyloidosis. CPs and CN regulate a wide array of proteins involved in the structural and functional organization of synapses and dendrites (Perrin & Huttenlocher 2002; Goll et al. 2003; Groth et al. 2003). Some postsynaptic elements, such as NMDA receptors (NMDARs), act as a source and a target for the activation of CPs and CN (Guttmann et al. 2001; Simpkins et al. 2003; Yuen et al. 2008). Recent evidence shows that the NR2A and NR2B subunits of the NMDA receptor complex are proteolyzed by CP through a pathway involving CN (Yuen et al., 2008). As such, these proteins may show increased sensitivity to alterations in CN proteolysis during MCI.
Based on these reports, we assessed protein levels for NR2A and NR2B in membrane fractions from post-mortem human hippocampus of the same control and MCI subjects used for CP/CN measurements (Figs. 1–3). The ubiquitously expressed Na+/K+ ATPase was used as a loading control. As shown in Figure 5A, we observed little if any proteolysis of the NR2A subunit, which appeared as an approximately 170 kDa band, and found no significant differences in the levels of this protein with MCI (not shown). In contrast, NR2B appeared as two primary bands with molecular weights of 180 and 115 kDa. The 115 kDa band has been characterized as a proteolytic product in other reports (Simpkins et al. 2003). Relative to controls, MCI subjects exhibited a more than two-fold reduction in levels for the 180 kDa band (p < 0.01), but showed a > 25% increase in the 115 kDa band (p = 0.05, Fig. 5B), suggesting an increase in NR2B proteolysis with MCI. Indeed, 85% of all membrane-associated NR2B was in the form of the 115 kDa product for the MCI subjects, compared to 61% in the control group (p < 0.01, see Fig. 5B inset). Interestingly, the 115 kDa band appeared at much lower levels in hippocampal synaptosomes, whether isolated from MCI or control subjects, suggesting that proteolysis may be more characteristic of extrasynaptic NR2B subunits (Fig. 5C). To determine if human NR2B is proteolyzed by CP I in vitro, we picked membrane samples from two control subjects that exhibited very little NR2B proteolysis and incubated them for 15 min in sample buffer with or without 1 mM Ca2+ and purified CP I . The results confirmed that NR2B is indeed vulnerable to CP-mediated proteolysis (Fig. 5D). Furthermore, the extent of NR2B proteolysis shown in Figure 5A and B was significantly correlated with levels for activated CP I (i.e. 76 kDa) and ΔCN-Aα48 in cytosolic fractions prepared from the same subjects (Fig. 5E and 5F). Additional studies on rat hippocampal cultures showed that NR2B undergoes proteolysis (p < 0.01) during prolonged (24 h) exposure to oligomeric Aβ peptides (Fig. 6). This proteolysis was reduced (p < 0.05) by co-application of the CP inhibitor, calpeptin (10μM) or a membrane-soluble CN auto-inhibitory peptide (10 μM). Together, these results suggest that changes in CP I and CN regulation help drive alterations in NR2B proteolysis during MCI and/or Aβ pathology.
Fig. 5. Proteolysis of NR2B is increased during MCI and correlated to activated CP I and ΔCN-Aα48 levels.

(A) Representative Western blots of NR2A and NR2B in human hippocampal membrane fractions from 10 control and 10 MCI subjects. (B) Mean ± SD NR2B levels in human hippocampal membrane fractions across all subjects. Note that data is from the same subjects used for CP and CN measures shown in Figures 1 and 2. Inset shows the levels of the 115 kDa NR2B proteolytic fragment relative to total NR2B levels (expressed as % of total NR2B) for the same membrane fractions (C) Representative Western blots of NR2B and PSD-95 in hippocampal synaptosomal fractions from three control and three MCI patients (D) Western blot of NR2B in human hippocampal membrane fractions digested in the presence or absence of Ca2+ and recombinant pig-calpain1 (CP I). (E–F) Scatterplots showing ranked proteolytic levels for NR2B in human hippocampal membrane fractions relative to ranked CP I (E) and ΔCN-Aα48 levels (F) measured in the cytosolic fractions of the same subjects (see Figures 1 and 2). #p<0.01.
Fig. 6. CP/CN-dependent proteolysis of NR2B in mixed hippocampal cultures treated with oligomeric Aβ.
