

Abstract
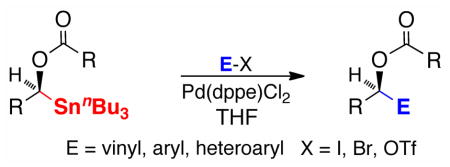
Racemic and scalemic α-(acyloxy)-tri-N-butylstannanes undergo Pd-catalyzed cross-couplings with alkenyl/aryl/heteroaryl iodides, bromides, and triflates in moderate to good yields in THF at 45 °C. Simple aryl iodides and unprotected aza-arenes, two classes of electrophiles that typically react sluggishly, are also good substrates. Cross-couplings proceed with retention of configuration at the alkenyl and stannyl-substituted stereocenters.
The Stille reaction,1 i. e., the transition metal-mediated cross-coupling of organostannanes with organic electrophiles, has achieved wide acceptance2 as an exceptionally mild and efficient method for the creation of C-C bonds, especially between sp- and/or sp2-hybridized centers.3 Not surprisingly, the apparent advantages that would accrue from expanding the traditional scope and structural confines of the Stille reaction have attracted much interest.4 In the early 1990’s, this laboratory5 and others6 explored the utility of tri-n-butylstannanes for the transfer of stereogenic carbons bearing heteroatoms and reported the stereospecific palladium/copper co-catalyzed cross-coupling of scalemic α-alkoxy- and α-aminoalkylstannanes with acid chlorides.7,8 Subsequent studies led to copper mediated cross-couplings with reactive electrophiles such as allylic and propargylic halides.9 The utility of this methodology for the construction of chiral ethers and alcohols was cogently demonstrated during asymmetric total syntheses of the anticancer agent (+)-goniofufurone10 and the potent endothelium-derived vasodilator 11,12,15-THETA.11 On the other hand, comparable unions between α-heteroatom-substituted triorganostannanes and alkenyl/aryl electrophiles were conspicuously absent12 and suitable methodology has been elusive.13 To address this methodological gap, we conducted an extensive survey of alternative reaction parameters including oxygen substituents14 and herein describe a practical, stereospecific cross-coupling capable of using a broad range of sp2-hybridized iodides/triflates/bromides (Scheme 1).
Scheme 1.
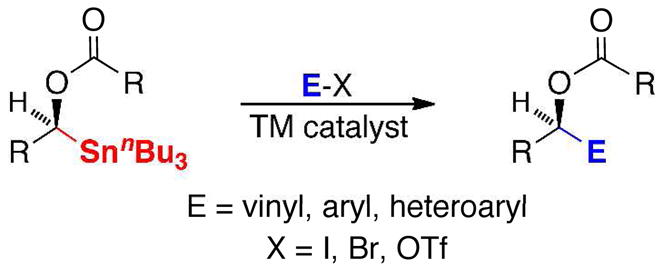
Proposed Cross-Coupling
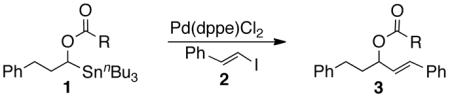
Since esters and alkenyl iodides were identified as the most promising pairing in our initial evaluations, α-(acetyloxy)stannane 1a and (E)-β-iodostyrene (2) were selected as the test system. Screening an extensive collection of transition metals salts and complexes, tested individually or in combination, revealed palladium complexes were especially efficacious, and in particular freshly recrystallized Pd(dppe)Cl2.15 The yield of adduct 3a increased proportionately with the amount of Pd(dppe)Cl2 up to 10 mol % (62%; Table 1), but thereafter neither more catalyst nor alterations in mode of addition improved the outcome.16 Yields of 3a were patently better in THF as well as the reaction rate compared to other common solvents, inter alia, toluene, DME, and DMF; there was no cross-coupling in NMP, acetone, EtOAc, and CH3CN. In contrast with the experience of others,17 sources of fluoride (LiF, TBAF, KF, AlF3, CsF) did not have a beneficial effect nor did Lewis acids [MgBr2, AlCl3, FeCl3, ZnBr2, BF3·Et2O, Sc(OTf)3].
Table 1.
Cross-Coupling of α-(Acyloxy)stannanes with (E)-β-Iodostyrenea
 | |||||||
|---|---|---|---|---|---|---|---|
| compd | R | time (h) | yield (%) | compd | R | time (h) | yield (%) |
| a |  |
12 | 62 | f | 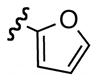 |
12 | 68 |
| b | 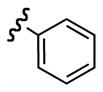 |
10 | 71 | g | 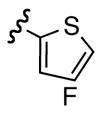 |
12 | 72 |
| c | 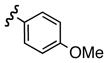 |
10 | 72 | h | 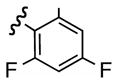 |
10 | 72 |
| d | 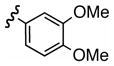 |
12 | 70 | i | 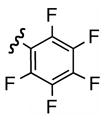 |
8 | 81 |
| e | 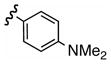 |
10 | 68 | j | 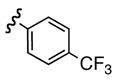 |
8 | 81 |
Reaction conditions: α-(acyloxy)stannane 1 (1 equiv), 2 (1.5 equiv), Pd(dppe)Cl2 (10 mol %), THF, 45 °C, 8–12 h.