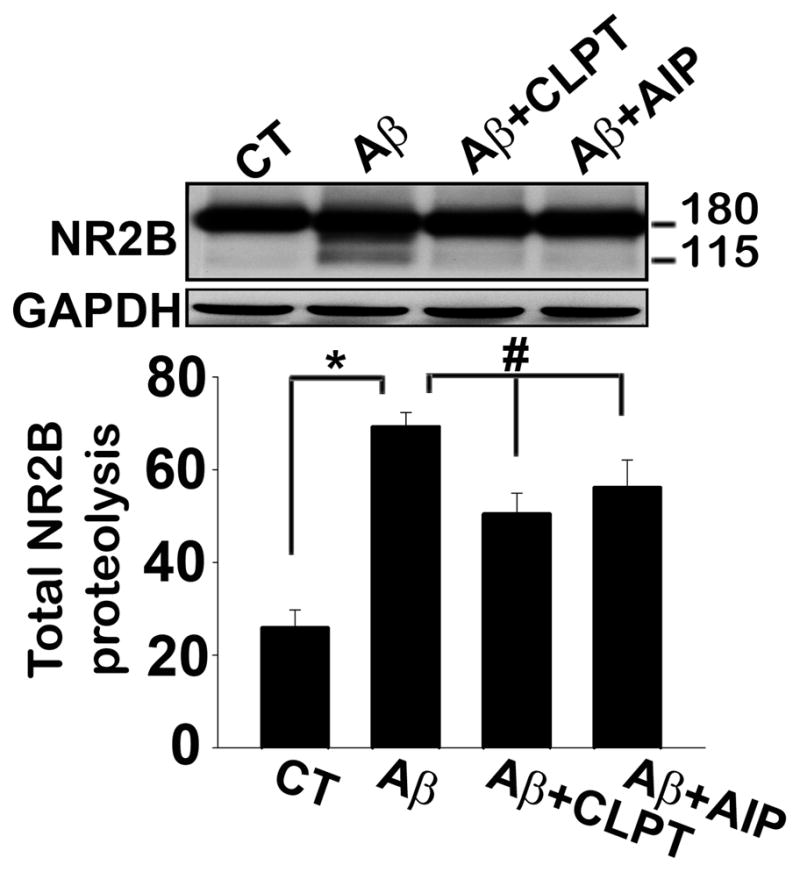
Top panel shows representative Western blot (top) of NR2B in individual mixed hippocampal cultures, and the bottom panel shows the average extent (mean ± SD) of NR2B proteolysis (115 kDa band expressed as percent of total NR2B) measured in all cultures treated with oligomeric Aβ(1-42) in the presence or absence of the CP inhibitor, calpeptin (CLPT), or the CN autoinhibitory peptide (AIP). CT refers to control (untreated) cells. *p<0.05; #p<0.01.
DISCUSSION
CPs and isoform-specific changes in CN proteolysis with MCI
Of all the Ca2+ sensitive proteins, CPs are perhaps most consistently linked to deleterious changes in neuronal function and viability due to injury and/or chronic disease. Here, we found that activated CP I levels are elevated in human hippocampus during MCI, demonstrating that CP alterations shown previously in AD tissue (Saito & Nixon 1993; Liu et al. 2005) arise early in disease progression. These results are consistent with numerous other studies that have proposed a causative role for CPs in AD (Kuwako et al. 2002; Trinchese et al. 2008; Nimmrich et al. 2010). CPs can initiate degenerative processes through actions on a variety of substrates, including cytoskeletal proteins and membrane receptors, to name a few (Nixon 2003; Bertipaglia & Carafoli 2007; Vosler et al. 2008). In addition, CP is increasingly recognized as a critical regulator of CN-mediated neuronal dysfunction in several disorders (Wu et al. 2007; Qu et al. 2010). CP-directed removal or shortening of the CN autoinhibitory domain, located in the C-terminus of the CN-A subunit, can generate CN fragments with constitutively high basal activity (Tallant et al. 1988; Wang et al. 1989; Wu et al. 2004). Recent studies have reported significant proteolysis and activation of CN in cortical tissue of humans with severe AD (Liu et al. 2005; Wu et al. 2010). These proteolytic changes may have been underestimated in earlier studies due to the use of C-terminus directed antibodies that do not detect most CN-A proteolytic fragments (Billingsley et al. 1994; Ladner et al. 1996; Lian et al. 2001). Using an N-terminus antibody, we observed isoform and subcellular specific elevations in CN proteolysis in MCI hippocampus. In particular, the ΔCN-Aα48 fragment made up more than 25% of the total CN-Aα available in the cytosol of MCI subjects and appeared in conjunction with elevated CP I levels. Why CN-Aα, but not CN-Aβ, undergoes elevated proteolysis with MCI is unclear. Few studies have directly investigated expressional differences between these isoforms in brain, and even fewer have focused on their functional differences. In healthy rat brain tissue, CN-Aα appears to be more abundantly expressed in area CA1 than in the dentate, while CN-Aβ shows a more even distribution across subregions (Kuno et al. 1992). It’s possible that these expressional differences could govern the exposure of each isoform to CP and other proteases. CPs or other proteases may also bind CN-Aβ with lesser affinity, and/or catalyze proteolysis with reduced efficiency. Regardless, this is the second recent study from our group that has found changes in CN-Aα, but not CN-Aβ with MCI or AD (Abdul et al. 2009), suggesting that CN-Aα has greater relevance to disease progression.
Implications of CN proteolysis for progression of AD
When overexpressed in experimental models, ΔCN-Aα48 has been shown to recapitulate numerous biobehavioral markers of aging and AD including synaptic impairments (Mansuy et al. 1998; Winder et al. 1998), dendritic atrophy (Wu et al. 2010), Ca2+ dysregulation (Norris et al. 2010), and glial activation/neuroinflammation (Norris et al. 2005). Moreover, CN inhibitors reduce memory deficits in AD mouse models (Dineley et al. 2007; Taglialatela et al. 2009) and negate many of the neurotoxic actions of Aβ (Chen et al. 2002; Cardoso & Oliveira 2005; Snyder et al. 2005; Shankar et al. 2007; Reese et al. 2008; Abdul et al. 2009; Li et al. 2009; Wu et al. 2010). It is noteworthy thatΔCN-Aα48 is also found at elevated levels in late stage AD (Wu et al. 2010). However, unlike in MCI, this increase is more prominent in nuclear, than in cytosolic fractions. Nuclear localization of CN may reflect a more toxic stage of Ca2+ dysregulation involving NFAT3-mediated transcriptional regulation (Abdul et al. 2010). Our previous research (Abdul et al. 2009) and work by Wu et al. (2010) showed that elevated nuclear levels of CN in late stage AD are strongly associated with increased nuclear NFAT3 levels. In animal models of injury, NFAT3 activation has been linked to neuronal death via induction of the Fas ligand (Shioda et al. 2007; Luoma & Zirpel 2008), whereas NFAT inhibition preserves dendritic integrity following exposure to Aβ (Wu et al., 2010). Combined with the findings of the present study, these observations suggest that the generation of excessiveΔCN-Aα48 in MCI is a critical change that may set off a host of other deleterious processes resulting in widespread neurodegeneration and dementia.
Aβ, CP/CN interactions, and NMDARs
Soluble oligomeric Aβ peptides, which are elevated during MCI (Murphy et al. 2007; Abdul et al. 2009; Okello et al. 2009), cause profound Ca2+ dysregulation (Green & LaFerla 2008) and stimulate CPs and CN in a variety of cell types (Cardoso & Oliveira 2005; Reese et al. 2008; Abdul et al. 2009; Wu et al. 2010). The present work suggests that Aβ-mediated activation of neurotoxic CN signaling partly results from extensive CP-directed proteolysis of the CN-Aα isoform. However, while CP and CN inhibitors each reduced neuronal degeneration in response to Aβ, CN inhibitors appeared to confer greater protection. As mentioned, FLCN is exquisitely sensitive to Ca2+ and might be expected to show rapid and direct activation in response to Aβ-dependent Ca2+ elevations with or without CP activation. In fact, CP inhibitors were much more effective at inhibiting CN proteolysis and signaling over a 24 hr period than within the first few hours of Aβ exposure. The initial Aβ-dependent increase in FLCN-A activity may therefore be sufficient to initiate degenerative processes, while CP-mediated proteolysis of CN exacerbates and/or perpetuates these processes. Regardless, these findings strongly suggest that CP and CN activation are not discrete outcomes of elevated Aβ, but instead are part of a common pathway involved in neurotoxicity.
Changes in CP/CN interactions during MCI appear to be intertwined with NMDARs, which are the primary target of memantine, an FDA-approved drug used in the treatment of AD (Parsons et al. 2007). Our results demonstrate a selective increase in the proteolysis of NR2B- type NMDARs with MCI concurrent with elevated CP and ΔCN-Aα48 levels. Similar proteolysis of NR2B was induced in rat hippocampal cultures by Aβ and was reduced by co-application of CP and CN inhibitors. These results are generally consistent with a recent study that observed CP/CN-dependent proteolysis of NR2B in primary cortical neuron cultures (Yuen et al, 2008). That we observed little proteolysis of NR2B in human hippocampal synaptosomes suggests that extrasynaptic NMDARs are a more likely target of elevated CP/CN signaling (at least, in regard to MCI and AD). This selectivity may have important functional implications as extrasynaptic NMDARs appear to play a greater role in neuronal degeneration (Hardingham et al. 2002; Xu et al. 2009), particularly after exposure to Aβ (Ronicke et al. 2010). Moreover, a recent report indicates that memantine preferentially acts at extrasynaptic as opposed to synaptically localized NMDARs (Xia et al. 2010). How CP-mediated proteolysis of extrasynaptic NR2B affects neurologic function in MCI is unclear. While proteolysis may help exacerbate NMDAR-mediated toxicity, it’s also possible that CP/CN interactions provide a negative feedback mechanism to dampen NMDAR activity during Ca2+ dysregulation (Yuen et al. 2008). Clearly, further work will be necessary to distinguish between these possibilities.
Role for other proteases in CN proteolysis during MCI and AD?
Caspases are another class of proteases that are strongly implicated in neurodegenerative processes associated with AD and amyloidosis (Cotman et al. 2005), with several family members showing increased expression/activity during MCI (Albrecht et al. 2007; Sultana et al. 2010). Similar to CPs, caspases have been shown to proteolyze CN in vitro and in vivo (Mukerjee et al. 2000; Mukerjee et al. 2001). However, the present study revealed little to no effect of the caspase inhibitor, Z-YVAD-FMK, on Aβ-mediated proteolysis of CN in primary hippocampal cultures. Nonetheless, even though our data demonstrate a close relationship between CPs and CN in human hippocampus, they do not rule out the possibility that caspases, or other proteases, interact with CN and/or contribute to altered CN proteolysis during MCI.