Replacement of the acetate of 3a with a benzoate, i.e., 3b, resulted in a modest improvement in yield. Electron donating substituents (3c–d) had little influence above that of the parent aromatic 3b and neither did electron-rich heterocycles like 2-carboxyfuran 3f and 2-carboxythiophene 3g despite the potential for intramolecular coordination in the transition state. In contrast, electron-withdrawing substituents (3h–j) offered an additional improvement in adduct yield and a modest rate enhancement. Because of its better handling characteristics and lower cost than the polyfluorinated aryls 3h,i, p-trifluoromethylbenzoate 3j was used in subsequent optimization studies.
Having standardized the reaction conditions, we next explored the scope of the cross-coupling of p-trifluoromethylbenzoate (TFMB) 1j with a panel of representative sp2-hybridized iodides, triflates, and bromides (Table 2). 3-Iodopropenoate 4, despite its proclivity towards loss of HI and/or isomerization, smoothly cross-coupled using Pd(dppe)Cl2 (catalyst A) to furnish 5 (entry 1) in excellent yield without complication or loss of the Z-configuration at the alkenyl center (>98% by 1H/13C NMR), thus precluding a conjugate addition mechanism. Under the same reaction conditions, 2-iodo-2-cyclohex-2-enone (6), (Z)-iodostyrene (9), (E)-triflate 12, and unconjugated alkenyl iodide 15 led to adducts 8 (entry 2), 11 (entry 3), 14 (entry 4), and 16 (entry 5), respectively, although the reaction rates were somewhat slower than for 4. TFMB-benzyl alcohols 19 (entry 6), 22 (entry 7), and 24 (entry 8) were obtained analogously from aryl/heteroaryl iodides 17, 20, and 23, respectively, in useful yields.
Table 2.
Scope of Cross-Coupling using α-(TFMB)stannane 1ja
| entry | electrophile | catalystb | time (h) | adduct | yield (%) |
|---|---|---|---|---|---|
| 1 |
4 |
A | 6 |
5 |
86 |
| 2 |
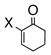 6: X = I 7: X = Br |
A B |
10 12 |
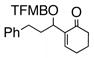 8 |
61 61 |
| 3 |
9: X = I 10: X = Br |
A B |
10 12 |
11 |
61 52 |
| 4 |
12: X = OTf 13: X = Br |
A | 10 12 |
14 |
60 72 |
| 5 |
15 |
A | 10 |
16 |
66 |
| 6 |
17: X = I 18: X = Br |
A B |
8 12 |
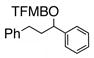 19 |
61 64 |
| 7 |
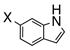 20: X = I 21: X = Br |
A B |
10 12 |
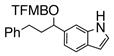 22 |
65 62 |
| 8 |
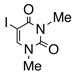 23 |
A | 10 |
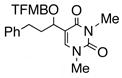 24 |
62 |
Reaction conditions: 1j (1 equiv), electrophile (1.5 equiv), Pd catalyst (10 mol %), THF, 45 °C, 8–12 h.
Catalyst A = recrystallized Pd(dppe)Cl2, catalyst B = chloro(2-di-tert-butylphosphino-2′,4′,6′-tri-iso-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl] palladium(II).
Comparable couplings of alkenyl and aryl bromides using Pd(dppe)Cl2 proved more challenging and only poor yields of adduct could be obtained. Fortunately, we discovered that the Buchwald pallacyclic catalyst18 chloro(2-di-tert-butylphosphino-2′,4′,6′-tri-iso-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl] palladium(II) (t-BuXphos) restored efficacy (Table 2). While reaction times were modestly increased for bromides 10, 13, 18, and 21, adduct yields were generally comparable or even slightly better than those seen with alkenyl iodides/triflates. The exception was (Z)-bromostyrene (10) which likely reflects this electrophile’s greater steric hindrance.