Summary and conclusions
The Ca2+ hypothesis of aging and AD, proposed more than 25 years ago (Khachaturian 1984; Landfield & Pitler 1984; Gibson & Peterson 1987), has been supported by abundant evidence obtained from multiple experimental models using diverse experimental approaches (Canzoniero & Snider 2005; Bezprozvanny & Mattson 2008; Green & LaFerla 2008; Small 2009). However, the present study is among the relatively few to directly link changes in Ca2+ regulation to the early stages of AD progression. Our results suggest that increased CN signaling observed previously during MCI (Abdul et al. 2009) and in multiple animal models of AD and amyloidosis (Dineley et al. 2007; Kuchibhotla et al. 2008; Wu et al. 2010) may arise from CP-mediated proteolysis of the CN-Aα isoform. Disrupting the structural and/or functional interaction of CPs and CN offers a promising novel approach for treating AD and other neurodegenerative disorders.
Experimental Procedures
Subjects
Post-mortem brain samples from the hippocampus and cerebellum were provided by the Neuropathology Core of the Alzheimer’s Disease Center (ADC) at the University of Kentucky. Specimens from the hippocampus were obtained at autopsy and snap-frozen in liquid nitrogen. Hippocampus of 10 MCI and 10 age-matched cognitively unimpaired subjects were used for this study (Table 1). All subjects were participants in the University of Kentucky’s ADC Autopsy program. At autopsy (~3 h after death), tissue from multiple brain regions was processed for neuropathological evaluations as described elsewhere (Nelson et al. 2007).
Preparation of cell extracts from human brain tissue
Autopsied human brain tissue samples were flash-frozen in liquid nitrogen and stored at -80° C until use. Membrane, cytosolic and nuclear fractions from hippocampal tissue samples were prepared as described previously (Abdul et al. 2009). The membrane fractions obtained were resuspended in sucrose buffer [in mM: 300 sucrose, 75 Nacl, 10 Tris (pH 7.4), 20 EDTA, 20 EGTA] containing phosphatase, protease, and CP inhibitor cocktails (EMD Chemicals, Gibbstown, NJ). Cytosolic and nuclear fractions were resuspended in buffer C [in mM: 50 HEPES (pH 7.6), 50 KCl, 0.1 EDTA, 10% glycerol, 1 dithiothreitol (DTT)], containing phosphatase, protease, and CP inhibitor cocktails and stored at −80° C until use.
In vitro proteolysis of CN by calpain1
Human whole brain extracts were prepared in HEPES homogenizing buffer (pH 7.5) with phosphatase inhibitor. The extracts were incubated in the presence of Ca2+ (1 mM), DTT (5mM) and/or recombinant Pig calpain 1 (1Unit) for 15 min at 37 °C. The reactions were terminated by addition of 2X concentrated SDS-PAGE sample buffer. The samples were boiled in water for 5 min and the products of proteolysis were analyzed by Western blots, probing with antibodies to CN-Aα, CN-Aβ and GAPDH (loading control).
Synaptosome preparation
Synaptosome fractions from control and MCI human hippocampus were isolated as described previously (Mohmmad Abdul & Butterfield 2005). In brief, hippocampal tissue was homogenized in isolation buffer (in mM: 320 sucrose, 0.2 PMSF, 2 EDTA, 2 EGTA, 20 HEPES, 4 μg/ml leupeptin, 4 μg/ml pepstatin, 5 μg/ml aprotinin, and 20 μg/ml trypsin inhibitor) and centrifuged at 1940 × g for 10 min at 0 °C. The supernatant was collected and centrifuged at 25,400 × g for 12 min at 0 °C. The resulting pellet was mixed with a small volume of cold isolation buffer and layered onto cold discontinuous sucrose gradients containing 10 ml each of 1.18 M (pH 8.5), 1.0 M (pH 8.0), and 0.85 M (pH 8.0) sucrose, as well as 2 mM EDTA, 2 mM EGTA, and 10 mM HEPES. Gradients were centrifuged in a Beckman-Coulter Optima L-90K ultracentrifuge at 82,500 × g for 1 h at 4 °C. Resulting purified synaptosomes were removed from the 1.18/1.0 M interface and washed three times in Locke's buffer (in mM: 0.15 NaCl, 5.6 KCl, 2.3 CaCl2, 1.0 MgCl2, 3.6 NaHCO3, 5.0 Glucose, and 5.0 HEPES; pH 7.4) at 15,500 rpm (29,100 × g) for 12 min at 0 °C.
Primary cell culture
Primary mixed (neurons and astrocytes) hippocampal cultures were prepared from embryonic day 18 Sprague-Dawley rat pups as described previously (Sama et al. 2008). Cells were investigated at between 14 and 21 days in vitro (DIV).
Western blot analysis
Western blots of postmortem human tissue and primary cell cultures were performed using ECL Plus reagents (Amersham, Piscataway, NJ) as described previously (Norris et al. 2005; Sama et al. 2008; Abdul et al. 2009). Primary antibodies used include: 1:3000 anti-CN-Aα or 1:3000 anti-CN-Aβ (EMD Biochemicals), 1:10000 anti-histone3 (Sigma-Aldrich, Saint Louis, MO,USA), 1:10000 anti-calpain1 or 1:10000 anti-Na+-K+-ATPase (Abcam, Cambridge, MA) or 1:1000 anti-NR2A/NR2B (Millipore, Billerica, MA), 1:10000 anti-spectrin-Ab38 (Abcam).
Oligomeric Aβ treatment of primary hippocampal cultures
Synthetic oligomeric Aβ (65nM) prepared as previously described (Abdul et al., 2009) was delivered to cell cultures in the presence or absence of the following inhibitors (from EMD): calpeptin (10μM), FK-506 (3 μM), CN autoinhibitory peptide (10μM), and Z-YVAD-FMK (1μM). All inhibitors were added 2h prior to oligomeric Aβ(1-42) treatments. Western blots, NFAT luciferase activity assays, and/or immunofluorescent labeling were then performed 3 or 24 h later using our previously published methods (Sama et al. 2008; Abdul et al. 2009; Furman et al. 2010). For immunofluorescent labeling of MAP2-positive neurons, ten 40X fields of cells were chosen at random within each condition and cell fluorescence was imaged using a Nikon CoolSnap ES digital camera.
NFAT activity assays
Methods for eliciting and measuring NFAT-dependent transcriptional activation in primary cultures were very similar to those used previously by our group (Sama et al. 2008; Abdul et al. 2009; Furman et al. 2010). At approximately 24 hr before treatment with oligomeric Aβ, 35 mm hippocampal culture dishes were infected (at a multiplicity of infection of 100) with recombinant adenovirus encoding an NFAT-dependent luciferase reporter construct (Ad-NFAT-Luc), provided by Dr. Jeff Molkentin at the University of Cincinnati. Ad-NFAT-luc encodes nine copies of an NFAT binding site (from the IL-4 promoter) and an additional minimal promoter upstream of a luciferase sequence, and has been described elsewhere (Wilkins et al. 2004). In our experience, transgene expression is achieved in > 90% of all astrocytes and 60–70% of all neurons in mixed hippocampal cultures when adenovirus is used at an MOI of 50 or more. At either 3 or 24 h after exposure to oligomeric Aβ, each culture dish was washed three times in PBS and cells were scraped free and pelleted at 13000 rpm. Supernatants from each 35 mm dish were removed and replaced with CAT buffer (250 mM Tris pH 8.0, 1mM EDTA) and pellets were stored at −20° C until use. Samples were freeze/thawed twice, re-suspended, centrifuged at 13000 rpm, and supernatants collected. To control for potential between-group variability in NFAT-luciferase expression, all sample volumes were normalized to the same protein concentration using the Lowry method. Luciferase expression was quantified using a luciferase detection kit (Tropix, luc Screen) and a plate reader. Typically, six or more dishes were analyzed per treatment condition, resulting in a well-powered experimental design.
Statistical Analysis
Unless otherwise stated, all results are expressed as means ± SD. Analysis of variance (ANOVA), followed by Fischer’s PLSD post hoc test, was used for most all statistical comparisons. For within-subject correlational analyses, each subject (regardless of diagnosis) was ranked in ascending order (from 1 to 20) on the basis of protein expression levels for each target protein (e.g. CP I, CN Aα 48 kDa, and NR2B) as determined by Western blot. Ranked values for any two target proteins were then compared using a simple regression analysis. Subjects used for these analyses were the same ones used to generate average group data shown in Figures 1, 2, and 5. Differences were considered significant at p < 0.05.
Supplementary Material
Representative Western blots for CN-Aα (A) and CN-Aβ (B) in cytosolic (C) and nuclear (N) fractions prepared from hippocampal tissue samples from two control and two MCI subjects. Note these blots were processed at a lower exposure time to clearly distinguish the full-length (60 kDa) from the 57 kDa CN-A fragment. Cytosolic and nuclear accumulation (mean ± SD) of full-length (FL) CN-Aα and CN-Aβ were measured across all subjects and no differences were observed, consistent with results from Figure 2C.
(A and B, top panels) Representative Western blots for membrane-associated CN-Aα (A) and CN-Aβ (B) in cytosolic (C) and nuclear (N) fractions prepared from hippocampal tissue samples from two control and two MCI subjects. Na+/K+-ATPase was used as a loading control. (A and B, bottom panels) Quantitation of membrane-associated full-length (FL) CN-Aα and CN-Aβ (mean ± SD) across all subjects. (C) Western blot showing membrane-associated CN-Aα (lanes 1 and 2) in comparison to CN-Aα products associated with human hippocampal nuclear and cytosolic extracts (lanes 3 and 4) and primary rat hippocampal culture (lane 5).
Acknowledgments
This work was supported by National Institutes of Health Grants AG024190, AG027297, AG028383, and AG010836 and a gift from the Kleberg Foundation.