To ascertain the stereochemistry of the C-C bond formation, enantioenriched benzoate 25 (98% ee), readily prepared from the corresponding aldehyde via the one-pot procedure of He and Falck,19 was added to phenyl iodide using the standard conditions (Figure 1). Saponification of the resultant benzoate 26 gave rise to the known20 1,3-diphenylpropan-1(R)-ol (98% ee via chiral HPLC), thus demonstrating complete retention of configuration at the stannyl-substituted stereogenic center. This is consistent with prior experience using other classes of electrophiles, but opposite to the inversion of configuration observed by Kells and Chong8 using scalemic α-(sulfonamido)organostannanes and Pd/Cu cocatalysis. Analogous coupling of the diastereomeric glyceryl stannane 2721 to give 28 (Figure 1) was also instructive and revealed the adjacent stereocenter had no apparent influence on the outcome.
Figure 1.
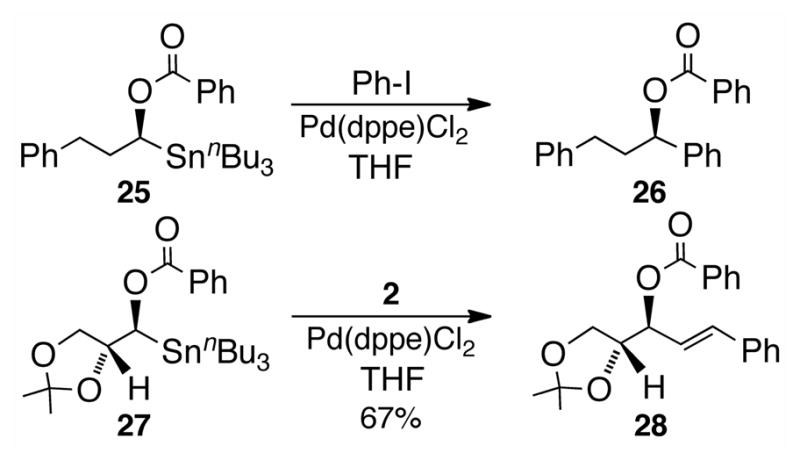
Cross-Coupling of Enantioenriched Stannanes
In conclusion, we have demonstrated the stereospecific Pd-mediated cross-coupling TFMB-protected α-hydroxystannanes with alkenyl/aryl/heteroaryl iodides/triflates/bromides including secondary cycloalkenyl and unactivated aryl iodides in THF under mild conditions.22 When combined with the recently introduced methodology for the synthesis of racemic and scalemic α-hydroxystannanes,19 we anticipate the foregoing methodology will find wide utility in the synthesis of heteroatom-substituted stereogenic centers.
Supplementary Material
Acknowledgments
Financial support provided by the Robert A. Welch Foundation (GL625910) and NIH (GM31278, DK38226). Dr. E. R. Fogel (CRO Labs, Inc.) is thanked for useful insights.
Footnotes
Supporting Information Available: Synthetic procedures, analytical data, chiral HPLC chromatograms, and 1H/13C spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Review: Mitchell TN. In: Metal-Catalyzed Cross-Coupling Reactions. 2. Chapter 3 de Meijere A, Diederich F, editors. Vol. 1. Wiley-VCH Verlag GmbH & Co; Weinheim: 2004.
- 2.Recent applications in natural products total synthesis: Masters KS, Flynn BL. Org Biomol Chem. 2010;8:1290–1292. doi: 10.1039/b924542a.Tang B, Bray CD, Pattenden G, Rogers J. Tetrahedron. 2010;66:2492–2500.Candy M, Audran G, Bienayme H, Bressy C, Pons JM. J Org Chem. 2010;75:1354–1359. doi: 10.1021/jo902582w.Amans D, Bareille L, Bellosta V, Cossy J. J Org Chem. 2009;74:7665–7674. doi: 10.1021/jo900945x.
- 3.Pascual S, Echavarren AM. In: Tin Chemistry. Davies AG, editor. John Wiley & Sons Ltd; Chichester: 2008. pp. 579–606. [Google Scholar]
- 4.For example: Jensen MS, Yang C, Hsiao Y, Rivera N, Wells KM, Chung JYL, Yasuda N, Hughes DL, Reider PJ. Org Lett. 2000;2:1081–1084. doi: 10.1021/ol005641d.Jarosz S. Tetrahedron Lett. 1996;37:3063–3066.
- 5.(a) Bhatt RK, Shin DS, Falck JR, Mioskowski C. Tetrahedron Lett. 1992;33:4885–4888. [Google Scholar]; (b) Belosludtsev YY, Bhatt RK, Falck JR. Tetrahedron Lett. 1995;36:5881–5882. [Google Scholar]; (c) Falck JR, Bhatt RK, Reddy KM, Ye J. Synlett. 1997:481–482. [Google Scholar]
- 6.Linderman RJ, Graves DM, Kwochka WR, Ghannam AF, Anklekar TV. J Am Chem Soc. 1990;112:7438–7439. [Google Scholar]
- 7.Ye J, Bhatt RK, Falck JR. J Am Chem Soc. 1994;116:1–5. [Google Scholar]
- 8.In 2004, α-sulfonamidoorganostannanes were introduced and shown to be the highly efficacious: Kells KW, Chong JM. J Am Chem Soc. 2004;126:15666–15667. doi: 10.1021/ja044354s.