Footnotes
Author contributions:
HMA: Performed experiments, prepared manuscript
IB: Produced oligomeric Abeta peptides
HL III: Provided oligomeric Abeta peptides; provided guidance for executing Abeta experiments
RPG: Provided calpain antibodies; provided expertise on proteolysis experiments and helped interpret proteolytic products in Western blots
CMN: supervised project; prepared manuscript
References
- Abdul HM, Furman JL, Sama MA, Mathis DM, Norris CM. NFATs and Alzheimer's Disease. Mol Cell Pharmacol. 2010;2:7–14. [PMC free article] [PubMed] [Google Scholar]
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, 3rd, Kraner SD, Norris CM. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostinho P, Lopes JP, Velez Z, Oliveira CR. Overactivation of calcineurin induced by amyloid-beta and prion proteins. Neurochem Int. 2008;52:1226–1233. doi: 10.1016/j.neuint.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol. 2007;170:1200–1209. doi: 10.2353/ajpath.2007.060974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova LG, Mosconi L, Thompson PM, Green AE, Hwang KS, Ramirez A, Mistur R, Tsui WH, de Leon MJ. Subregional hippocampal atrophy predicts Alzheimer's dementia in the cognitively normal. Neurobiol Aging. 2010;31:1077–1088. doi: 10.1016/j.neurobiolaging.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertipaglia I, Carafoli E. Calpains and human disease. Subcell Biochem. 2007;45:29–53. doi: 10.1007/978-1-4020-6191-2_2. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley ML, Ellis C, Kincaid RL, Martin J, Schmidt ML, Lee VM, Trojanowski JQ. Calcineurin immunoreactivity in Alzheimer's disease. Exp Neurol. 1994;126:178–184. doi: 10.1006/exnr.1994.1056. [DOI] [PubMed] [Google Scholar]
- Canzoniero LM, Snider BJ. Calcium in Alzheimer's disease pathogenesis: too much, too little or in the wrong place? J Alzheimers Dis. 2005;8:147–154. doi: 10.3233/jad-2005-8207. discussion 209–115. [DOI] [PubMed] [Google Scholar]
- Cardoso SM, Oliveira CR. The role of calcineurin in amyloid-beta-peptides-mediated cell death. Brain Res. 2005;1050:1–7. doi: 10.1016/j.brainres.2005.04.078. [DOI] [PubMed] [Google Scholar]
- Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002;77:354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol. 2005;64:104–112. doi: 10.1093/jnen/64.2.104. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007;88:217–224. doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Artiushin IA, Norris CM. Disparate effects of serum on basal and evoked NFAT activity in primary astrocyte cultures. Neurosci Lett. 2010;469:365–369. doi: 10.1016/j.neulet.2009.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GE, Peterson C. Calcium and the aging nervous system. Neurobiol Aging. 1987;8:329–343. doi: 10.1016/0197-4580(87)90072-8. [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Green KN, LaFerla FM. Linking calcium to Abeta and Alzheimer's disease. Neuron. 2008;59:190–194. doi: 10.1016/j.neuron.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Groth RD, Dunbar RL, Mermelstein PG. Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun. 2003;311:1159–1171. doi: 10.1016/j.bbrc.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Guttmann RP, Baker DL, Seifert KM, Cohen AS, Coulter DA, Lynch DR. Specific proteolysis of the NR2 subunit at multiple sites by calpain. J Neurochem. 2001;78:1083–1093. doi: 10.1046/j.1471-4159.2001.00493.x. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Huang W, Fileta J, Rawe I, Qu J, Grosskreutz CL. Calpain activation in experimental glaucoma. Invest Ophthalmol Vis Sci. 2010;51:3049–3054. doi: 10.1167/iovs.09-4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Miller RJ. Role of calpain-and interleukin-1 beta converting enzyme-like proteases in the beta-amyloid-induced death of rat hippocampal neurons in culture. J Neurochem. 1997;68:1612–1621. doi: 10.1046/j.1471-4159.1997.68041612.x. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Towards theories of brain aging. In: Kay DW, Burrows GW, editors. Handbook of Studies on Psychiatry and Old Age. Amsterdam: Elsevier; 1984. pp. 7–30. [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno T, Mukai H, Ito A, Chang CD, Kishima K, Saito N, Tanaka C. Distinct cellular expression of calcineurin A alpha and A beta in rat brain. J Neurochem. 1992;58:1643–1651. doi: 10.1111/j.1471-4159.1992.tb10036.x. [DOI] [PubMed] [Google Scholar]
- Kuwako K, Nishimura I, Uetsuki T, Saido TC, Yoshikawa K. Activation of calpain in cultured neurons overexpressing Alzheimer amyloid precursor protein. Brain Res Mol Brain Res. 2002;107:166–175. doi: 10.1016/s0169-328x(02)00489-8. [DOI] [PubMed] [Google Scholar]
- Ladner CJ, Czech J, Maurice J, Lorens SA, Lee JM. Reduction of calcineurin enzymatic activity in Alzheimer's disease: correlation with neuropathologic changes. J Neuropathol Exp Neurol. 1996;55:924–931. doi: 10.1097/00005072-199608000-00008. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science. 1984;226:1089–1092. doi: 10.1126/science.6494926. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Q, Ladner CJ, Magnuson D, Lee JM. Selective changes of calcineurin (protein phosphatase 2B) activity in Alzheimer's disease cerebral cortex. Exp Neurol. 2001;167:158–165. doi: 10.1006/exnr.2000.7534. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- Lopes JP, Oliveira CR, Agostinho P. Neurodegeneration in an Abeta-induced model of Alzheimer's disease: the role of Cdk5. Aging Cell. 2010;9:64–77. doi: 10.1111/j.1474-9726.2009.00536.x. [DOI] [PubMed] [Google Scholar]
- Luoma JI, Zirpel L. Deafferentation-induced activation of NFAT (nuclear factor of activated T-cells) in cochlear nucleus neurons during a developmental critical period: a role for NFATc4-dependent apoptosis in the CNS. J Neurosci. 2008;28:3159–3169. doi: 10.1523/JNEUROSCI.5227-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansuy IM, Winder DG, Moallem TM, Osman M, Mayford M, Hawkins RD, Kandel ER. Inducible and reversible gene expression with the rtTA system for the study of memory. Neuron. 1998;21:257–265. doi: 10.1016/s0896-6273(00)80533-4. [DOI] [PubMed] [Google Scholar]
- Marcum JL, Mathenia JK, Chan R, Guttmann RP. Oxidation of thiol-proteases in the hippocampusof Alzheimer's disease. Biochem Biophys Res Commun. 2005;334:342–348. doi: 10.1016/j.bbrc.2005.06.089. [DOI] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Butterfield DA. Protection against amyloid beta-peptide (1-42)-induced loss of phospholipid asymmetry in synaptosomal membranes by tricyclodecan-9-xanthogenate (D609) and ferulic acid ethyl ester: implications for Alzheimer's disease. Biochim Biophys Acta. 2005;1741:140–148. doi: 10.1016/j.bbadis.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Mukerjee N, McGinnis KM, Gnegy ME, Wang KK. Caspase-mediated calcineurin activation contributes to IL-2 release during T cell activation. Biochem Biophys Res Commun. 2001;285:1192–1199. doi: 10.1006/bbrc.2001.5278. [DOI] [PubMed] [Google Scholar]
- Mukerjee N, McGinnis KM, Park YH, Gnegy ME, Wang KK. Caspase-mediated proteolytic activation of calcineurin in thapsigargin-mediated apoptosis in SH-SY5Y neuroblastoma cells. Arch Biochem Biophys. 2000;379:337–343. doi: 10.1006/abbi.2000.1889. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, St Clair DK, LeVine H, 3rd , Keller JN. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66:1136–1146. doi: 10.1097/nen.0b013e31815c5efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmrich V, Reymann KG, Strassburger M, Schoder UH, Gross G, Hahn A, Schoemaker H, Wicke K, Moller A. Inhibition of calpain prevents NMDA-induced cell death and beta-amyloid-induced synaptic dysfunction in hippocampal slice cultures. Br J Pharmacol. 2010;159:1523–1531. doi: 10.1111/j.1476-5381.2010.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. The calpains in aging and aging-related diseases. Ageing Res Rev. 2003;2:407–418. doi: 10.1016/s1568-1637(03)00029-1. [DOI] [PubMed] [Google Scholar]
- Norris CM, Blalock EM, Chen KC, Porter NM, Thibault O, Kraner SD, Landfield PW. Hippocampal 'zipper' slice studies reveal a necessary role for calcineurin in the increased activity of L-type Ca(2+) channels with aging. Neurobiol Aging. 2010;31:328–338. doi: 10.1016/j.neurobiolaging.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggersreactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, Walker Z, Kennedy A, Fox N, Rossor M, Brooks DJ. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72:56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CG, Stoffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system--too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699–723. doi: 10.1016/j.neuropharm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Perrin BJ, Huttenlocher A. Calpain. Int J Biochem Cell Biol. 2002;34:722–725. doi: 10.1016/s1357-2725(02)00009-2. [DOI] [PubMed] [Google Scholar]
- Qu J, Wang D, Grosskreutz CL. Mechanisms of retinal ganglion cell injury and defense in glaucoma. Exp Eye Res. 2010;91:48–53. doi: 10.1016/j.exer.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MV, Mohan PS, Peterhoff CM, Yang DS, Schmidt SD, Stavrides PH, Campbell J, Chen Y, Jiang Y, Paskevich PA, Cataldo AM, Haroutunian V, Nixon RA. Marked calpastatin (CAST) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J Neurosci. 2008;28:12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese LC, Zhang W, Dineley KT, Kayed R, Taglialatela G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell. 2008;7:824–835. doi: 10.1111/j.1474-9726.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronicke R, Mikhaylova M, Ronicke S, Meinhardt J, Schroder UH, Fandrich M, Reiser G, Kreutz MR, Reymann KG. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Nixon RA. Specificity of calcium-activated neutral proteinase (CANP) inhibitors for human mu CANP and mCANP. Neurochem Res. 1993;18:231–233. doi: 10.1007/BF01474689. [DOI] [PubMed] [Google Scholar]
- Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, Norris CM. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem. 2008;283:21953–21964. doi: 10.1074/jbc.M800148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- See V, Loeffler JP. Oxidative stress induces neuronal death by recruiting a protease and phosphatase-gated mechanism. J Biol Chem. 2001;276:35049–35059. doi: 10.1074/jbc.M104988200. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating anNMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda N, Han F, Moriguchi S, Fukunaga K. Constitutively active calcineurin mediates delayed neuronal death through Fas-ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J Neurochem. 2007;102:1506–1517. doi: 10.1111/j.1471-4159.2007.04600.x. [DOI] [PubMed] [Google Scholar]
- Shioda N, Moriguchi S, Shirasaki Y, Fukunaga K. Generation of constitutively active calcineurin by calpain contributes to delayed neuronal death following mouse brain ischemia. J Neurochem. 2006;98:310–320. doi: 10.1111/j.1471-4159.2006.03874.x. [DOI] [PubMed] [Google Scholar]
- Simpkins KL, Guttmann RP, Dong Y, Chen Z, Sokol S, Neumar RW, Lynch DR. Selective activation induced cleavage of the NR2B subunit by calpain. J Neurosci. 2003;23:11322–11331. doi: 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH. Dysregulationof calcium homeostasis in Alzheimer's disease. Neurochem Res. 2009;34:1824–1829. doi: 10.1007/s11064-009-9960-5. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer's disease. J Neurosci Res. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg C, Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci. 2009;29:14439–14450. doi: 10.1523/JNEUROSCI.3590-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela G, Hogan D, Zhang WR, Dineley KT. Intermediate-and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav Brain Res. 2009 doi: 10.1016/j.bbr.2008.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallant EA, Brumley LM, Wallace RW. Activation of a calmodulin-dependent phosphatase by a Ca2+-dependent protease. Biochemistry. 1988;27:2205–2211. doi: 10.1021/bi00406a059. [DOI] [PubMed] [Google Scholar]
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell. 2007;6:307–317. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A. The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell. 2007;6:267–273. doi: 10.1111/j.1474-9726.2007.00296.x. [DOI] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A, Landfield PW. Ca(2+) regulation and gene expression in normal brain aging. Trends Neurosci. 2004;27:614–620. doi: 10.1016/j.tins.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118:2796–2807. doi: 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisid T, Kosower NS, Katzav A, Chapman J, Barnoy S. Calpastatin levels affect calpain activationand calpain proteolytic activity in APP transgenic mouse model of Alzheimer's disease. Neurochem Int. 2007;51:391–397. doi: 10.1016/j.neuint.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Veeranna, Kaji T, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA. Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer's disease. Am J Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KK, Roufogalis BD, Villalobo A. Characterization of the fragmented forms of calcineurin produced by calpain I. Biochem Cell Biol. 1989;67:703–711. doi: 10.1139/o89-105. [DOI] [PubMed] [Google Scholar]
- Wei Z, Song MS, MacTavish D, Jhamandas JH, Kar S. Role of calpain and caspase in beta-amyloid-induced cell death in rat primary septal cultured neurons. Neuropharmacology. 2008;54:721–733. doi: 10.1016/j.neuropharm.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Matsui H. Calpain-calcineurin signaling in the pathogenesis of calcium-dependent disorder. Acta Med Okayama. 2007;61:123–137. doi: 10.18926/AMO/32905. [DOI] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Ren Y, Yan Z. Postsynaptic density-95 (PSD-95) and calcineurin control the sensitivity of N-methyl-D-aspartate receptors to calpain cleavage in cortical neurons. Mol Pharmacol. 2008;74:360–370. doi: 10.1124/mol.108.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, Stone DJ, Ferrer M, Townsend M, Wolfe AL, Seager MA, Kinney GG, Shughrue PJ, Ray WJ. Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem. 2010;285:7619–7632. doi: 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative Western blots for CN-Aα (A) and CN-Aβ (B) in cytosolic (C) and nuclear (N) fractions prepared from hippocampal tissue samples from two control and two MCI subjects. Note these blots were processed at a lower exposure time to clearly distinguish the full-length (60 kDa) from the 57 kDa CN-A fragment. Cytosolic and nuclear accumulation (mean ± SD) of full-length (FL) CN-Aα and CN-Aβ were measured across all subjects and no differences were observed, consistent with results from Figure 2C.
(A and B, top panels) Representative Western blots for membrane-associated CN-Aα (A) and CN-Aβ (B) in cytosolic (C) and nuclear (N) fractions prepared from hippocampal tissue samples from two control and two MCI subjects. Na+/K+-ATPase was used as a loading control. (A and B, bottom panels) Quantitation of membrane-associated full-length (FL) CN-Aα and CN-Aβ (mean ± SD) across all subjects. (C) Western blot showing membrane-associated CN-Aα (lanes 1 and 2) in comparison to CN-Aα products associated with human hippocampal nuclear and cytosolic extracts (lanes 3 and 4) and primary rat hippocampal culture (lane 5).