- 9.Falck JR, Bhatt RK, Ye J. J Am Chem Soc. 1995;117:5973–5982. [Google Scholar]
- 10.Ye J, Bhatt RK, Falck JR. Tetrahedron Lett. 1993;34:8007–8010. [Google Scholar]
- 11.Falck JR, Barma D, Mohapatra S, Bandyopadhyay A, Reddy KM, Qi J, Campbell W. Bioorg Med Chem Lett. 2004;14:4987–4990. doi: 10.1016/j.bmcl.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 12.A catalyst-free cross-coupling of α-sulfur-substituted alkyltriorganostannanes with acid chlorides has been described: Kagoshima H, Takahashi N. Chem Lett. 2007;36:14–15.
- 13.The copper(I) thiophene-2-carboxylate catalyzed cross-coupling of α-(thiocarbamoyl)tri-n-butylstannanes with sp2-electrophiles was subsequently found to be accompanied by a facile Newman-Kwart O → S rearrangement that in many cases generated the corresponding thiolcarbamate as the major product: Falck JR, Patel PK, Bandyopadhyay A. J Am Chem Soc. 2007;129:790–793. doi: 10.1021/ja064948q. Correction: J. Am. Chem. Soc. 2008, 130, 2372.
- 14.Alternative sulfur-containing derivatives (e.g., thioesters, thiocarbonates, thiooxamides, thiophosphates were evaluated, but it was not possible to completely suppress the rearrangement and still achieve acceptable yields of cross-coupled adduct (see Supporting Information).
- 15.Catalyst quality varied widely with the source as revealed in cross-coupling yield of 3j, e.g., Aldrich (brown color) mp 222 °C, 10%; TCI (light brown) mp 255 °C, 40%; Strem (pale yellow) mp 285 °C, 68%; Strem (recrystallized from EtOH, pale yellow) mp 298 °C, 78%; Strem (recrystallized from DMF/Et2O, pale yellow) mp 301 °C, 81%; lit. mp >300, pale yellow: Paquette LA. Encyclopedia of Reagents for Organic Synthesis. Vol. 3. John Wiley & Sons; New York, NY: 1985. pp. 1675–1676.
- 16.Modifications evaluated included reversing the order of reagent addition and slow addition of reagents via syringe pump.
- 17.Mee SPH, Lee V, Baldwin JE. Chem Eur J. 2005;11:3294–3308. doi: 10.1002/chem.200401162. [DOI] [PubMed] [Google Scholar]
- 18.Biscoe MR, Fors BP, Buchwald SL. J Am Chem Soc. 2008;130:6686–6687. doi: 10.1021/ja801137k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He A, Falck JR. Angew Chem Int Ed. 2008;47:6586–6589. doi: 10.1002/anie.200802313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Node M, Nishide K, Shigeta Y, Shiraki H, Obata K. J Am Chem Soc. 2000;122:1927–1936. [Google Scholar]; (b) Anthony R, Stoner HEJ, Peterson MJ, Grover VK. J Org Chem. 2003;68:8092–8096. doi: 10.1021/jo0301907. [DOI] [PubMed] [Google Scholar]
- 21.Mohapatra S, Bandyopadhyay A, Barma DK, Capdevila JH, Falck JR. Org Lett. 2003;5:4759–4762. doi: 10.1021/ol035458v. [DOI] [PubMed] [Google Scholar]
- 22.Cross-Coupling Procedure: A Schlenk tube is charged with α-(acyloxy)-tri-n-butylstannane (0.16 mmol) and recrystallized Pd(dppe)Cl2 (0.016 mmol, 10 mol %), when using an alkenyl/aryl/heteroaryl iodide/triflate, or chloro(2-di-tert-butylphosphino-2′,4′,6′-tri-iso-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl] palladium(II) (t-BuXphos), when using an aryl/heteroaryl/alkenyl bromide, and then degassed via four alternating high vacuum-argon cycles. After dissolving in anhydrous THF (3 mL) under an argon atmosphere, a solution of electrophile (0.24 mmol) in THF (2 mL) is added via syringe and the mixture is warmed to 45 °C. After 6–12 h, the reaction mixture is diluted with Et2O (10 mL) and filtered through a short pad of neutral alumina. The filter cake is washed with Et2O (2 × 20 mL) and the combined organic filtrates are washed with water, brine, and evaporated in vacuo. Chromatographic purification (SiO2) of the residue furnishes the cross-coupled adduct in the indicated yield (Tables 1 and 2).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.